
Rational Design and Synthesis of AT1R Antagonists


 and
and 
Abstract
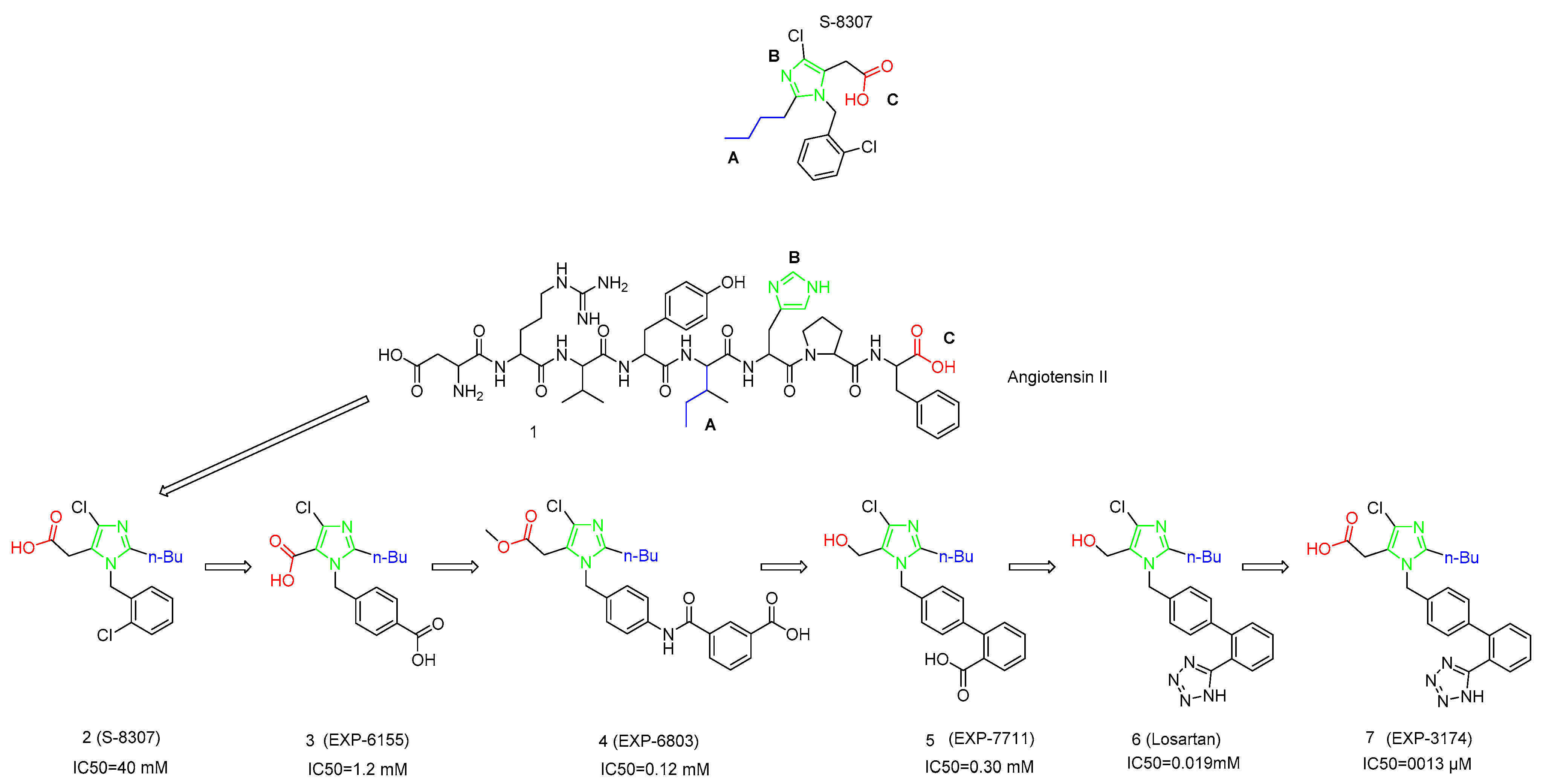
1. Introduction


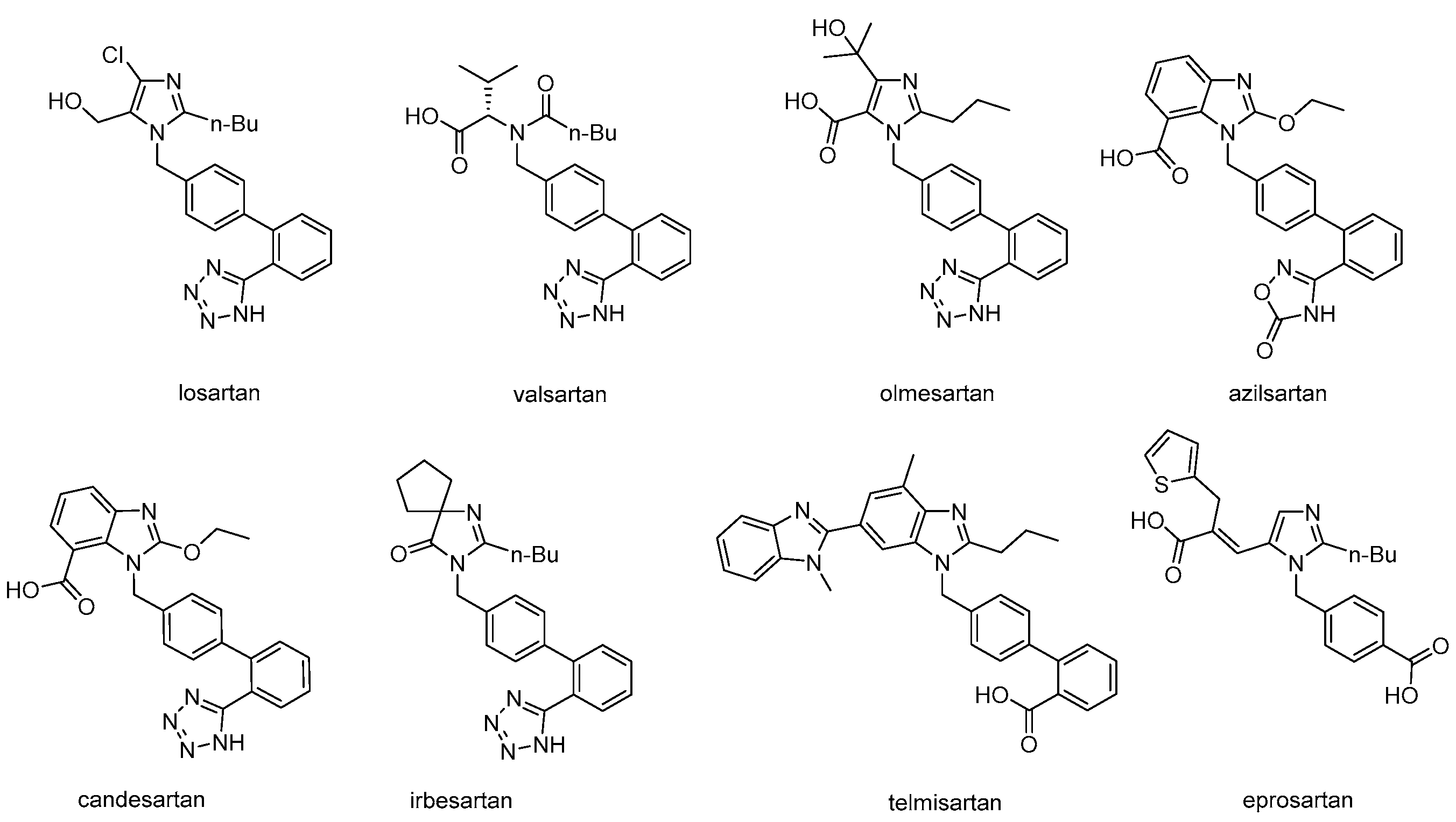
