
Identification of Kaurane-Type Diterpenes as Inhibitors of Leishmania Pteridine Reductase I

 , ,
, ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. A Combined Ligand-/Structure-Based Virtual Screening Approach Using LmPTR1
2.1.1. Ligand-Based VS
2.1.2. Structure-Based VS
2.1.3. Consensus Analysis of the Two VS Approaches
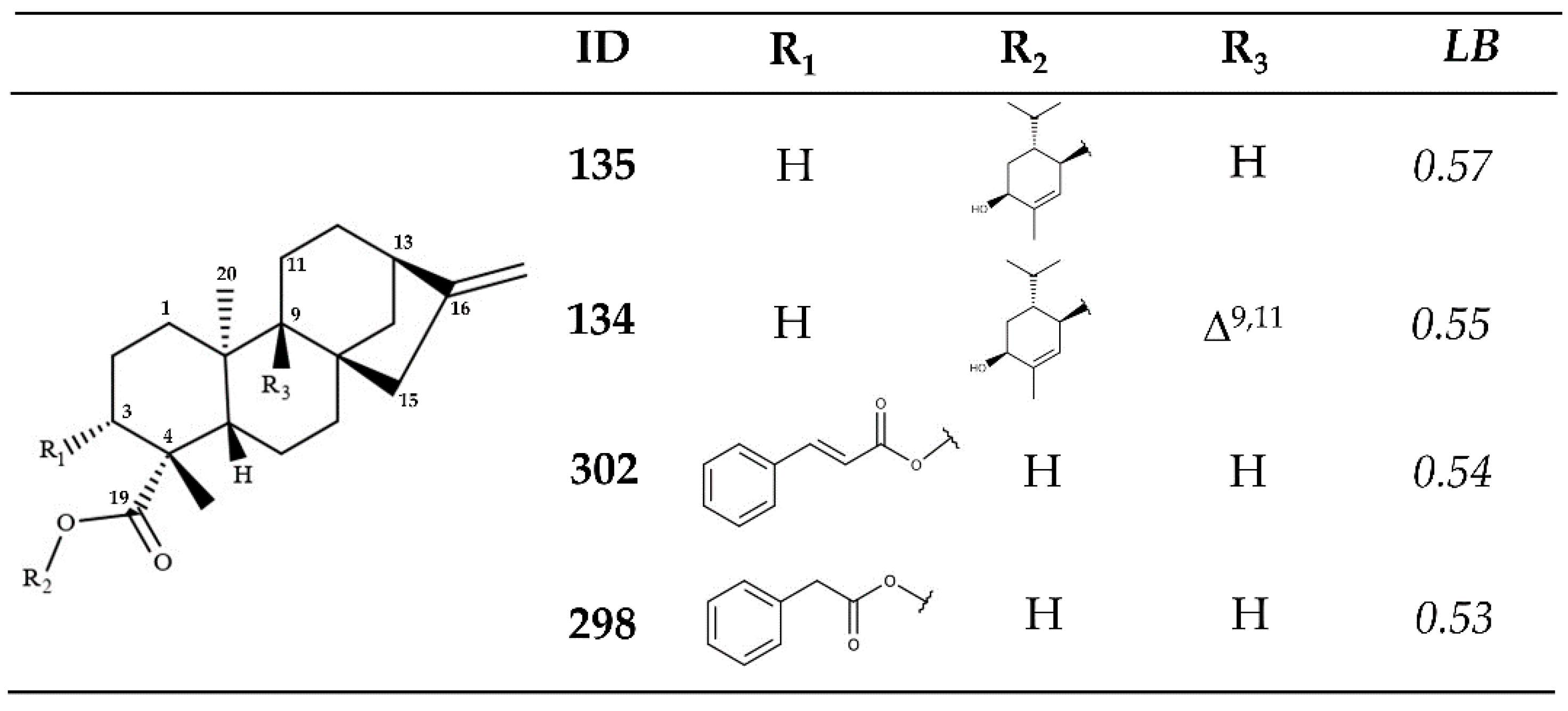
2.2. In Vitro Enzymatic Activity Inhibition for VS-Selected Kauranes against Lmptr1
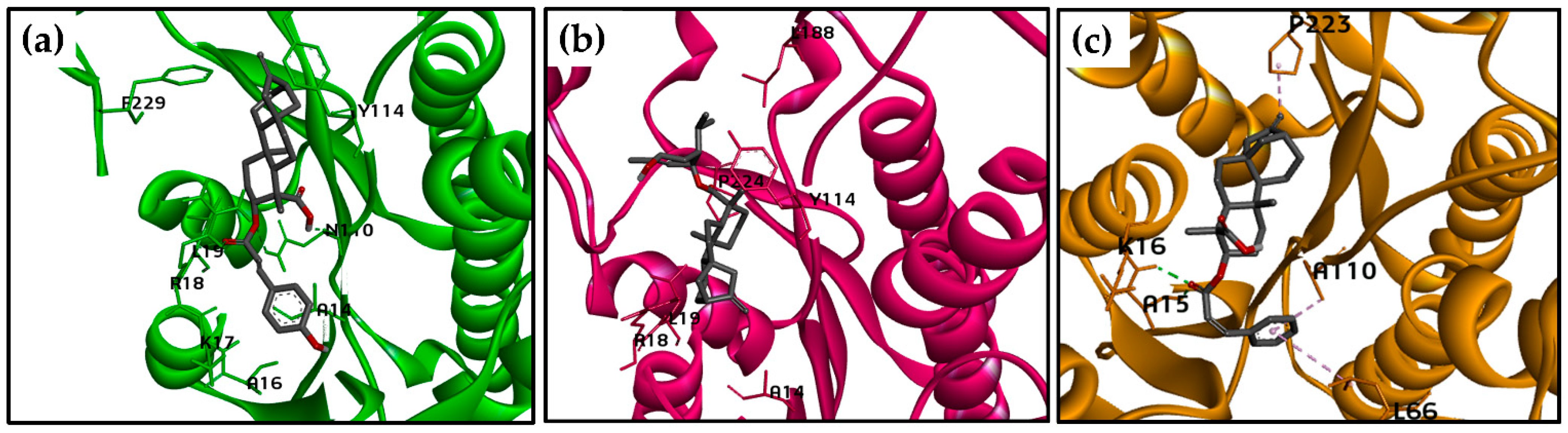
2.3. Molecular Docking Calculations for the Kaurane Dataset Using Hybrid Models of La, Lb, and Lpptr1
2.3.1. Hybrid Models of La, Lb, and LpPTR1
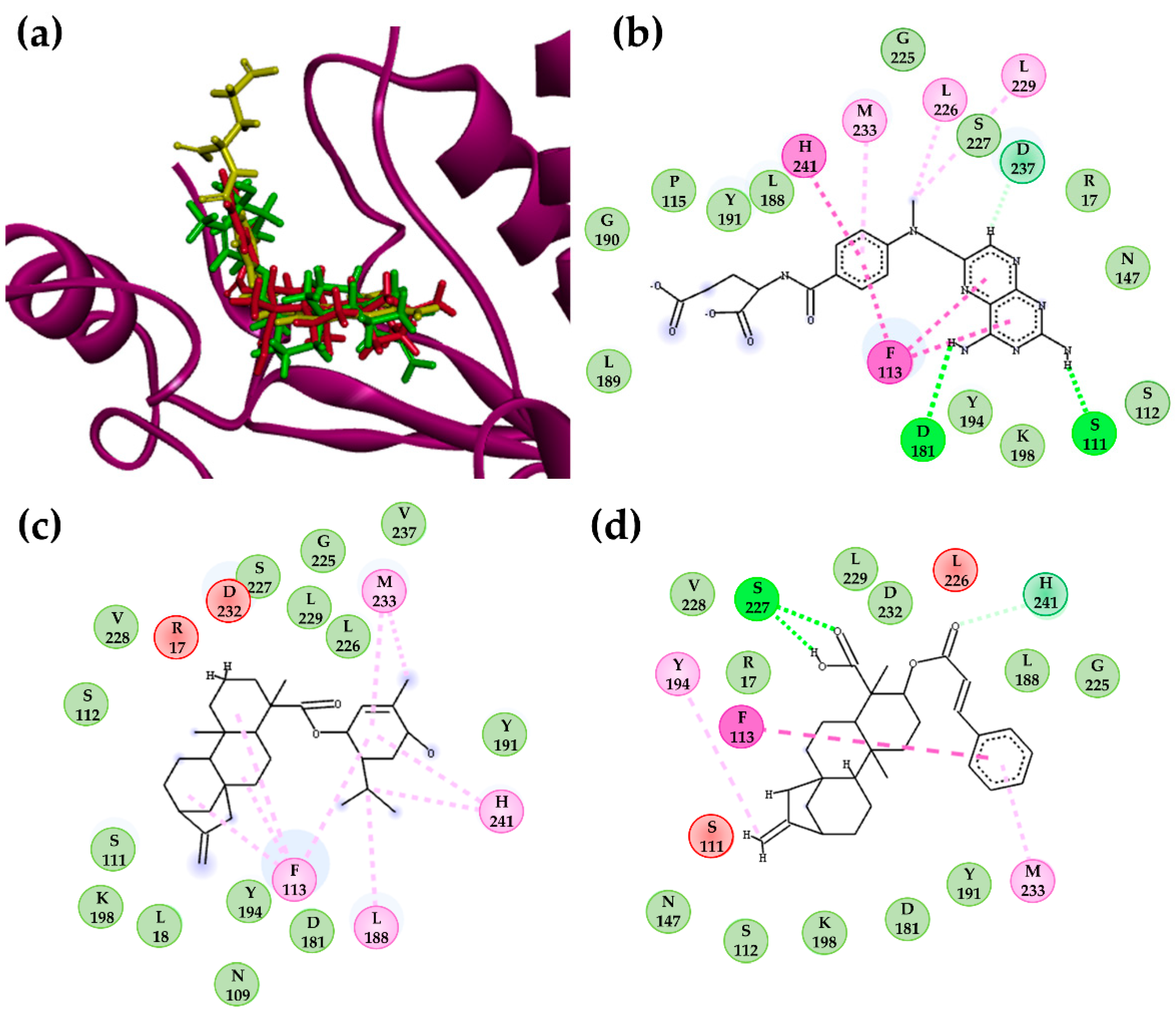
2.3.2. Molecular Docking Calculations for Kauranes Dataset
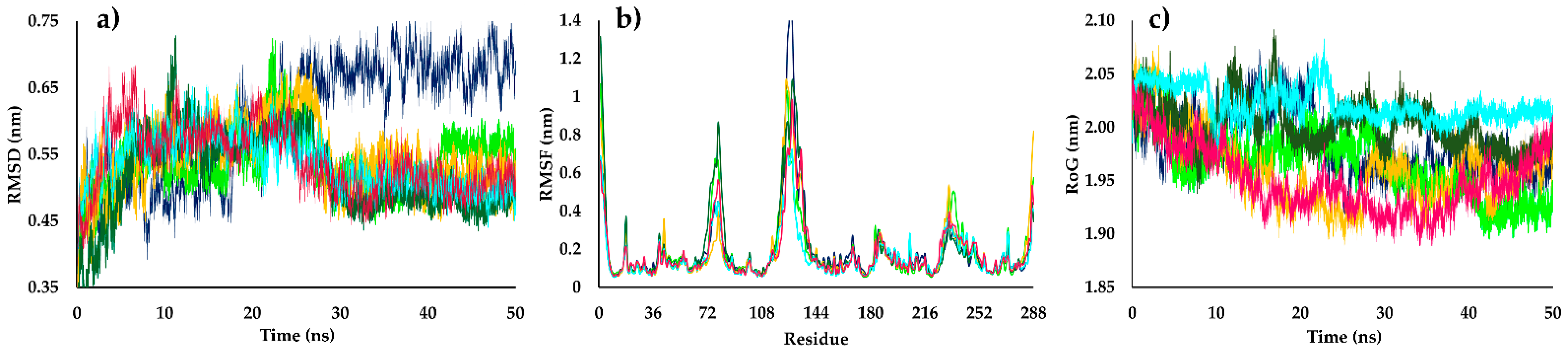
2.4. Molecular Dynamics Simulations
2.5. Prediction of ADMET Properties
3. Materials and Methods
3.1. Database
3.2. Volsurf+ Descriptors
3.3. RF Models
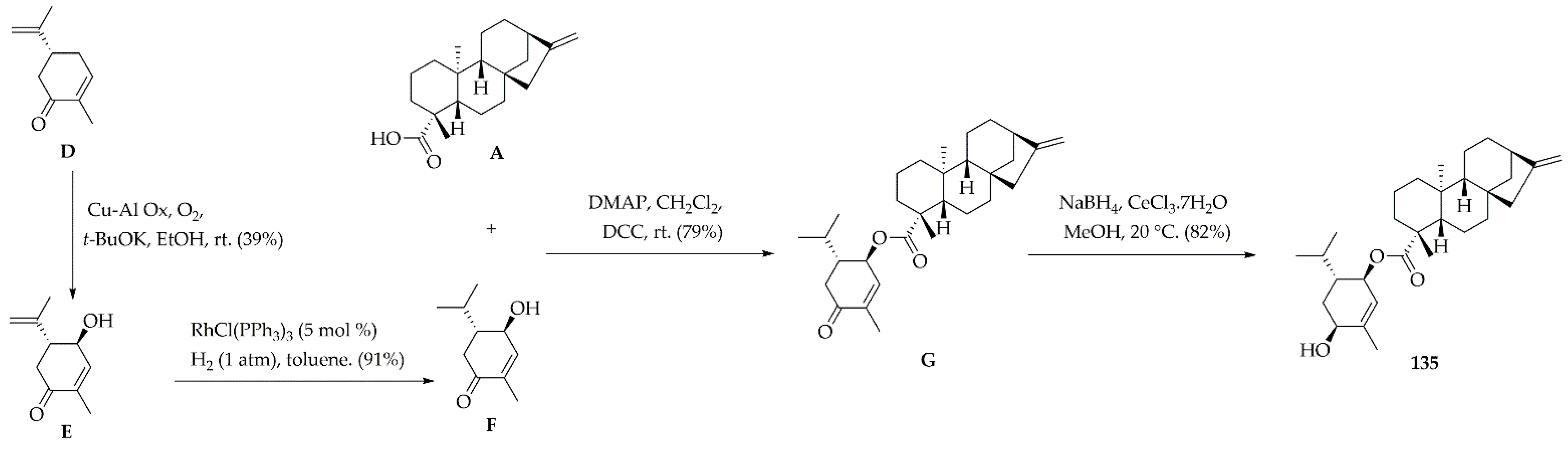
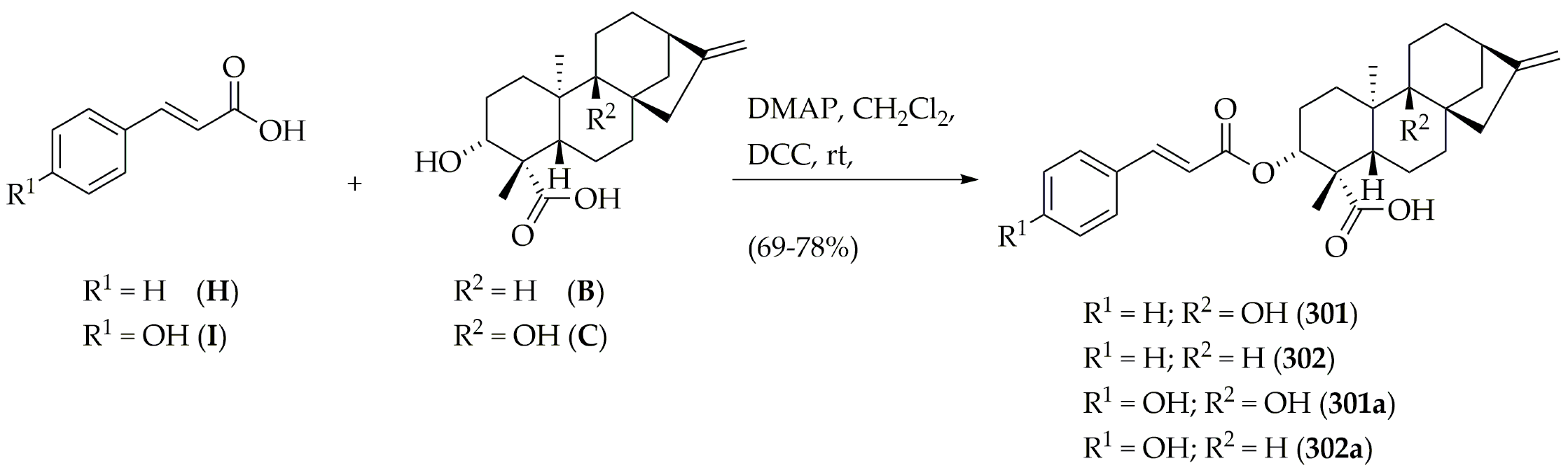
3.4. Synthesis of VS-Selected Diterpenes
3.4.1. Materials and Reagents
3.4.2. Isolation of Compounds A–C
3.4.3. Synthesis of 2β-Hydrohy-menth-6(1)-en-5β-yl ent-kaurenoate (135)
Synthesis of 5β-Hydroxy-(R)-carvone (E)
Synthesis of 2-Oxo-menth-6-en-5β-ol (F)
Synthesis of 2-Oxo-menth-6-en-5β-yl ent-kaurenoate (G)
Synthesis of 2β-Hydroxy-menth-6-en-5β-yl ent-kaurenoate (135)
3.4.4. Synthesis of 3α-Cinnamoyloxy-9β-hydroxy-ent-kaur-16-en-19-oic Acid (301), 3α-cinnamoyloxy-ent-kaur-16-en-19-oic Acid (302), 3α-p-coumaroyloxy-9β-hydroxy-ent-kaur-16-en-19-oic Acid (301a), 3α-p-coumaroyloxy-ent-kaur-16-en-19-oic Acid (302a)
3.5. LmPTR1 Enzyme Inhibition Assay
3.6. Hybrid Models of L. braziliensis, L. panamensis and L. amazonensis
3.7. Molecular Docking Calculations
3.8. Molecular Dynamics Simulations
3.9. Prediction of ADMET Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Utzinger, J.; Becker, S.L.; Knopp, S.; Blum, J.; Neumayr, A.L.; Keiser, J.; Hatz, C.F. Neglected tropical diseases: Diagnosis, clinical management, treatment and control. Swiss Med. Wkly. 2012, 22, 142. [Google Scholar] [CrossRef]
