
Molecular Tailoring Approach for the Estimation of Intramolecular Hydrogen Bond Energy



Abstract
1. Introduction
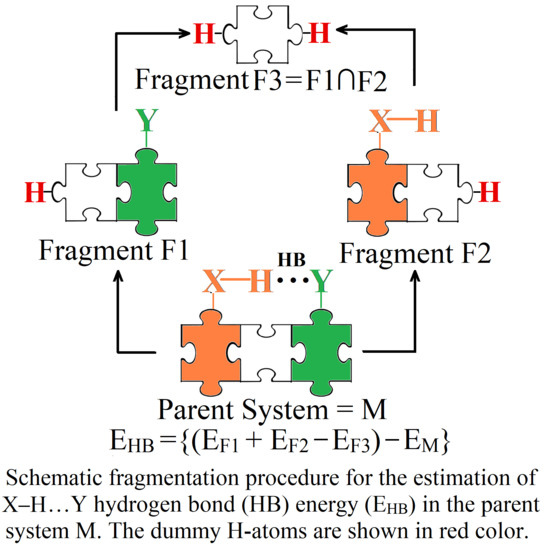
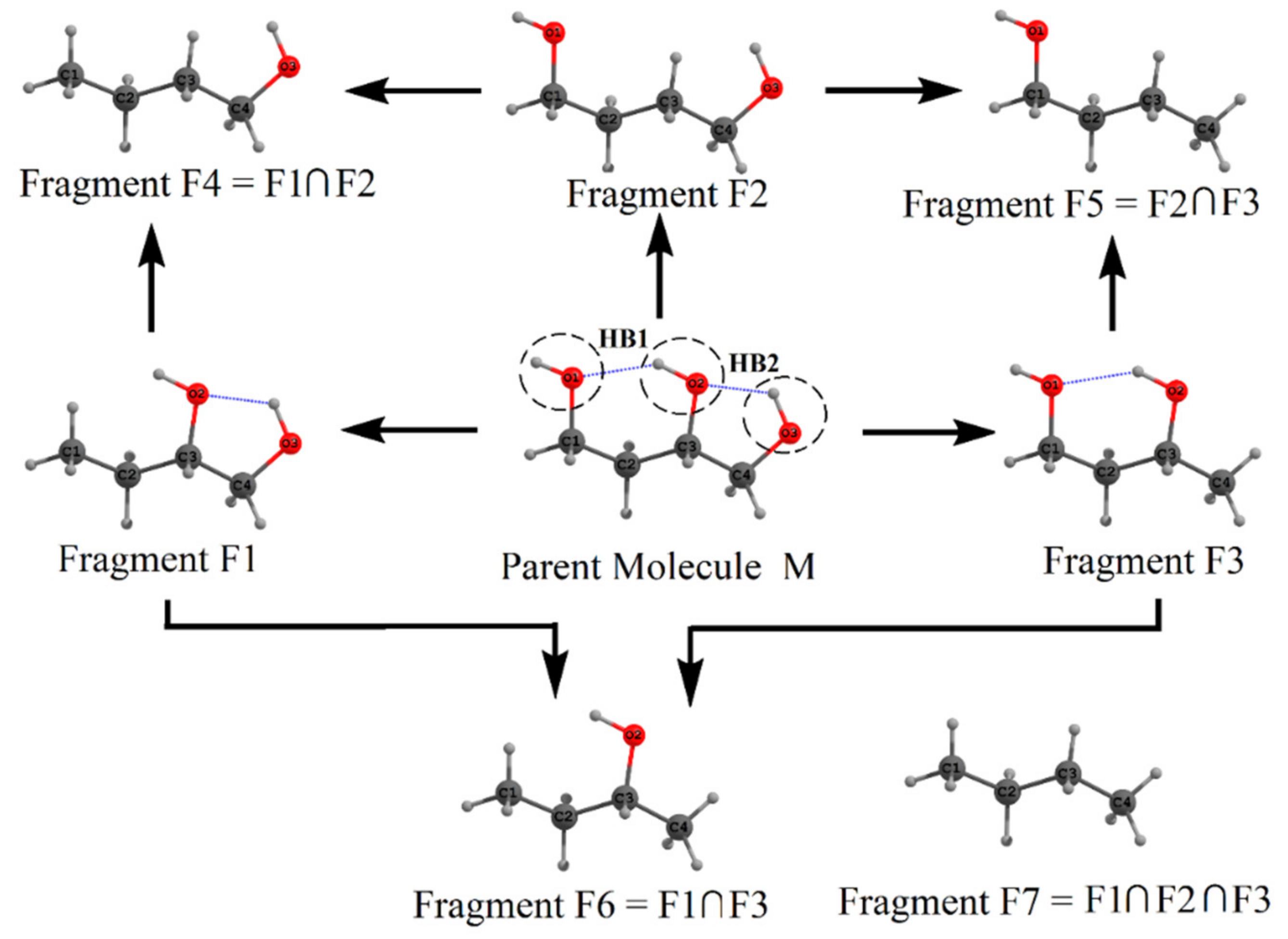
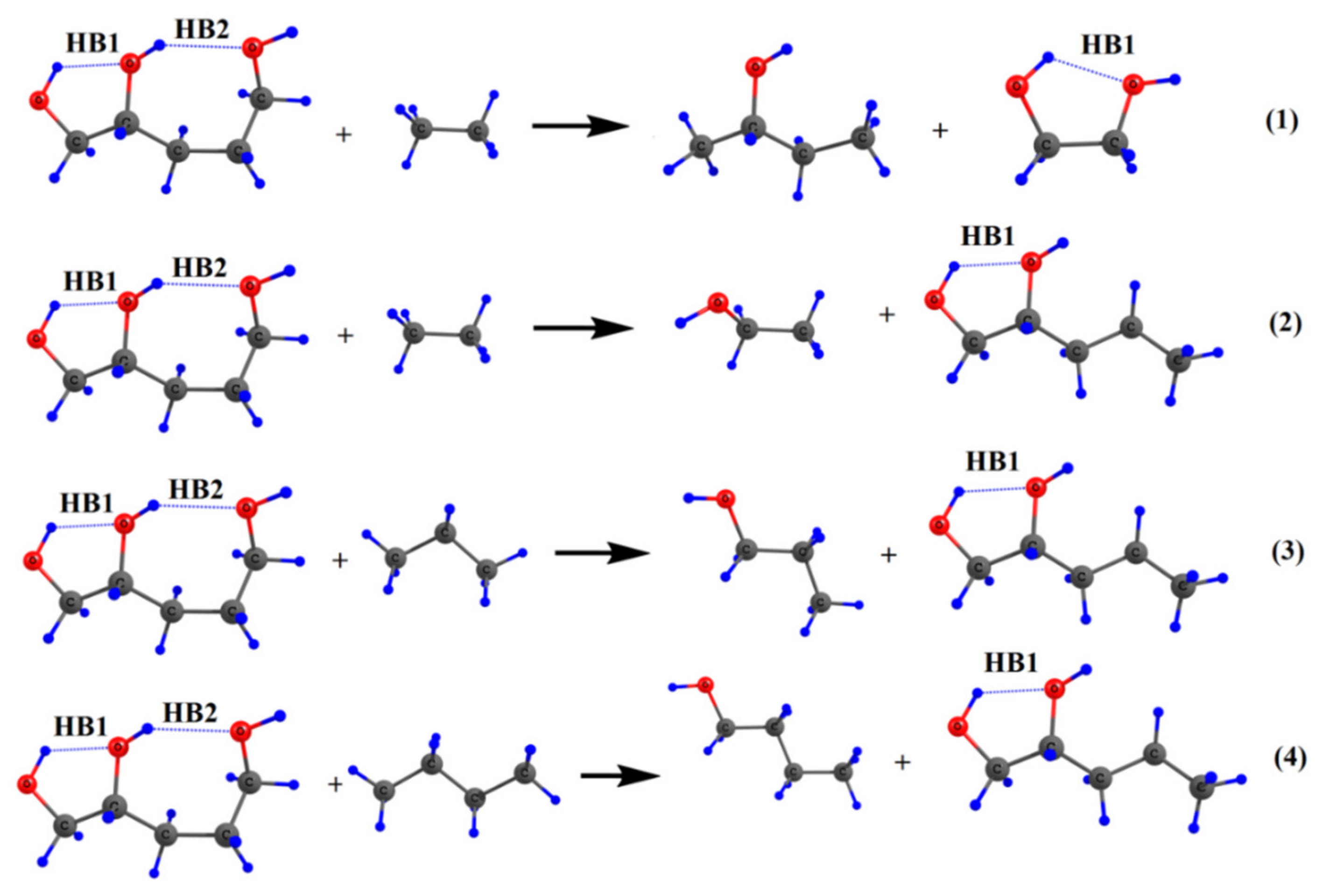
2. Molecular Tailoring Approach
3. Intramolecular Hydrogen Bond Energy Estimation by Molecular Tailoring Approach
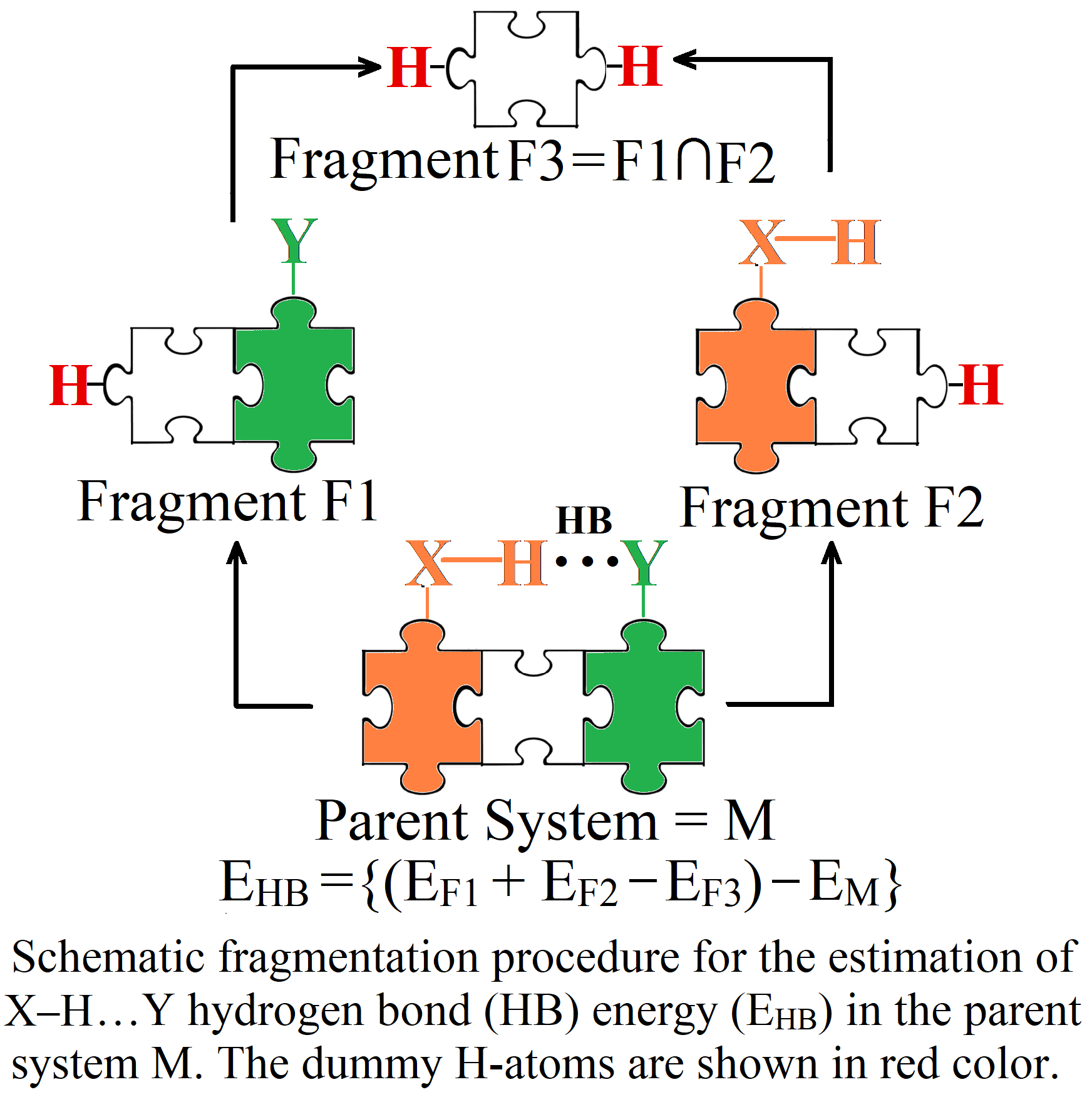
4. Critical Comparison of MTA with Other Methods
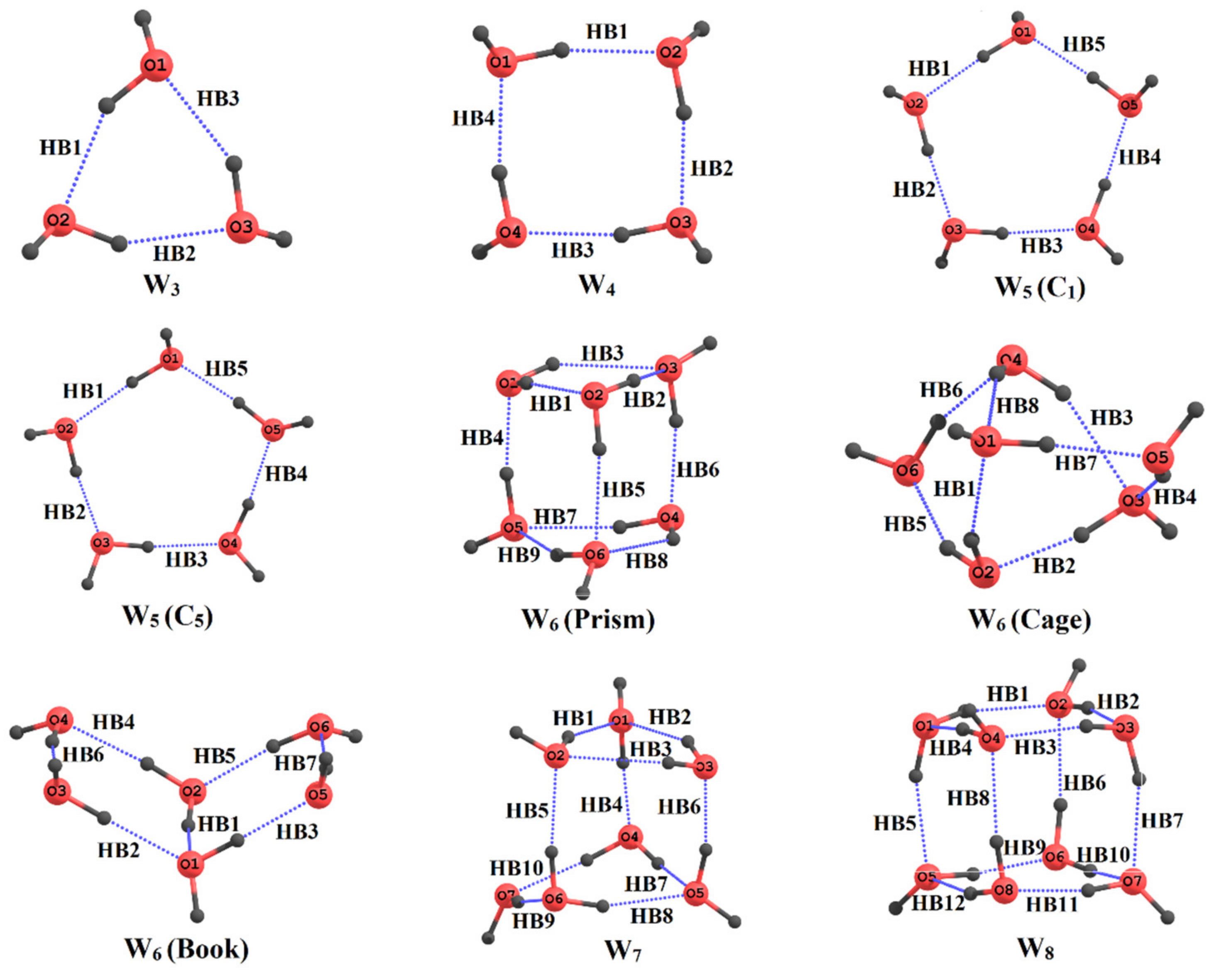
5. Application to Large Molecules and Clusters with Multiple Hydrogen Bonds
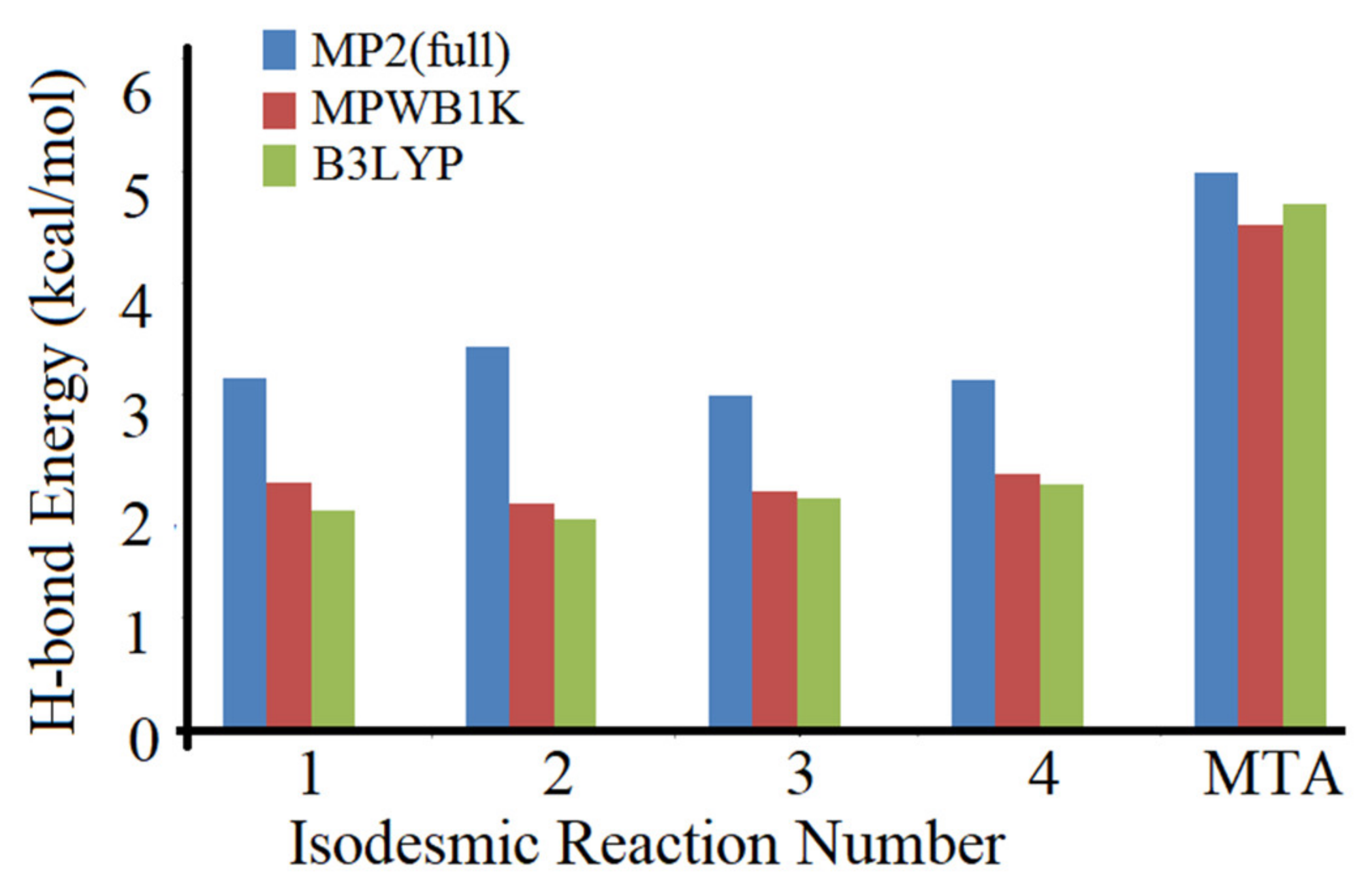
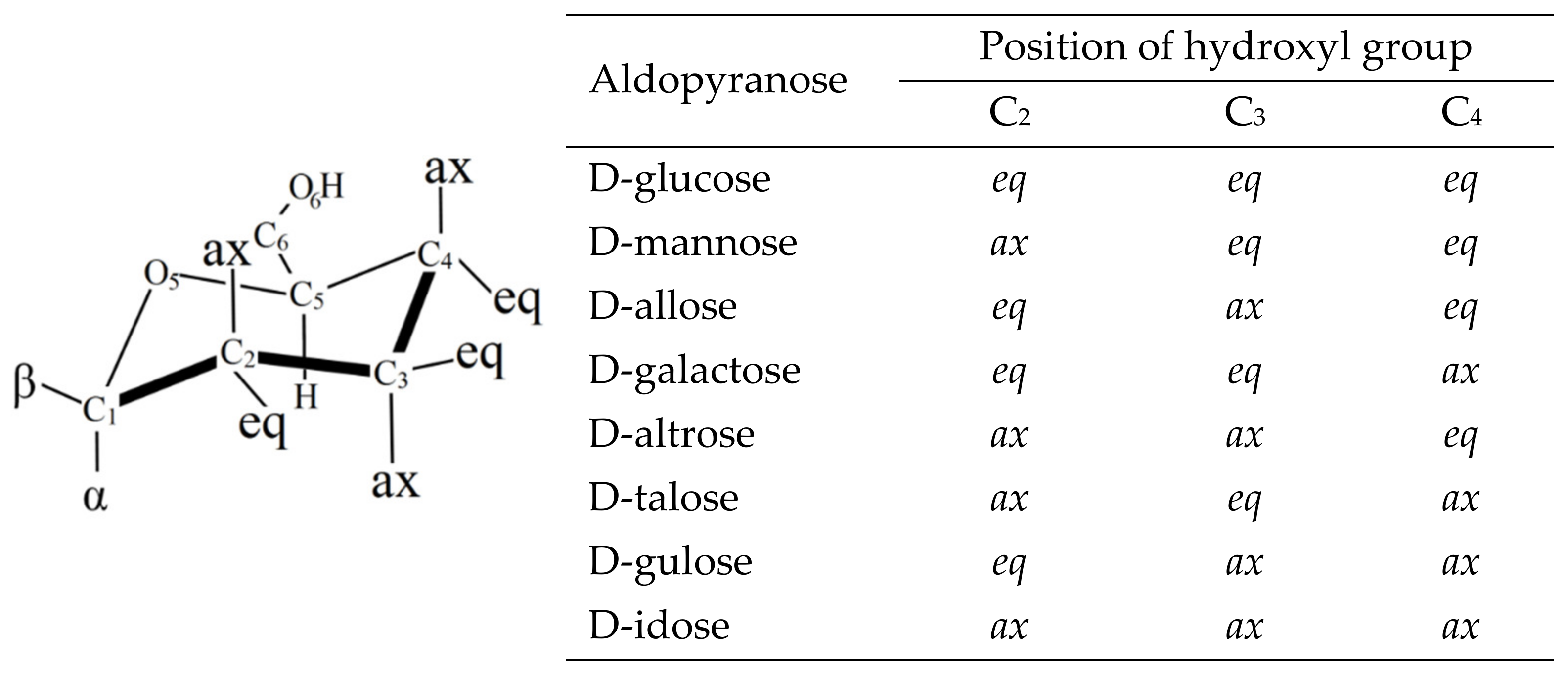
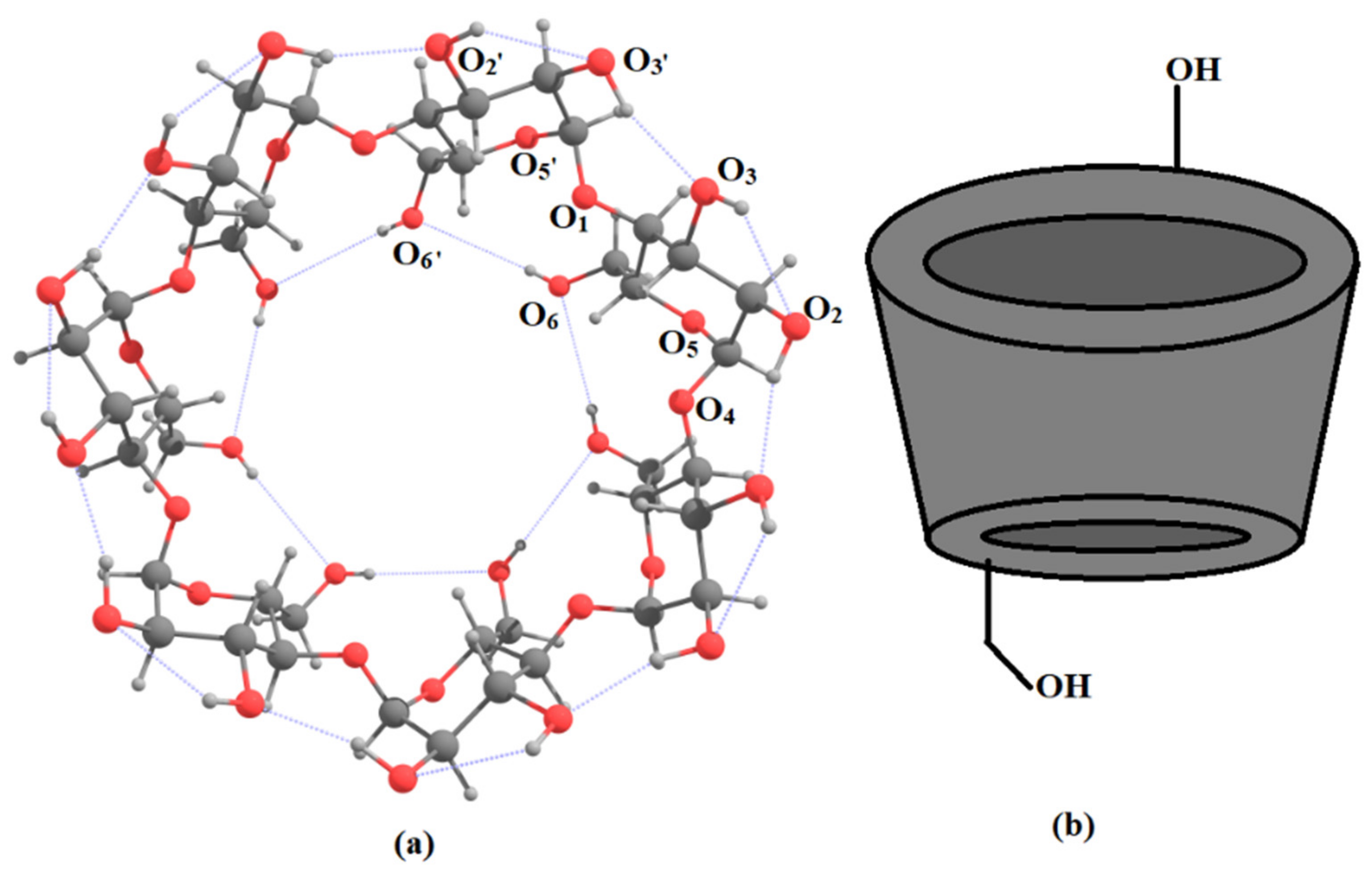
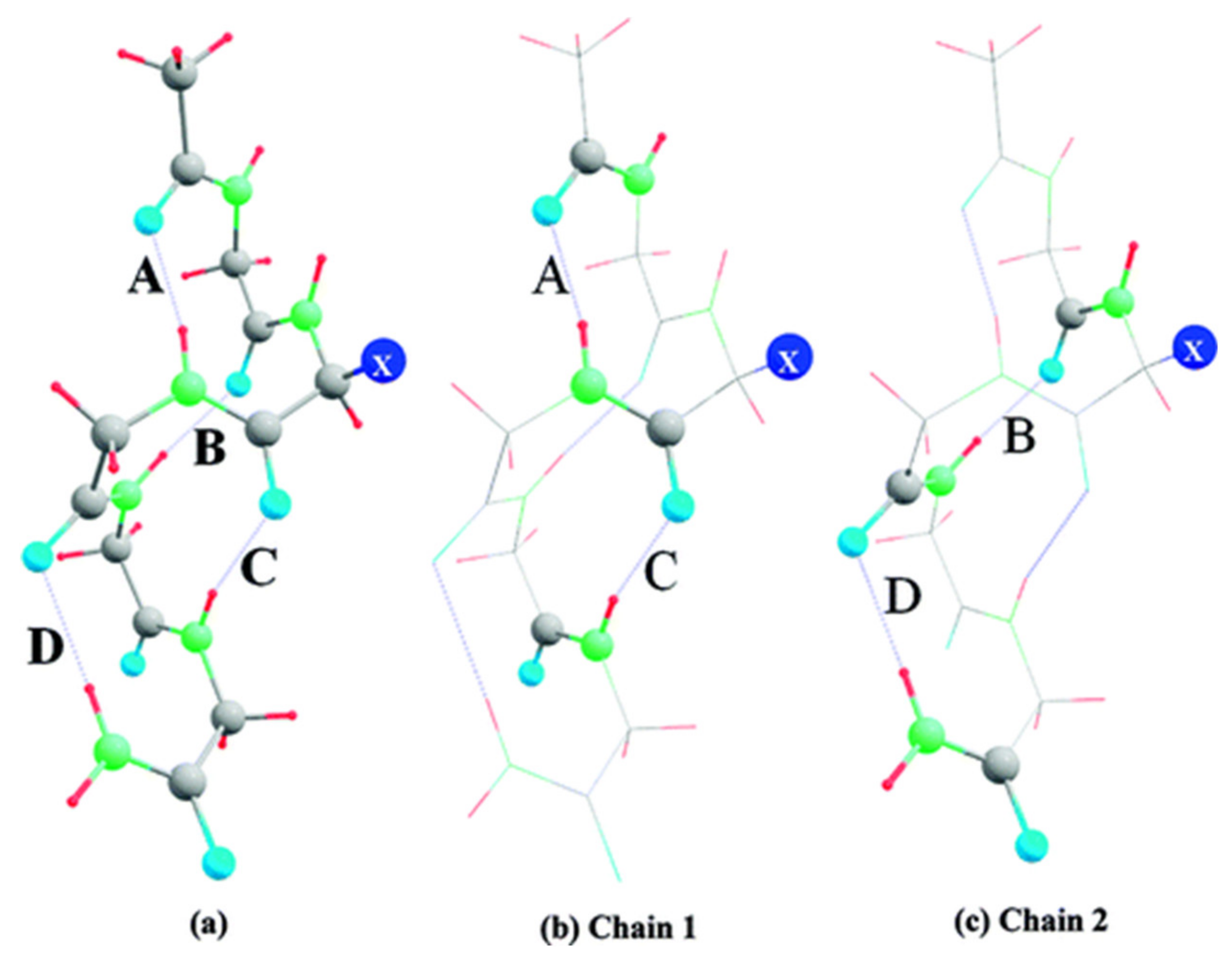
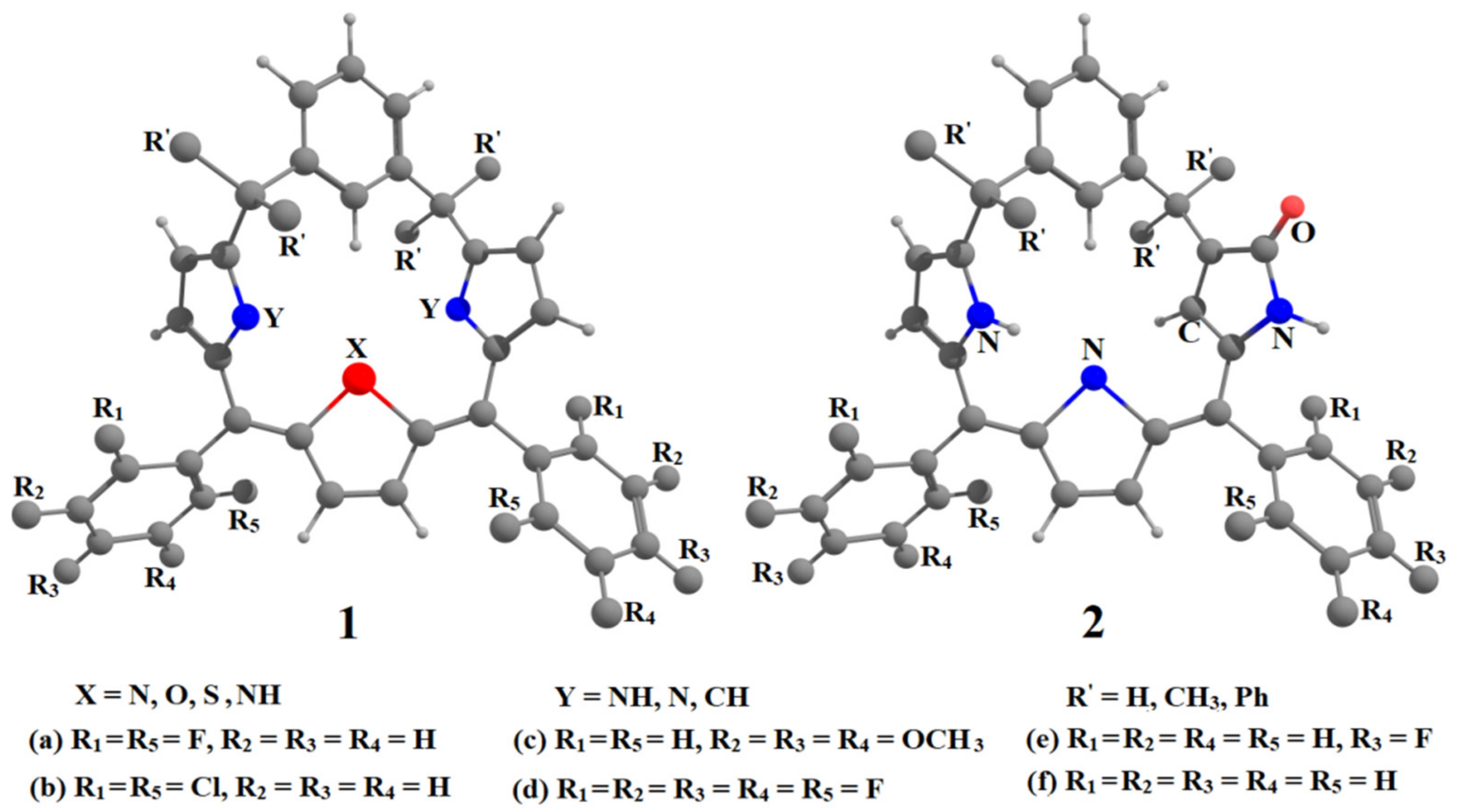
6. Applications to Biomolecules
7. Use of the MTA-Based Method by Other Researchers
8. Summary and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pauling, L. The Nature of the Chemical Bond; Oxford University Press: London, UK, 1939. [Google Scholar]
- Jeffery, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond. In Structural Chemistry and Biology; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Scheiner, S. Hydrogen Bond: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Huggins, M.L. Hydrogen Bridges in Ice and Liquid Water. J. Phys. Chem. 1936, 40, 723–731. [Google Scholar] [CrossRef]
- Huggins, M.L. Hydrogen Bridges in Organic Compounds. J. Org. Chem. 1936, 1, 407–456. [Google Scholar] [CrossRef]
- Nernst, W. Über die Löslichkeit von Mischkrystallen. Z. Phys. Chem. 1892, 9, 137–142. [Google Scholar] [CrossRef]
- Werner, A. Ueber Haupt und Nebenvalenzen und die Constitution der Ammoniumverbindungen. Liebigs Ann. Chem. 1902, 322, 261–296. [Google Scholar] [CrossRef]
- Moore, T.S.; Winmill, T.F. The State of Amines in Aqueous Solution. J. Chem. Soc. 1912, 101, 1635–1676. [Google Scholar] [CrossRef]
- Latimer, W.M.; Rodebush, W.H. Polarity and Ionization from the Standpoint of the Lewis Theory of Valence. J. Am. Chem. Soc. 1920, 42, 1419–1443. [Google Scholar] [CrossRef]
- Barnes, W.H. The Crystal Structure of Ice between 0 °C and −183 °C. Proc. R. Soc. Lond. A 1929, 125, 670–693. [Google Scholar]
- Huggins, M.L. 50 Years of Hydrogen Bond Theory. Angew. Chem. Int. Ed. Engl. 1971, 10, 147–152. [Google Scholar] [CrossRef]
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; Freeman: San Francisco, CA, USA, 1960. [Google Scholar]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Defining the Hydrogen Bond: An Account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond. Applications of Results Obtained from the Quantum Mechanics and from a Theory of Paramagnetic Susceptibility of the Structure of Molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [Google Scholar] [CrossRef]
- Chaplin, M.F. Water and Life: The Unique Properties of H2O; Lynden-Bell, R.M., Morris, S.M., Barrow, J.D., Finney, J.L., Harper, C., Eds.; CRC Press: Boca Raton, FL, USA, 2010; pp. 69–86. [Google Scholar]
- Laage, D.; Elsaesser, T.; Hynes, J.T. Water Dynamics in the Hydration Shells of Biomolecules. Chem. Rev. 2017, 117, 10694–10725. [Google Scholar] [CrossRef] [PubMed]
- Bellissent-Funel, M.-C.; Hassanali, A.; Havenith, M.; Henchman, R.; Pohl, P.; Sterpone, F.; van der Spoel, D.; Xu, Y.; Garcia, A.E. Water Determines the Structure and Dynamics of Proteins. Chem. Rev. 2016, 116, 7673–7697. [Google Scholar] [CrossRef] [PubMed]