2. Synthetic Routes of Different Sartans
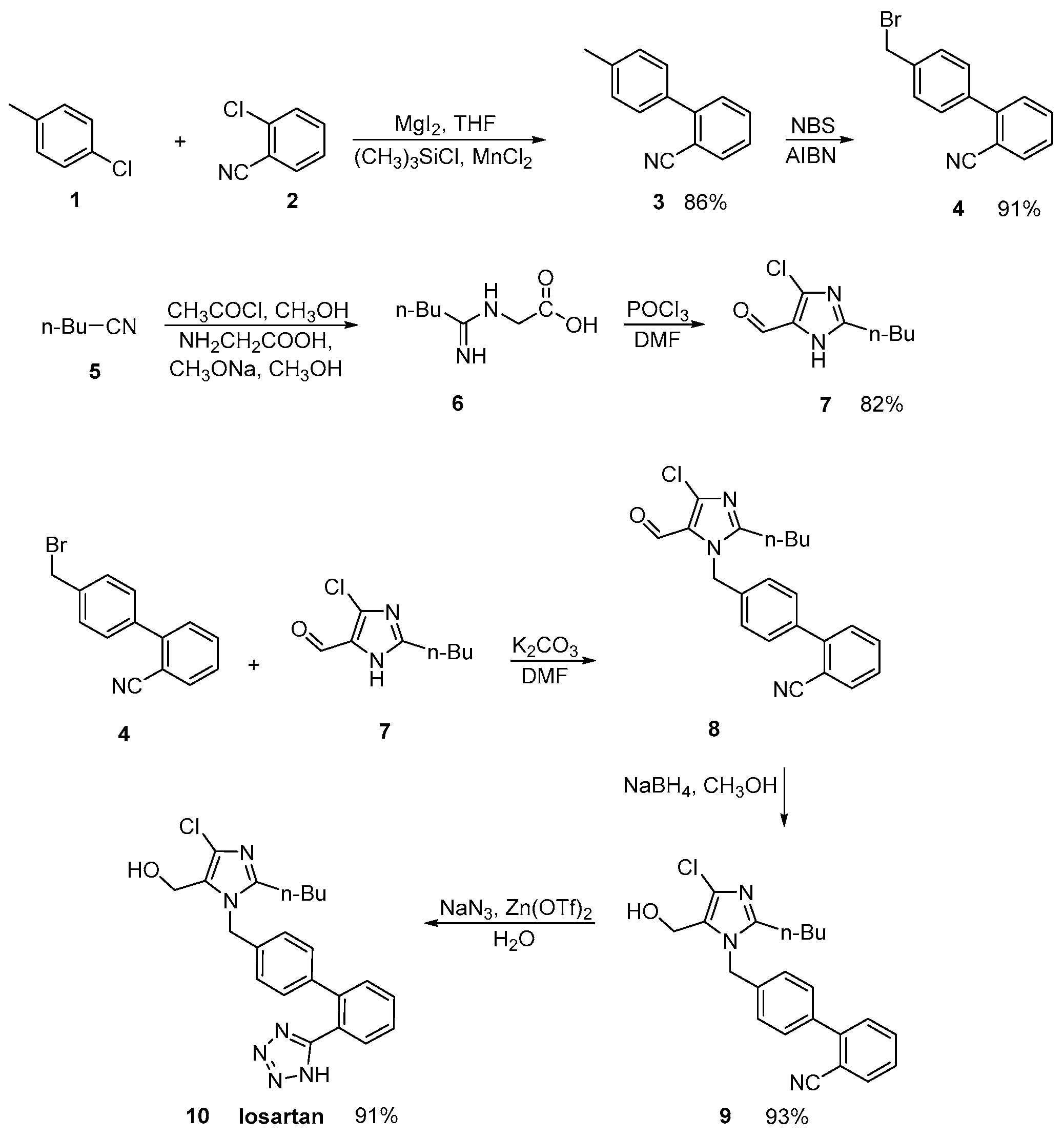