- López-Arencibia, A.; Bethencourt-Estrella, C.J.; Freijo, M.B.; Reyes-Batlle, M.; Sifaoui, I.; San Nicolás-Hernández, D.; McNaughton-Smith, G.; Lorenzo-Morales, J.; Abad-Grillo, T.; Piñero, J.E. New phenalenone analogues with improved activity against Leishmania species. Biomed. Pharmacother. 2020, 132, 110814. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Leishmaniasis. Available online: https://www.who.int/health-topics/leishmaniasis (accessed on 11 July 2020).
- Herrera Acevedo, C.; Scotti, L.; Feitosa Alves, M.; Formiga Melo Diniz, M.D.F.; Scotti, M.T. Computer-aided drug design using sesquiterpene lactones as sources of new structures with potential activity against infectious neglected diseases. Molecules 2017, 22, 79. [Google Scholar] [CrossRef] [PubMed]
- De Brito, R.C.F.; de Oliveira Aguiar-Soares, R.D.; de Oliveira Cardoso, J.M.; Coura-Vital, W.; Roatt, B.M.; Reis, A.B. Recent advances and new strategies in Leishmaniasis diagnosis. Appl. Microbiol. Biot. 2020, 104, 1–12. [Google Scholar] [CrossRef]
- Akilov, O.E.; Khachemoune, A.; Hasan, T. Clinical manifestations and classification of Old World cutaneous leishmaniasis. Int. J. Dermatol. 2007, 46, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Ghatee, M.A.; Taylor, W.R.; Karamian, M. The geographical distribution of cutaneous leishmaniasis causative agents in Iran and its neighboring countries, a review. Front. Public Health 2020, 8, 11. [Google Scholar] [CrossRef]
- Paz, C.; Samake, S.; Anderson, J.M.; Faye, O.; Traore, P.; Tall, K.; Cisse, M.; Keita, S.; Valenzuela, J.G.; Doumbia, S. Leishmania major, the predominant Leishmania species responsible for cutaneous leishmaniasis in Mali. Am. J. Trop. Med. Hyg. 2013, 8, 583–585. [Google Scholar]
- Vojtkova, B.; Spitzova, T.; Votypka, J.; Lestinova, T.; Kominkova, I.; Hajkova, M.; Santos-Mateus, D.; Miles, M.A.; Volf, P.; Sadlova, J. Central Asian rodents as model animals for Leishmania major and Leishmania donovani research. Microorganisms 2020, 8, 1440. [Google Scholar] [CrossRef]
- Sánchez-Suárez, J.; Bernal, F.A.; Coy-Barrera, E. Colombian contributions fighting Leishmaniasis: A systematic review on antileishmanials combined with chemoinformatics analysis. Molecules 2020, 25, 5704. [Google Scholar] [CrossRef]