- Keutsch, F.N.; Cruzan, J.D.; Saykally, R.J. The Water Trimer. Chem. Rev. 2003, 103, 2533–2578. [Google Scholar] [CrossRef]
- Sánchez-Garcȋa, E.; George, L.; Montero, L.A.; Sander, W. 1:2 Formic Acid/Acetylene Complexes: Ab Initio and Matrix Isolation Studies of Weakly Interacting Systems. J. Phys. Chem. A 2004, 108, 11846–11854. [Google Scholar] [CrossRef]
- Munshi, P.; Row, T.N.G. Exploring the Lower Limit in Hydrogen Bonds: Analysis of Weak C−H···O and C−H···π Interactions in Substituted Coumarins from Charge Density Analysis. J. Phys. Chem. A 2005, 109, 659–672. [Google Scholar] [CrossRef]
- Cockroft, S.L.; Hunter, C.A.; Lawso, K.R.; Perkins, J.; Urch, C.J. Electrostatic Control of Aromatic Stacking Interactions. J. Am. Chem. Soc. 2005, 127, 8594–8595. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Hydrogen Bonding Strength-Measures Based on Geometric and Topological Parameters. J. Phys. Org. Chem. 2004, 17, 18–31. [Google Scholar] [CrossRef]
- Bellamy. Advances in Infrared Group Frequencies; Methuen: London, UK, 1968; p. 241. [Google Scholar]
- Glasel, J.A. Water: A Comprehensive Treatise; Frank, F., Ed.; Plenum Press: New York, NY, USA; London, UK, 1982; Volume 1, p. 223, Chapter 6. [Google Scholar]
- Hobza, P.; Havlas, Z. Blue-Shifting Hydrogen Bonds. Chem. Rev. 2000, 100, 4253–4264. [Google Scholar] [CrossRef]
- Fuster, F.; Silvi, B. Does the Topological Approach Characterize the Hydrogen Bond? Theor. Chem. Acc. 2000, 104, 13–21. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X–H⋯F–Y Systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Klein, R.A. Hydrogen Bonding in Diols and Binary Diol–Water Systems Investigated using DFT Methods. II. Calculated Infrared OH-Stretch Frequencies, Force Constants, and NMR Chemical Shifts Correlate with Hydrogen Bond Geometry and Electron Density Topology. A Reevaluation of Geometrical Criteria for Hydrogen Bonding. J. Comput. Chem. 2003, 24, 1120–1131. [Google Scholar]
- Deshmukh, M.M.; Sastry, N.V.; Gadre, S.R. Molecular Interpretation of Water Structuring and Destructuring Effects: Hydration of Alkanediols. J. Chem. Phys. 2004, 121, 12402–12410. [Google Scholar] [CrossRef]
- Kovács, A.; Szabo, A.; Hargittai, I. Structural Characteristics of Intramolecular Hydrogen Bonding in Benzene Derivatives. Acc. Chem. Res. 2002, 35, 887–894. [Google Scholar] [CrossRef]
- Chung, G.; Kwon, O.; Kwon, Y. Theoretical Study on 1,2-Dihydroxybenzene and 2-Hydroxythiophenol: Intramolecular Hydrogen Bonding. J. Phys. Chem. A 1997, 101, 9415–9420. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Intramolecular Hydrogen Bonds in ortho-Substituted Hydroxybenzenes and in 8-Susbtituted 1-Hydroxynaphthalenes: Can a Methyl Group Be an Acceptor of Hydrogen Bonds? J. Phys. Chem. A 2001, 105, 10462–10467. [Google Scholar] [CrossRef]
- Lipkowski, P.; Koll, A.; Karpfen, A.; Wolschann, P. An Approach to Estimate the Energy of the Intramolecular Hydrogen Bond. Chem. Phys. Lett. 2002, 360, 256–263. [Google Scholar] [CrossRef]
- Buemi, G.; Zuccarello, F. Is the Intramolecular Hydrogen Bond Energy Valuable from Internal Rotation Barriers? J. Mol. Struct. Theochem. 2002, 581, 71–85. [Google Scholar] [CrossRef]