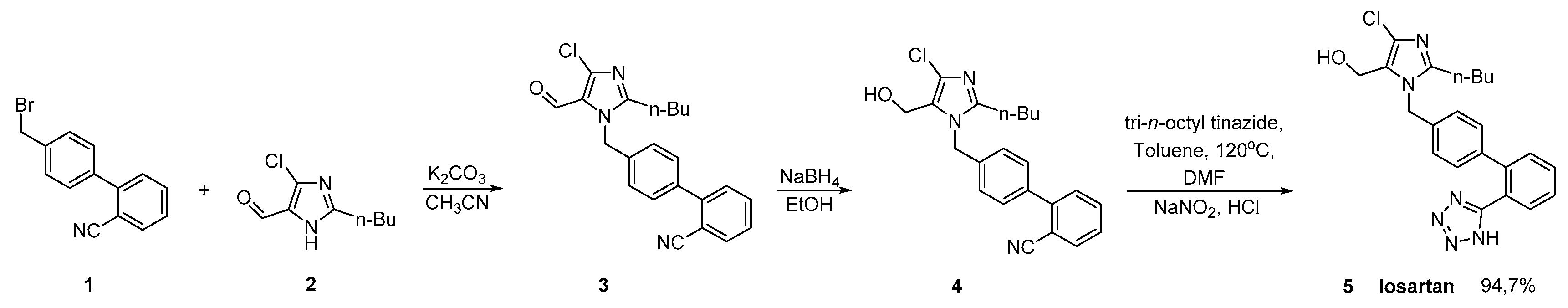
2.1. Synthetic Routes of Losartan