- Anversa, L.; Tiburcio, M.G.S.; Richini-Pereira, V.B.; Ramirez, L.E. Human leishmaniasis in Brazil: A general review. Rev. Assoc. Med. Bras. 2018, 64, 281–289. [Google Scholar] [CrossRef]
- Gervazoni, L.F.O.; Barcellos, G.B.; Ferreira-Paes, T.; Almeida-Amaral, E.E. Use of natural products in Leishmaniasis chemotherapy: An overview. Front. Chem. 2020, 8, 1031. [Google Scholar] [CrossRef]
- Rodrigues, B.C.; Ferreira, M.F.; Barroso, D.H.; da Motta, J.O.C.; de Paula, C.D.R.; Porto, C.; Martins, S.S.; Gomes, C.M.; Sampaio, R.N.R. A retrospective cohort study of the effectiveness and adverse events of intralesional pentavalent antimonials in the treatment of cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2020, 14, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, C.H.; Scotti, L.; Scotti, M.T. In silico studies designed to select sesquiterpene lactones with potential antichagasic activity from an in-house asteraceae database. ChemMedChem 2018, 13, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Varela, M.T.; Fernandes, J.P.S. Natural products: Key prototypes to drug discovery against neglected diseases caused by trypanosomatids. Curr. Med. Chem. 2020, 27, 2133–2146. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Herrera-Acevedo, C.; Perdomo-Madrigal, C.; Muratov, E.N.; Scotti, L.; Scotti, M.T. Discovery of alternative chemotherapy options for Leishmaniasis via computational studies of asteraceae. ChemMedChem 2020, 16, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.T.; Herrera-Acevedo, C.; Oliveira, T.B.; Costa, R.P.O.; Santos, S.Y.K.d.O.; Rodrigues, R.P.; Scotti, L.; Da-Costa, F.B. SistematX, an online web-based cheminformatics tool for data management of secondary metabolites. Molecules 2018, 23, 103. [Google Scholar] [CrossRef] [PubMed]
- Bernal, F.A.; Coy-Barrera, E. In-silico analyses of sesquiterpene-related compounds on selected Leishmania enzyme-based targets. Molecules 2014, 19, 5550–5569. [Google Scholar] [CrossRef]