- Korth, H.-G.; de Heer, M.I.; Mulder, P. A DFT Study on Intramolecular Hydrogen Bonding in 2-Substituted Phenols: Conformations, Enthalpies, and Correlation with Solute Parameters. J. Phys. Chem. A 2002, 106, 8779–8789. [Google Scholar] [CrossRef]
- Kjaergaard, H.G.; Howard, D.L.; Schofield, D.P.; Robinson, T.W.; Ishiuchi, S.; Fujii, M. OH- and CH-Stretching Overtone Spectra of Catechol. J. Phys. Chem. A 2002, 106, 258–266. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Sun, Y.-M.; Wang, X.-L. Substituent Effects on O-H Bond Dissociation Enthalpies and Ionization Potentials of Catechols: A DFT Study and Its Implications in the Rational Design of Phenolic Antioxidants and Elucidation of Structure–Activity Relationships for Flavonoid Antioxidants. Chem. Eur. J. 2003, 9, 502–508. [Google Scholar] [CrossRef]
- Bakalbassis, E.G.; Lithoxoidou, A.T.; Vafiadis, A.P. Theoretical Calculation of Accurate Absolute and Relative Gas- and Liquid-Phase O-H Bond Dissociation Enthalpies of 2-Mono- and 2,6-Disubstituted Phenols, Using DFT/B3LYP. J. Phys. Chem. A 2003, 107, 8594–8606. [Google Scholar] [CrossRef]
- Jabloński, M.A. Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions. Molecules 2020, 25, 5512. [Google Scholar] [CrossRef]
- Jabloński, M.; Kaczmarek, A.; Sadlej, A.J. Estimates of the Energy of Intramolecular Hydrogen Bonds. J. Phys. Chem. A 2006, 110, 10890–10898. [Google Scholar] [CrossRef] [PubMed]
- Jabloński, M. Full vs. Constrained Geometry Optimization in the Open-Closed Method in Estimating the Energy of Intramolecular Charge-Inverted Hydrogen Bonds. Chem. Phys. 2010, 376, 76–83. [Google Scholar] [CrossRef]
- Estacio, S.G.; Cabral do Counta, P.; Costa Cabral, B.J.; Minas Da Piedade, M.E.; Martinho Simoes, J.A. Energetics of Intramolecular Hydrogen Bonding in Di-substituted Benzenes by the ortho-para Method. J. Phys. Chem. A 2004, 108, 10834–10843. [Google Scholar] [CrossRef]
- Hammett, L.P. The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc. 1937, 59, 96–103. [Google Scholar] [CrossRef]
- Hammett, L.P. Physical Organic Chemistry; McGraw-Hill: New York, NY, USA, 1940. [Google Scholar]
- Suresh, C.H.; Gadre, S.R. A Novel Electrostatic Approach to Substituent Constants: Doubly Substituted Benzenes. J. Am. Chem. Soc. 1998, 120, 7049–7055. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Radom, L.; Pople, J.A. Molecular Orbital Theory of the Electronic Structure of Organic Compounds. V. Molecular Theory of Bond Separation. J. Am. Chem. Soc. 1970, 92, 4796–4801. [Google Scholar] [CrossRef]
- Suresh, C.H.; Koga, N. An Isodesmic Reaction Based Approach to Aromaticity of a Large Spectrum of Molecules. Chem. Phys. Lett. 2006, 419, 550–556. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Houk, K.N.; Schleyer, P.; Allen, W.D. A Hierarchy of Homodesmotic Reactions for Thermochemistry. J. Am. Chem. Soc. 2009, 131, 2547–2560. [Google Scholar] [CrossRef]
- George, P.; Trachtman, M.; Brett, A.M.; Bock, C.W. Comparison of Various lsodesmic and Homodesmotic Reaction Heats with Values derived from Published Ab initio Molecular Orbital Calculations. J. Chem. Soc. Perkin Trans. 1977, 2, 1036–1047. [Google Scholar] [CrossRef]
- Howard, S.T. Relationship between Basicity, Strain, and Intramolecular Hydrogen-Bond Energy in Proton Sponges. J. Am. Chem. Soc. 2000, 122, 8238–8244. [Google Scholar] [CrossRef]