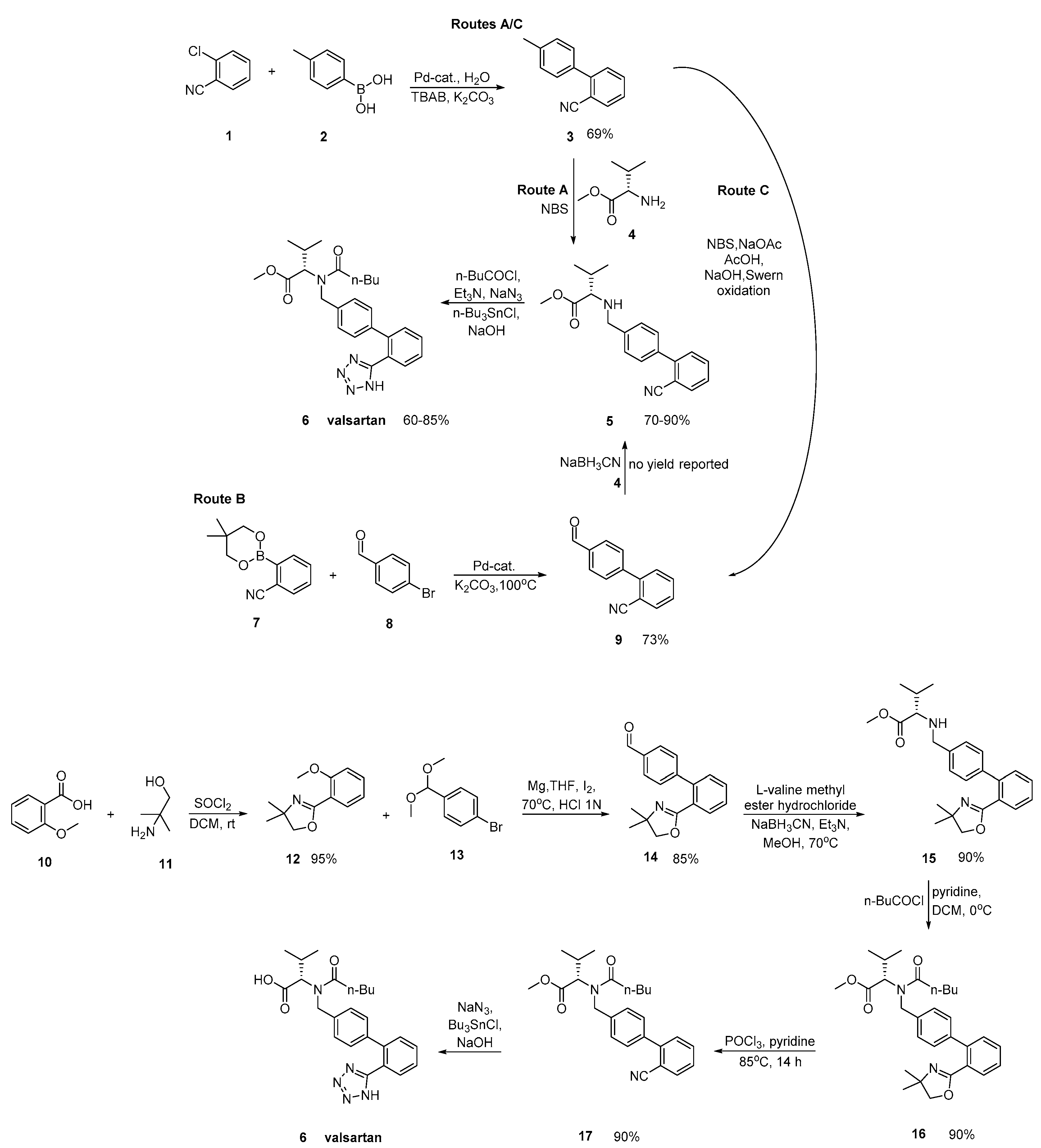
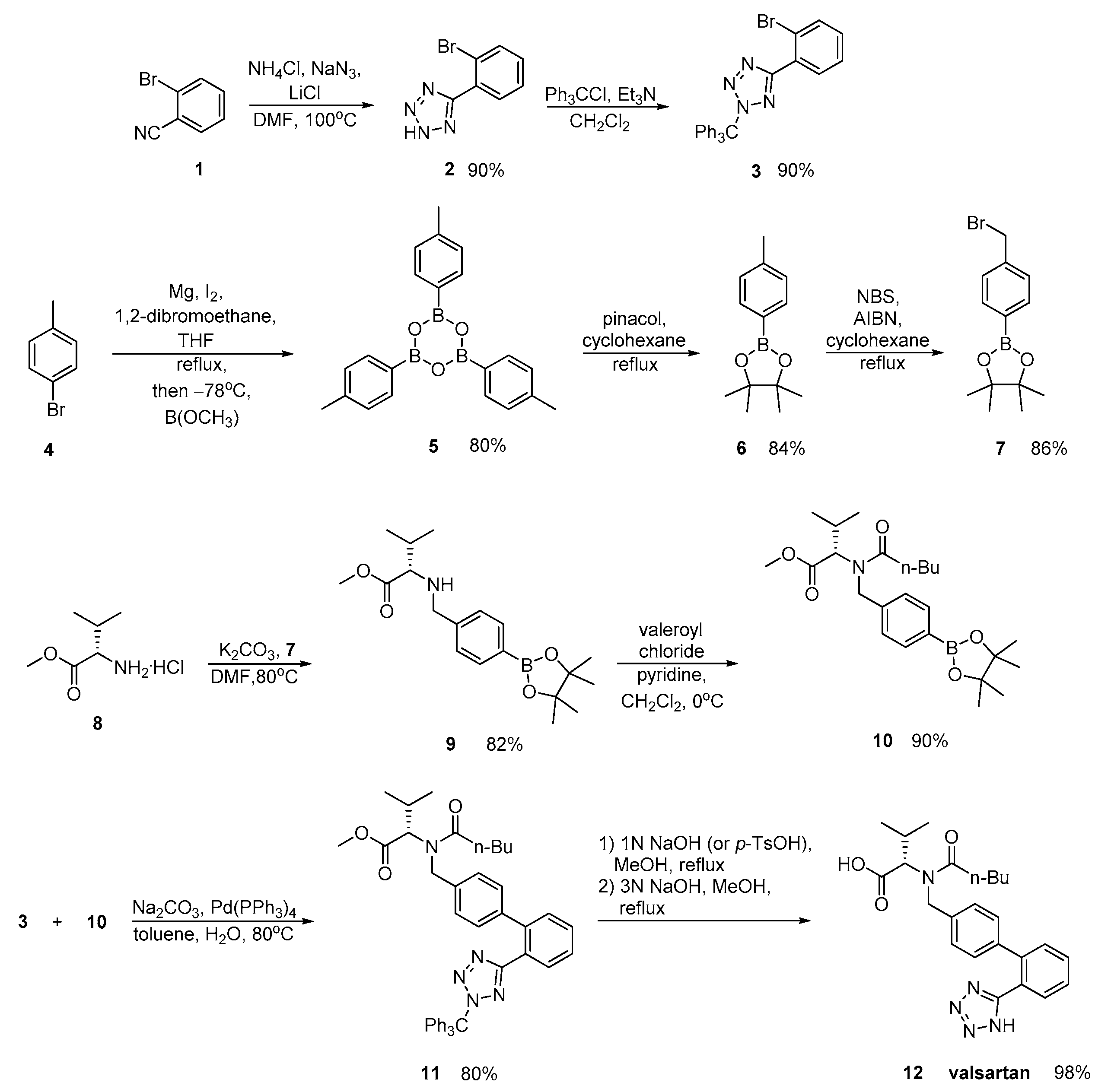