- Herrera-Acevedo, C.; Maia, M.D.S.; Cavalcanti, É.B.V.S.; Coy-Barrera, E.; Scotti, L.; Scotti, M.T. Selection of antileishmanial sesquiterpene lactones from SistematX database using a combined ligand-/structure-based virtual screening approach. Mol. Divers. 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Melo, T.S.; Gattass, C.R.; Soares, D.C.; Cunha, M.R.; Ferreira, C.; Tavares, M.T.; Saraiva, E.; Parise-Filho, R.; Braden, H.; Delorenzi, J.C. Oleanolic acid (OA) as an antileishmanial agent: Biological evaluation and in silico mechanistic insights. Parasitol. Int. 2016, 65, 227–237. [Google Scholar] [CrossRef]
- Shah, S.M.; Ullah, F.; Ayaz, M.; Sadiq, A.; Hussain, S.; Shah, S.A.A.; Nadhman, A. β-Sitosterol from Ifloga spicata (Forssk.) Sch. Bip. as potential anti-leishmanial agent against leishmania tropica: Docking and molecular insights. Steroids 2019, 148, 56–62. [Google Scholar] [CrossRef]
- García, P.A.; De Oliveira, A.B.; Batista, R. Occurrence, biological activities and synthesis of kaurane diterpenes and their glycosides. Molecules 2007, 12, 455–483. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, M.; Xia, Y.-X.; Liang, Z.-M.; Tsang, S.W.; Zhang, H.-J. Mechanistic pathways and molecular targets of plant-derived anticancer ent-kaurane diterpenes. Biomolecules 2020, 10, 144. [Google Scholar] [CrossRef]
- Duarte, N.; Ramalhete, C.; Lourenço, L. Plant Terpenoids as Lead Compounds against Malaria and Leishmaniasis. Stud. Nat. Prod. Chem. 2019, 62, 243–306. [Google Scholar]
- Seaman, F.; Bohlmann, F.; Zdero, C.; Mabry, T.J. Diterpenes of Flowering Plants: Compositae (Asteraceae); Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Nogueira, M.S.; Da Costa, F.B.; Brun, R.; Kaiser, M.; Schmidt, T.J. Ent-Pimarane and ent-kaurane diterpenes from Aldama discolor (Asteraceae) and their antiprotozoal activity. Molecules 2016, 21, 1237. [Google Scholar] [CrossRef] [PubMed]
- Orduz Díaz, L.; Bernal, F.; Coy Barrera, E. Kaurane-related diterpenes as Leishmania pteridine redutase inhibitors: An in-silico study. Revista Facultad de Ciencias Básicas 2013, 9, 142–153. [Google Scholar] [CrossRef]
- Nichol, C.A.; Smith, G.K.; Duch, D.S. Biosynthesis and metabolism of tetrahydrobiopterin and molybdopterin. Annu. Rev. Bichem. 1985, 54, 729–764. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, G.; Crivori, P.; Carrupt, P.A.; Testa, B. Molecular fields in quantitative structure–permeation relationships: The VolSurf approach. J. Mol. Struct. 2000, 503, 17–30. [Google Scholar] [CrossRef]