- Deshmukh, M.M.; Suresh, C.H.; Gadre, S.R. Intramolecular Hydrogen Bond Energy in Polyhydroxy Systems: A Critical Comparison of Molecular Tailoring and Isodesmic Approaches. J. Phys. Chem. A 2007, 111, 6472–6480. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Haaland, A.; Shorokhov, D.J.; Tverdova, N.V. Topological Analysis of Electron Densities: Is the Presence of an Atomic Interaction Line in an Equilibrium Geometry a Sufficient Condition for the Existence of a Chemical Bond? Chem. Eur. J. 2004, 10, 4416–4421. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Bickelhaupt, F.M. A Model of the Chemical Bond Must Be Rooted in Quantum Mechanics, Provide Insight, and Possess Predictive Power. Chem. Eur. J. 2006, 12, 2902–2905. [Google Scholar] [CrossRef]
- Dem’yanov, P.; Polestshuk, P. A Bond Path and an Attractive Ehrenfest Force Do Not Necessarily Indicate Bonding Interactions: Case Study on M2X2 (M = Li, Na, K; X = H, OH, F, Cl). Chem. Eur. J. 2012, 18, 4982–4993. [Google Scholar] [CrossRef]
- Jabłoński, M. Bond Paths Between Distant Atoms Do Not Necessarily Indicate Dominant Interactions. J. Comput. Chem. 2018, 39, 2183–2195. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strength Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Francisco, F.; Rocha-Rinza, T.; Pendás, A.M. Interacting Quantum Atoms-Review. Molecules 2020, 25, 4028. [Google Scholar] [CrossRef]
- Gatti, C.; May, E.; Destro, R.; Cargnoni, F. Fundamental Properties and Nature of CH···O Interactions in Crystals on the Basis of Experimental and Theoretical Charge Densities. The Case of 3,4-Bis(dimethylamino)-3-cyclobutene-1,2-dione (DMACB) Crystal. J. Phys. Chem. A 2002, 106, 2707–2720. [Google Scholar] [CrossRef]
- Nikolaienko, T.Y.; Bulavin, L.A.; Hovorun, D.M. Bridging QTAIM with Vibrational Spectroscopy: The Energy of Intramolecular Hydrogen Bonds in DNA-related Biomolecules. Phys. Chem. Chem. Phys. 2012, 14, 7441–7447. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.A. Electron Density Topological Analysis of Hydrogen Bonding in Glucopyranose and Hydrated Glucopyranose. J. Am. Chem. Soc. 2002, 124, 13931–13937. [Google Scholar] [CrossRef]
- Klein, R.A. Ab initio conformational studies on diols and binary diol-water systems using DFT methods. Intramolecular hydrogen bonding and 1:1 complex formation with water. J. Comput. Chem. 2002, 23, 585–599. [Google Scholar]
- Howard, D.L.; Jørgensen, P.; Kjaergaard, H.G. Weak Intramolecular Interactions in Ethylene Glycol Identified by Vapor Phase OH-Stretching Overtone Spectroscopy. J. Am. Chem. Soc. 2005, 127, 17096–17103. [Google Scholar] [CrossRef]
- Deshmukh, M.M.; Gadre, S.R.; Bartolotti, L.J. Estimation of Intramolecular Hydrogen Bond Energy via Molecular Tailoring Approach. J. Phys. Chem. A 2006, 110, 12519–12523. [Google Scholar]
- Deshmukh, M.M.; Bartolotti, L.J.; Gadre, S.R. Intramolecular Hydrogen Bonding and Cooperative Interactions in Carbohydrates via the Molecular Tailoring Approach. J. Phys. Chem. A 2008, 112, 312–321. [Google Scholar] [CrossRef]
- Gadre, S.R.; Shirsat, R.N.; Limaye, A.C. Molecular Tailoring Approach for Simulation of Electrostatic Properties. J. Phys. Chem. 1994, 98, 9165–9169. [Google Scholar] [CrossRef]