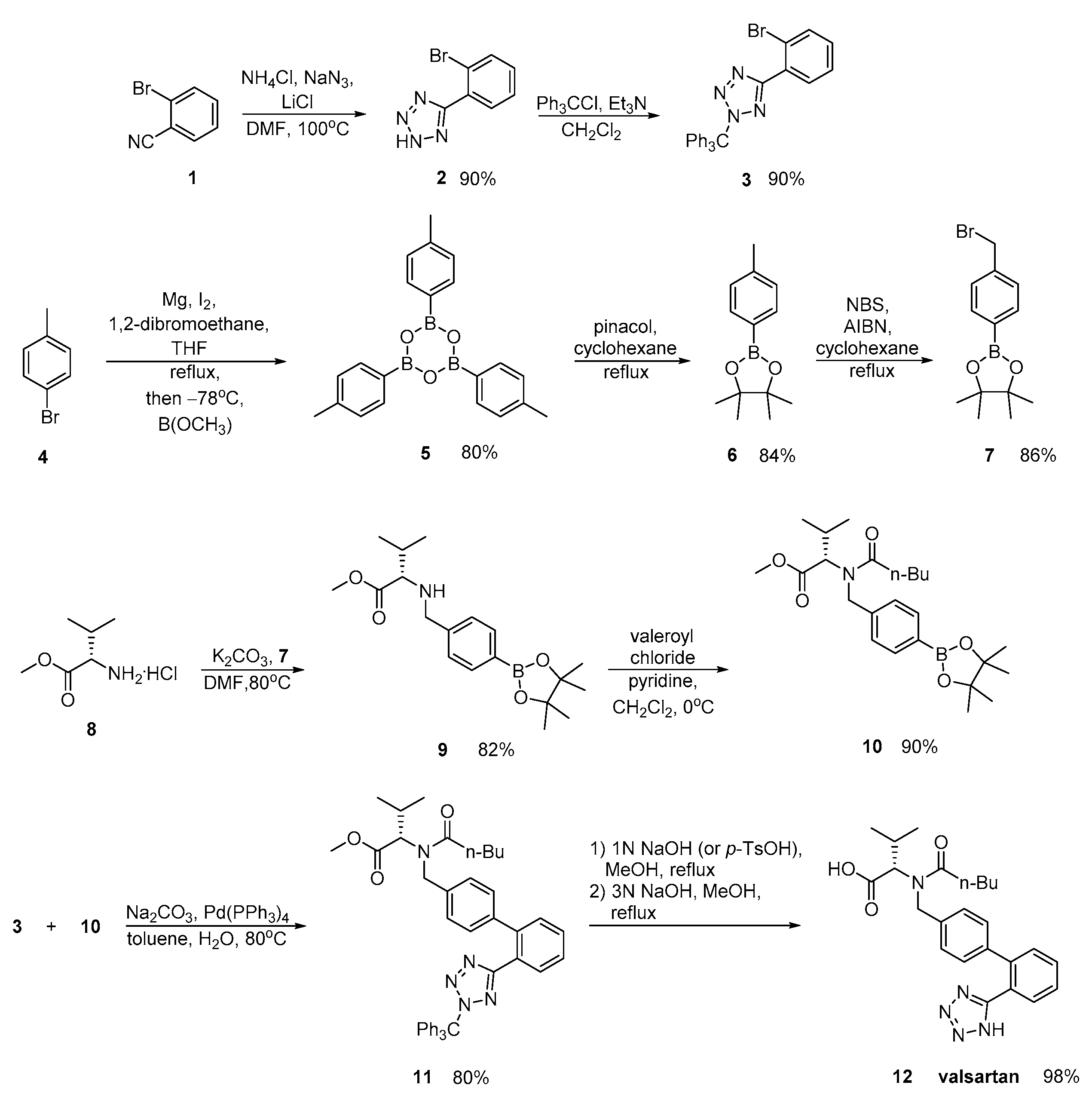
2.2. Synthetic Routes for Valsartan