- Cruciani, G.; Pastor, M.; Guba, W. VolSurf: A new tool for the pharmacokinetic optimization of lead compounds. Eur. J. Pharm. Sci.s 2000, 11, S29–S39. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME-the Konstanz information miner: Version 2.0 and beyond. SIGKDD Explor. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Fourches, D.; Pu, D.; Tassa, C.; Weissleder, R.; Shaw, S.Y.; Mumper, R.J.; Tropsha, A. Quantitative nanostructure−activity relationship modeling. ACS Nano 2010, 4, 5703–5712. [Google Scholar] [CrossRef]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Cai, C.; Zhang, Y.; Yang, D.; Hao, X.; Li, S. Two new kaurane-type diterpenoids from Wedelia chinensis (Osbeck.) Merr. Nat. Prod. Res. 2017, 31, 2531–2536. [Google Scholar] [CrossRef]
- Li, S.-F.; Ding, J.-Y.; Li, Y.-T.; Hao, X.-J.; Li, S.-L. Antimicrobial diterpenoids of Wedelia trilobata (L.) Hitchc. Molecules 2016, 21, 457. [Google Scholar] [CrossRef]
- Gourley, D.G.; Schüttelkopf, A.W.; Leonard, G.A.; Luba, J.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat. Struct. Bio. 2001, 8, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Gobu, F.-R.; Chen, J.-J.; Zeng, J.; Wei, W.-J.; Wang, W.-F.; Lin, C.-J.; Gao, K. Isolation, structure elucidition, and immunosuppressive activity of diterpenoids from Ligularia fischeri. J. Nat. Prod. 2017, 80, 2263–2268. [Google Scholar] [CrossRef]
- Moreira, I.C.; Roque, N.F.; Vilegas, W.; Zalewski, C.A.; Lago, J.H.G.; Funasaki, M. Genus Xylopia (Annonaceae): Chemical and biological aspects. Chem. Biodiverse. 2013, 10, 1921–1943. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.M.; Costa, E.V.; de Lima Nogueira, P.C.; de Souza Moraes, V.R.; de Holanda Cavalcanti, S.C.; Salvador, M.J.; Ribeiro, L.H.G.; Gadelha, F.R.; Barison, A.; Ferreira, A.G. Ent-kaurane diterpenoids and other constituents from the stem of Xylopia laevigata (Annonaceae). Quím. Nova 2012, 35, 1570–1576. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Hung, Y.-C.; Chang, F.-R.; Cosentino, M.; Wang, H.-K.; Lee, K.-H. Identification of ent-16β, 17-dihydroxykauran-19-oic acid as an anti-HIV principle and isolation of the new diterpenoids annosquamosins A and B from Annona squamosa. J. Nat. Prod. 1996, 59, 635–637. [Google Scholar] [CrossRef]