- Ganesh, V.; Dongare, R.K.; Balanarayan, P.; Gadre, S.R. Molecular Tailoring Approach for Geometry Optimization of Large Molecules: Energy Evaluation and Parallelization Strategies. J. Chem. Phys. 2006, 125, 104109–104110. [Google Scholar] [CrossRef]
- Elango, M.; Subramanian, V.; Rahalkar, A.P.; Gadre, S.R.; Sathyamurthy, N. Structure, Energetics, and Reactivity of Boric Acid Nanotubes: A Molecular Tailoring Approach. J. Phys. Chem. A 2008, 112, 7699–7704. [Google Scholar] [CrossRef]
- Gadre, S.R.; Ganesh, V. Molecular Tailoring Approach: Towards PC-based ab Initio Treatment of Large Molecules. J. Theor. Comput. Chem. 2006, 5, 835–855. [Google Scholar] [CrossRef]
- Rahalkar, A.P.; Ganesh, V.; Gadre, S.R. Enabling Ab-initio Hessian and Frequency Calculations of Large Molecules. J. Chem. Phys. 2008, 129, 234101. [Google Scholar] [CrossRef]
- Sahu, N.; Gadre, S.R. Accurate Vibrational Spectra via Molecular Tailoring Approach: A Case Study of Water Clusters at MP2 Level. J. Chem. Phys. 2015, 142, 014107. [Google Scholar] [CrossRef]
- Sahu, N.; Gadre, S.R. Vibrational Infrared and Raman Spectra of Polypeptides: Fragments-in-fragments Within Molecular Tailoring Approach. J. Chem. Phys. 2016, 144, 114113. [Google Scholar] [CrossRef]
- Furtado, J.P.; Rahalkar, A.P.; Shankar, S.; Bandyopadhyay, P.; Gadre, S.R. Facilitating Minima Search for Large Water Clusters at the MP2 Level via Molecular Tailoring. J. Phys. Chem. Lett. 2012, 3, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.; Singh, G.; Nandi, A.; Gadre, S.R. Toward an Accurate and Inexpensive Estimation of CCSD (T)/CBS Binding Energies of Large Water Clusters. J. Phys. Chem. A 2016, 120, 5706–5714. [Google Scholar] [CrossRef]
- Gadre, S.R.; Yeole, S.D.; Sahu, N. Quantum Chemical Investigations on Molecular Clusters. Chem. Rev. 2014, 114, 12132–12173. [Google Scholar] [CrossRef]
- Singh, G.; Nandi, A.; Gadre, S.R. Breaking the Bottleneck: Use of Molecular Tailoring Approach for the Estimation of Binding Energies at MP2/CBS Limit for Large Water Clusters. J. Chem. Phys. 2016, 144, 104102. [Google Scholar] [CrossRef] [PubMed]
- Yeole, S.D.; Gadre, S.R. On the Applicability of Fragmentation Methods to Conjugated π-Systems within Density Functional Framework. J. Chem. Phys. 2010, 132, 094102. [Google Scholar] [CrossRef]
- Yeole, S.D.; Gadre, S.R. Molecular Cluster Building Algorithm: Electrostatic Guidelines and Molecular Tailoring Approach. J. Chem. Phys. 2011, 134, 084111. [Google Scholar] [CrossRef]
- Khire, S.S.; Gadre, S.R. Pragmatic Many-body Approach for Economic MP2 Energy Estimation of Molecular Clusters. J. Phys. Chem. A 2019, 123, 5005–5011. [Google Scholar] [CrossRef] [PubMed]
- Khire, S.S.; Bartolotti, L.J.; Gadre, S.R. Harmonizing Accuracy and Efficiency: A Pragmatic Approach to Fragmentation of Large Molecules. J. Chem. Phys. 2018, 149, 064112. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Siddiqui, N.; Singh, V.; Deshmukh, M.M.; Gurunath, R. Structures, Stability and Hydrogen Bonding in Inositol Conformers. Phys. Chem. Chem. Phys. 2015, 17, 18514–18523. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, M.M.; Gadre, S.R.; Cocinero, E.M. Stability of Conformationally Locked Free Fructose: Theoretical and Computational Insights. New J. Chem. 2015, 39, 9006–9018. [Google Scholar] [CrossRef]