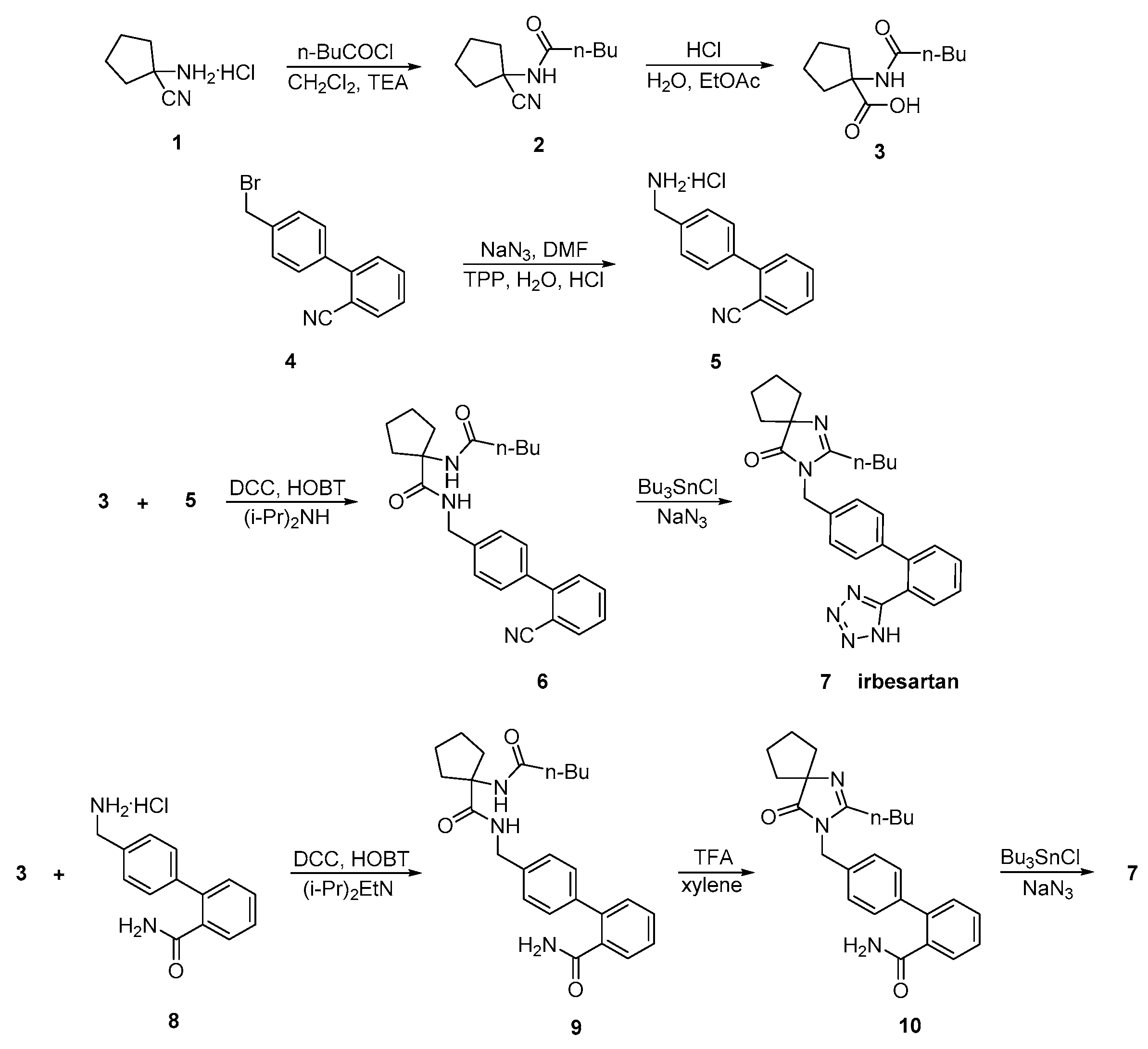
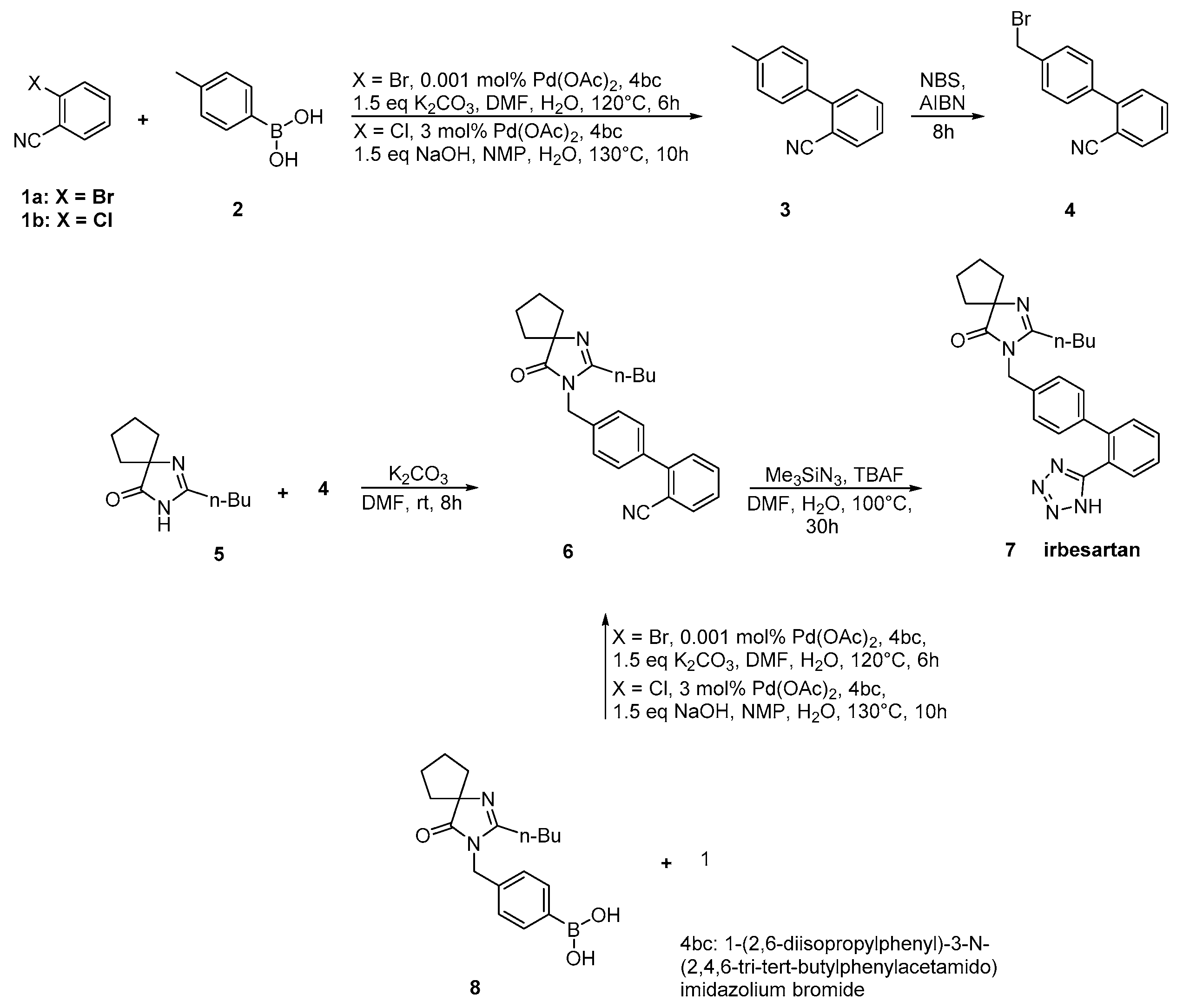