- Murakami, T.; Iida, H.; Tanaka, N.; Saiki, Y.; Chen, C.; Iitaka, Y. Chemische und chemotaxonomische Untersuchungen von Filices. XXXIII. Chemische Untersuchungen der Inhaltsstoffe von Pteris longipes DON. Chem. Pharm. Bull. 1981, 29, 657–662. [Google Scholar] [CrossRef]
- Zhang, P.; Li, Y.; Yan, Z.; Gong, J.; Yang, Z. Asymmetric total synthesis of (−)-pavidolide B via a thiyl-radical-mediated [3 + 2] annulation reaction. J. Org. Chem. 2019, 84, 15958–15971. [Google Scholar] [CrossRef]
- Neises, B.; Steglich, W. Simple method for the esterification of carboxylic acids. Angew. Chem. Int. Ed. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Šťastná, E.; Černý, I.; Pouzar, V.; Chodounská, H. Stereoselectivity of sodium borohydride reduction of saturated steroidal ketones utilizing conditions of Luche reduction. Steroids 2010, 75, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Prusoff, W.H. the concentration of inhibitor which causes 50 percent inhibition (I) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Jeronimo, S.M.B.; De Queiroz Sousa, A.; Pearson, R.D. Leishmaniasis. In Tropical Infectious Diseases, 2nd ed.; Guerrant, R.L., Walker, D.H., Weller, P.F., Eds.; Churchill Livingstone: Philadelphia, PL, USA, 2006; Volume 94, pp. 1095–1113. [Google Scholar]
- De Souza Lima, B.S.; Esteves, B.B.; Fialho-Júnior, L.C.; de Oliveira Mendes, T.A.; da Fonseca Pires, S.; Chapeourouge, A.; Perales, J.; de Andrade, H.M. Study of the differentially abundant proteins among Leishmania amazonensis, L. braziliensis, and L. infantum. PLoS ONE 2020, 15, e0240612. [Google Scholar]
- Krieger, E.; Vriend, G. YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall Iii, W.B.; De Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Krieger, E.; Hooft, R.W.W.; Nabuurs, S.; Vriend, G. PDBFinderII—A Database for Protein Structure Analysis and Prediction. Submitted: 2004. Available online: http://swift.cmbi.ru.nl/gv/pdbfinder/ (accessed on 9 March 2021).