- Deshmukh, M.M.; Bartolotti, L.J.; Gadre, S.R. Intramolecular Hydrogen Bond Energy and Cooperative Interactions in α-, β-, and γ-cyclodextrin Conformers. J. Comput. Chem. 2011, 32, 2996–3004. [Google Scholar] [CrossRef]
- Khedkar, J.K.; Deshmukh, M.M.; Gejji, S.P.; Gadre, S.R. Intramolecular Hydrogen Bonding and Cooperative Interactions in Calix[n]arenes (n = 4,5). J. Phys. Chem. A 2012, 116, 3739–3744. [Google Scholar] [CrossRef]
- Ahirwar, M.B.; Gadre, S.R.; Deshmukh, M.M. Direct and Reliable Method for Estimating the Hydrogen Bond Energies and Cooperativity in Water Clusters Wn, n = 3 to 8. J. Phys. Chem. A 2020, 124, 6699–6706. [Google Scholar] [CrossRef]
- Deshmukh, M.M.; Gadre, S.R. Estimation of N-H···O=C Intramolecular Hydrogen Bond Energy in Polypeptides. J. Phys. Chem. A 2009, 113, 7927–7932. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Hung, C.-H.; Rana, S.; Deshmukh, M.M. Study on the Structure, Stability, and Tautomerism of meta-Benziporphodimethenes and N-Confused Isomers Containing γ-Lactam Ring. J. Mol. Struct. 2019, 1187, 138–150. [Google Scholar] [CrossRef]
- Ahluwalia, D.; Kumar, A.; Warkar, S.G.; Deshmukh, M.M. Effect of Substitutions on the Geometry and Intramolecular Hydrogen Bond Strength in meta-benziporphodimethenes: A New Porphyrin Analogue. J. Mol. Struct. 2020, 1220, 128773. [Google Scholar] [CrossRef]
- Singh, V.; Ibnusaud, I.; Gadre, S.R.; Deshmukh, M.M. Fragmentation Method Reveals a Wide Spectrum of Intramolecular Hydrogen Bond Energies in Antioxidant Natural Products. New J. Chem. 2020, 44, 5841–5849. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenkoa, A.V.; Sigalov, M.V. Estimating the energy of intramolecular hydrogen bonds from 1H NMR and QTAIM calculations. Org. Biomol. Chem. 2016, 14, 11199–11211. [Google Scholar] [CrossRef]
- Lipkowitz, K.B. Applications of Computational Chemistry to the Study of Cyclodextrins. Chem. Rev. 1998, 98, 1829–1874. [Google Scholar] [CrossRef]
- Crini, G. Review: A History of Cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef] [PubMed]
- Pinjari, R.V.; Joshi, K.A.; Gejji, S.P. Theoretical Studies on Hydrogen Bonding, NMR Chemical Shifts and Electron Density Topography in α, β and γ-Cyclodextrin Conformers. J. Phys. Chem. A 2007, 111, 13583–13589. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Connors, K.A. Advances in Analytical Chemistry and Instrumentation; Reilly, C.N., Ed.; Wiley-Interscience: New York, NY, USA, 1965; Volume 4, pp. 117–212. [Google Scholar]
- Loftsson, T.; Brewster, M.E. Pharmaceutical Applications of Cyclodextrins. 1. Drug Solubilization and Stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Jarho, P.; Másson, M.; Järvine, T. Cyclodextrins in Drug Delivery. Expert Opin. Drug Deliv. 2005, 2, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Aprà, E.; Zeng, X.C.; Xantheas, S.S. High-Level Ab Initio Electronic Structure Calculations of Water Clusters (H2O)16 and (H2O)17: A New Global Minimum for (H2O)16. J. Phys. Chem. Lett. 2010, 1, 3122–3127. [Google Scholar] [CrossRef]
- Maheshwary, S.; Patel, N.; Sathyamurthy, N.; Kulkarni, A.D.; Gadre, S.R. Structure and Stability of Water Clusters (H2O)n, n =8−20: An Ab Initio Investigation. J. Phys. Chem. A 2001, 105, 10525–10537. [Google Scholar] [CrossRef]
- Kazachenko, S.; Thakkar, A.J. Improved Minima-Hopping. TIP4P Water Clusters, (H2O)n with n ≤ 37. Chem. Phys. Lett. 2009, 476, 120–124. [Google Scholar] [CrossRef]
- Sahu, N.; Gadre, S.R.; Rakshit, A.; Bandyopadhyay, P.; Miliordos, E.; Xantheas, S.S. Low Energy Isomers of (H2O)25 from a Hierarchical Method Based on Monte Carlo Temperature Basin Paving and Molecular Tailoring Approaches Benchmarked by MP2 Calculations. J. Chem. Phys. 2014, 141, 164304. [Google Scholar] [CrossRef] [PubMed]
- Foti, M.C.; Johnson, E.R.; Vinqvist, M.R.; Wright, J.S.; Barclay, L.R.C.; Ingold, K.U. Naphthalene Diols: A New Class of Antioxidants Intramolecular Hydrogen Bonding in Catechols, Naphthalene Diols, and Their Aryloxyl Radicals. J. Org. Chem. 2002, 67, 5190–5196. [Google Scholar] [CrossRef] [PubMed]
- Foti, M.C.; Barclay, L.R.C.; Ingold, K.U. The Role of Hydrogen Bonding on the H-Atom-Donating Abilities of Catechols and Naphthalene Diols and on a Previously Overlooked Aspect of Their Infrared Spectra. J. Am. Chem. Soc. 2002, 124, 12881–12888. [Google Scholar] [CrossRef] [PubMed]
- Amorati, R.; Lucarini, M.; Mugnaini, V.; Pedulli, G.F. Antioxidant Activity of o-Bisphenols: The Role of Intramolecular Hydrogen Bonding. J. Org. Chem. 2003, 68, 5198–5204. [Google Scholar] [CrossRef] [PubMed]
- Martίnez-Cifuentes, M.; Weiss-López, B.E.; Santos, L.S.; Araya-Maturana, R. Intramolecular Hydrogen Bond in Biologically Active o-Carbonyl Hydroquinones. Molecules 2014, 19, 9354–9368. [Google Scholar] [CrossRef]
- Llano, S.; Gómez, S.; Londoño, J.; Restropo, A. Antioxidant activity of Curcuminoids. Phys. Chem. Chem. Phys. 2019, 21, 3752–3760. [Google Scholar] [CrossRef]
- Lopes Jesus, A.J.; Rosado, M.T.S.; Reva, I.; Fausto, R.; Eusébio, M.E.S.; Redinha, J.S. Structure of Isolated 1,4-Butanediol: Combination of MP2 Calculations, NBO Analysis, and Matrix-Isolation Infrared Spectroscopy. J. Phys. Chem. A 2008, 112, 4669–4678. [Google Scholar] [CrossRef]
- Iogansen, A.V. Direct Proportionality of the Hydrogen Bonding Energy and the Intensification of the Stretching ν(XH) Vibration in Infrared Spectra. Spectrochim. Acta Part A 1999, 55, 1585–1612. [Google Scholar] [CrossRef]
- Rusinska-Roszak, D.; Sowinski, G. Estimation of the Intramolecular O−H···O=C Hydrogen Bond Energy via the Molecular Tailoring Approach. Part I: Aliphatic Structures. J. Chem. Inf. Model. 2014, 54, 1963–1977. [Google Scholar] [CrossRef]
- Rusinska-Roszak, D. Intramolecular O−H···O=C Hydrogen Bond Energy via the Molecular Tailoring Approach to RAHB Structures. J. Phys. Chem. A 2015, 119, 3674–3687. [Google Scholar] [CrossRef] [PubMed]
- Rusinska-Roszak, D. Energy of Intramolecular Hydrogen Bonding in ortho-Hydroxybenzaldehydes, Phenones and Quinones. Transfer of Aromaticity from ipso-Benzene Ring to the Enol System(s). Molecules 2017, 22, 481. [Google Scholar] [CrossRef]
- Afonin, A.V.; Rusinska-Roszak, D. Molecular Tailoring Approach–New Guide to Quantify the Energy of Push-Pull Effect: Case Study on the (E) 3-(1H-pyrrol-2-yl)prop-2-enones. Phys. Chem. Chem. Phys. 2020, 22, 22190–22194. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenko, A.V. Quantitative Decomposition of Resonance-assisted Hydrogen Bond Energy in β-diketones into Resonance and Hydrogen Bonding (π- and σ-) Components using Molecular Tailoring and Function-based Approaches. J. Comput. Chem. 2020, 41, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Keykhaei, A.; Nowroozi, A. On the Performance of Molecular Tailoring Approach for Estimation of the Intramolecular Hydrogen Bond Energies of RAHB Systems: A Comparative Study. Struct. Chem. 2020, 31, 423–433. [Google Scholar] [CrossRef]