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Almario, J.; Hernández, C.A.; Ovalle-Bracho, C. Geographical distribution of Leishmania species in Colombia, 1985–2017. Biomédica 2019, 39, 278–290. [Google Scholar] [CrossRef]
- Belli, A.A.; Miles, M.A.; Kelly, J.M. A putative Leishmania panamensis/Leishmania braziliensis hybrid is a causative agent of human cutaneous leishmaniasis in Nicaragua. Parasitology 1994, 109, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Rozo-Lugo, C.; Cuca-Suárez, L.E.; Schmidt, T.J.; Coy-Barrera, E. Tetrahydrobenzofuran-6 (2 H)-one neolignans from Ocotea heterochroma: Their platelet activating factor (PAF) antagonistic activity and in silico insights into the PAF receptor binding mode. J. Nat. Prod. 2018, 81, 1968–1975. [Google Scholar] [CrossRef]
- Crivori, P.; Cruciani, G.; Carrupt, P.-A.; Testa, B. Predicting blood− brain barrier permeation from three-dimensional molecular structure. J. Med. Chem. 2000, 43, 2204–2216. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M. Design, synthesis and ADMET prediction of bis-benzimidazole as anticancer agent. Bioorg. Chem. 2020, 96, 103576. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Xu, L.; Li, D.; Hou, T. Recent developments in computational prediction of HERG blockage. Curr. Top. Med. Chem. 2013, 13, 1317–1326. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Dubovskaja, V.I.; Rudik, A.V.; Pogodin, P.V.; Druzhilovskiy, D.S.; Gloriozova, T.A.; Filimonov, D.A.; Sastry, N.G.; Poroikov, V.V. CLC-Pred: A freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLoS ONE 2018, 13, e0191838. [Google Scholar] [CrossRef]
- Ji, C.; Svensson, F.; Zoufir, A.; Bender, A. eMolTox: Prediction of molecular toxicity with confidence. Bioinformatics 2018, 34, 2508–2509. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef]
- Fourches, D.; Muratov, E.; Tropsha, A. Curation of chemogenomics data. Nat. Chem. Biol. 2015, 11, 535. [Google Scholar] [CrossRef]
- Muratov, E.N.; Varlamova, E.V.; Artemenko, A.G.; Polishchuk, P.G.; Kuz’min, V.E. Existing and developing approaches for QSAR analysis of mixtures. Mol. Inform. 2012, 31, 202–221. [Google Scholar] [CrossRef]
- Fruhmann, P.; Hametner, C.; Mikula, H.; Adam, G.; Krska, R.; Fröhlich, J. Stereoselective luche reduction of deoxynivalenol and three of its acetylated derivatives at C8. Toxins 2014, 6, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.-J.; Wen, C.-N.; Gao, Y.; Ren, F.-C.; Wang, F.; Liu, J.-K. ent-Kaurane diterpenoids from the plant Wedelia trilobata. Nat. Prod. Bioprospect. 2013, 3, 107–111. [Google Scholar] [CrossRef]
- Borsari, C.; Luciani, R.; Pozzi, C.; Poehner, I.; Henrich, S.; Trande, M.; Cordeiro-da-Silva, A.; Santarem, N.; Baptista, C.; Tait, A. Profiling of flavonol derivatives for the development of antitrypanosomatidic drugs. J. Med. Chem. 2016, 59, 7598–7616. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Modeling Environment. San Diego: DassaultSystèmes. 2016. Available online: https://3ds.com/products-services/biovia/products (accessed on 1 September 2016).
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Docking Score (kJ/mol) | SD | RMSD | SB |
|---|---|---|---|---|
| 101 | −449.5 | 2.8 | 1.5 | 1.00 |
| 270 | −437.6 | 7.4 | 1.6 | 0.97 |
| 302 | −423.0 | 9.4 | 1.3 | 0.94 |
| 299 | −422.7 | 9.2 | 1.3 | 0.94 |
| 175 | −421.8 | 18.0 | 1.0 | 0.94 |
| 298 | −420.2 | 20.1 | 1.6 | 0.93 |
| 174 | −419.9 | 9.7 | 1.4 | 0.93 |
| 173 | −419.7 | 7.4 | 1.3 | 0.93 |
| 135 | −416.7 | 9.1 | 1.1 | 0.93 |
| MTX | −560.4 | 17.6 | 0.4 | - |
| Kaurane | SB | LB | CALm |
|---|---|---|---|
| 135 | 0.93 | 0.57 | 0.70 |
| 101 | 1.00 | 0.51 | 0.69 |
| 302 | 0.94 | 0.54 | 0.68 |
| 134 | 0.90 | 0.55 | 0.68 |
| 298 | 0.93 | 0.53 | 0.68 |
| Compound | 135 | 302 | 301 | 302a | 301a | PMA |
|---|---|---|---|---|---|---|
| IC50 (µM) | 8.6 | 9.6 | 21.2 | 6.1 | 23.2 | 1.11 |