- Henry, L. Formation synthétique d’alcools nitrés. Ir. J. Med. Sci. 1895, 120, 1265–1268. [Google Scholar]
- Alegre-Requena, J.V.; Marqueés-Loópez, E.; Herrera, R.P. “Push−Pull π+/π−” (PPππ) Systems in Catalysis. ACS Catal. 2017, 7, 6430–6439. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Honda, K.; Uchimaru, T.; Mikami, M.; Tanabe, K. Origin of Attraction and Directionality of the π/π Interaction: Model Chemistry Calculations of Benzene Dimer Interaction. J. Am. Chem. Soc. 2002, 124, 104–112. [Google Scholar] [CrossRef]
- Mishra, B.K.; Deshmukh, M.M.; Ramanathan, V. C-H···π interactions and the nature of the donor carbon atom. J. Org. Chem. 2014, 79, 8599–8606. [Google Scholar] [CrossRef]
- Desiraju, G.; Ho, P.S.; Kloo, L.; Anthony, C.L.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Grabowski, S.J. Dihydrogen bond and X-H···σ interaction as sub-classes of hydrogen bond. J. Phys. Org. Chem. 2013, 26, 452–459. [Google Scholar] [CrossRef]
- Biswal, H.S.; Wategaonkar, S. Sulfur, not too far behind O, N, and C: SH···π hydrogen bond. J. Phys. Chem. A 2009, 113, 12774–12782. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, D.K.; Jena, S.; Dutta, J.; Rana, A.; Biswal, H.S. Nature and strength of M–H···S and M–H···Se (M = Mn, Fe, & Co) hydrogen bond. J. Phys. Chem. A 2019, 123, 2227–2236. [Google Scholar] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | HB Label | HB Distances a (in Å) | HB Energy (kcal/mol) | ΔE (kcal/mol) | O–H Stretch Frequency (cm−1) | MED at (3, −1) BCP (a.u.) |
|---|---|---|---|---|---|---|
| 1,2,3-propanetriol b | HB1 HB2 HB3 | 2.16 2.08 2.58 | 1.90 2.47 1.63 | 0.50 | 3784 3765 3845 | 0.0201 |
| 1,2,3-butanetriol b | HB1 HB2 HB3 | 2.13 2.05 2.58 | 2.13 2.72 1.60 | 0.50 | 3768 3745 3844 | 0.0211 |
| 1,2,4-butanetriol | HB1 HB2 | 1.98 2.22 | 2.90 1.75 | 0.40 | 3789 3828 3875 | 0.0219 |
| 1,2,5-pentanetriol | HB1 HB2 | 1.80 2.25 | 4.97 1.78 | 0.55 | 3669 3825 3865 | 0.0334 |
| 1,3,5-pentanetriol | HB1 HB2 | 1.94 1.96 | 2.91 2.90 | 0.58 | 3763 3792 3875 | 0.0225 0.0239 |
| 2,3,4-pentanetriol b | HB1 HB2 HB3 | 2.12 2.02 2.56 | 2.18 2.94 1.50 | 0.52 | 3759 3731 3820 | 0.0223 |
| 2,4,6-heptanetriol | HB1 HB2 | 1.92 1.93 | 3.02 2.94 | 0.65 | 3753 3773 3857 | 0.0250 0.0242 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deshmukh, M.M.; Gadre, S.R. Molecular Tailoring Approach for the Estimation of Intramolecular Hydrogen Bond Energy. Molecules 2021, 26, 2928. https://doi.org/10.3390/molecules26102928
Deshmukh MM, Gadre SR. Molecular Tailoring Approach for the Estimation of Intramolecular Hydrogen Bond Energy. Molecules. 2021; 26(10):2928. https://doi.org/10.3390/molecules26102928
Chicago/Turabian StyleDeshmukh, Milind M., and Shridhar R. Gadre. 2021. "Molecular Tailoring Approach for the Estimation of Intramolecular Hydrogen Bond Energy" Molecules 26, no. 10: 2928. https://doi.org/10.3390/molecules26102928
APA StyleDeshmukh, M. M., & Gadre, S. R. (2021). Molecular Tailoring Approach for the Estimation of Intramolecular Hydrogen Bond Energy. Molecules, 26(10), 2928. https://doi.org/10.3390/molecules26102928