| Confidence Interval (95%) | 9.4–7.9 | 10.7–8.6 | 23.4–18.9 | 7.1–5.2 | 26.3–20.4 | 1.20–1.01 |
| Kiapp | 1.88 | 2.10 | 4.64 | 1.33 | 5.08 | 0.24 |
| Kaurane | Lb SB | Lp SB | La SB | CA |
|---|---|---|---|---|
| 302a | 0.87 | 1.00 | 1.00 | 0.96 |
| 301a | 0.86 | 0.97 | 0.98 | 0.94 |
| 175 | 0.90 | 0.95 | 0.92 | 0.92 |
| 69 | 0.93 | 0.94 | 0.88 | 0.92 |
| 135 | 1.00 | 0.88 | 0.82 | 0.90 |
| 134 | 0.93 | 0.80 | 0.87 | 0.87 |
| 302 | 0.85 | 0.89 | 0.82 | 0.86 |
| Protein | Ligand | VINA Score (kcal/mol) | Interacting Residues |
|---|---|---|---|
| LpPTR1 | Structure 302 | −9.73 | Van der Waals: A14, G20, L19, N110, S112, Y114, M179, I180, Q186, P187, Y194, G225, L226, S227, L228, F229, Y283; Carbon H-bond: K198; Alkyl: R18, L19; π-alkyl: M183; π-sigma: L188; π-anion: D181. |
| PMA | −7.92 | H-bond N110, I180; Van der Waals: R18, L19, S112, M179, Y194, K198, G225, L226, S227, L228; π-alkyl: Y114, F229; π-π T-shaped: Y114; π-anion: D181. | |
| DHB | −8.33 | H-bond: M179, D181, K198, G224; Van der Waals: L19, S112, Y194, P224, L226, S227, F229; Carbon H-bond: I180; π-π T-shaped: Y114 π-anion: D181. | |
| LbPTR1 | Structure 302 | −11.1 | H-bond G225; Van der Waals: K17, R18, N110, S112, Y114 I180, D181, L188, Y194, K198, S227, L228, F229, P230, Y241; π-sigma: M233, L226; Alkyl: L19 |
| PMA | −7.41 | H-bond: R14, L19, N110; Van der Waals: G20, C21, A111, S112, S227, L228; π-alkyl: R18, Y194; π-sigma: Y114. | |
| DHB | −7.75 | H-bond: L19, N110, P224; Van der Waals: A14, K17, R18, G20, C21, S112, I179, I180, D181, A182, Y194, S227, L228. | |
| LaPTR1 | Structure 302 | −9.87 | H-bond: A15, K16; Van der Waals: T12, G13, A14, R17, L18, H36, Y37, H38, R39, S40, N109, S111, S146, Y193, K197; Alkyl: P223 π-alkyl: A110, L66. |
| PMA | −7.19 | H-bond: G224; Van der Waals: S111, M178, V179, A181, Y193, L228, M232; π-alkyl: P223; Alkyl: F113, L187, L225, Y240 π-anion: D180. | |
| DHB | −7.61 | H-bond:K16, R17, N109; Van der Waals: G13, G19, M178, V179, D180, A181, Y193, K197, P223, G224, L225; π-alkyl: R17, L18. |
| 135 | 302 | 302a | PMA | DHB | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Energy Contribution | kJ/mol | SD | kJ/mol | SD | kJ/mol | SD | kJ/mol | SD | kJ/mol | SD |
| Van der Waals | −210.7 | 6.0 | −170.8 | 7.9 | −208.6 | 7.6 | −138.8 | 1.7 | −121.3 | 3.0 |
| Electrostatic | −2.9 | 1.5 | −26.7 | 3.4 | −9.7 | 3.0 | −145.0 | 2.5 | −194.6 | 10.3 |
| Polar solvation | 103.6 | 4.1 | 95.5 | 9.9 | 100.7 | 13.1 | 186.4 | 5.9 | 221.4 | 12.0 |
| SASA | −22.7 | 0.5 | −19.4 | 0.9 | −20.6 | 0.4 | −12.7 | 0.4 | −12.9 | 0.3 |
| Binding energy | −132.7 | 7.6 | −121.4 | 6.1 | −138.3 | 9.3 | −110.0 | 4.2 | −107.4 | 6.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrera-Acevedo, C.; Flores-Gaspar, A.; Scotti, L.; Mendonça-Junior, F.J.B.; Scotti, M.T.; Coy-Barrera, E. Identification of Kaurane-Type Diterpenes as Inhibitors of Leishmania Pteridine Reductase I. Molecules 2021, 26, 3076. https://doi.org/10.3390/molecules26113076
Herrera-Acevedo C, Flores-Gaspar A, Scotti L, Mendonça-Junior FJB, Scotti MT, Coy-Barrera E. Identification of Kaurane-Type Diterpenes as Inhibitors of Leishmania Pteridine Reductase I. Molecules. 2021; 26(11):3076. https://doi.org/10.3390/molecules26113076
Chicago/Turabian StyleHerrera-Acevedo, Chonny, Areli Flores-Gaspar, Luciana Scotti, Francisco Jaime Bezerra Mendonça-Junior, Marcus Tullius Scotti, and Ericsson Coy-Barrera. 2021. "Identification of Kaurane-Type Diterpenes as Inhibitors of Leishmania Pteridine Reductase I" Molecules 26, no. 11: 3076. https://doi.org/10.3390/molecules26113076
APA StyleHerrera-Acevedo, C., Flores-Gaspar, A., Scotti, L., Mendonça-Junior, F. J. B., Scotti, M. T., & Coy-Barrera, E. (2021). Identification of Kaurane-Type Diterpenes as Inhibitors of Leishmania Pteridine Reductase I. Molecules, 26(11), 3076. https://doi.org/10.3390/molecules26113076