2.3. Synthetic Routes of Irbesartan

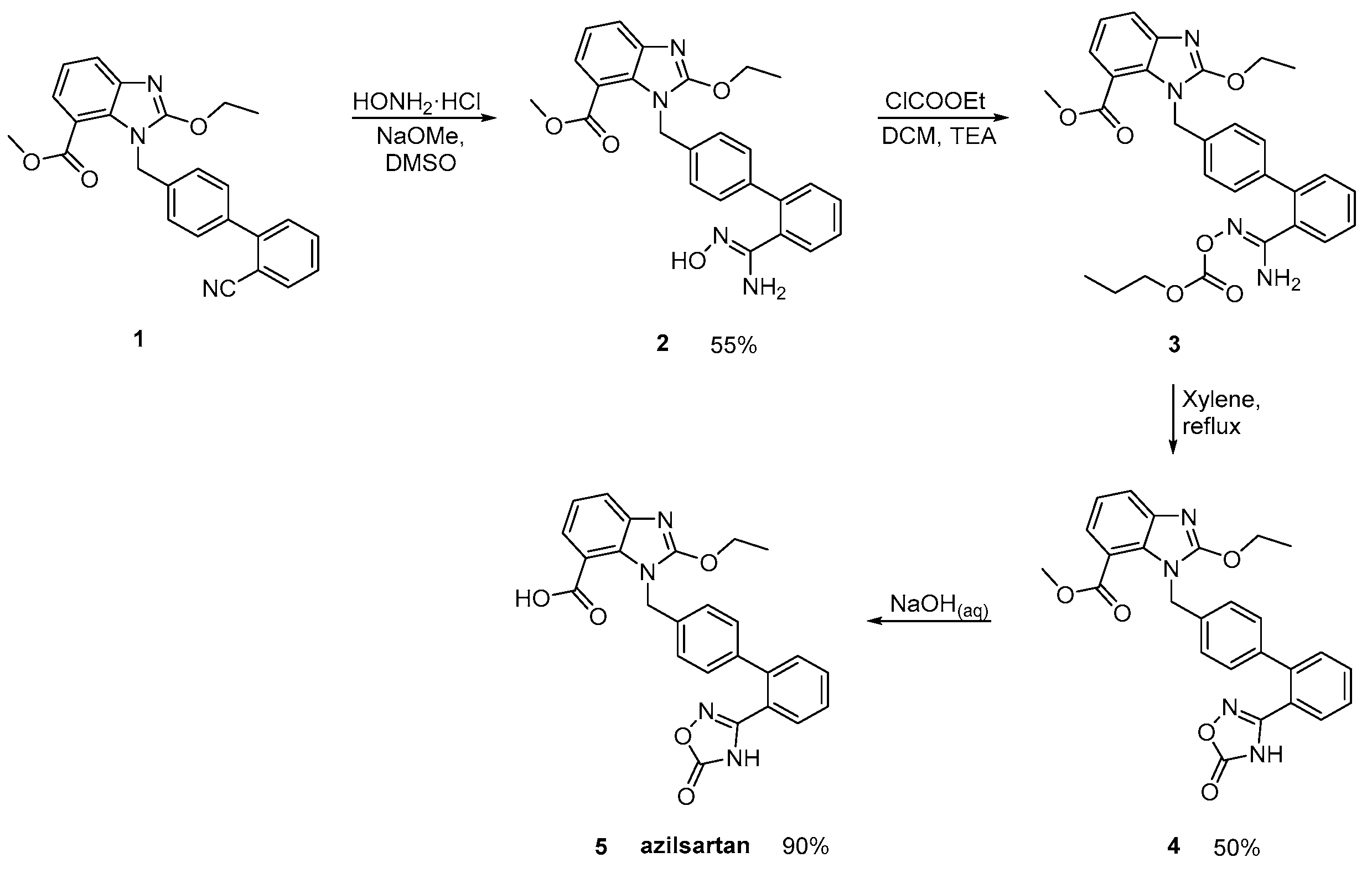
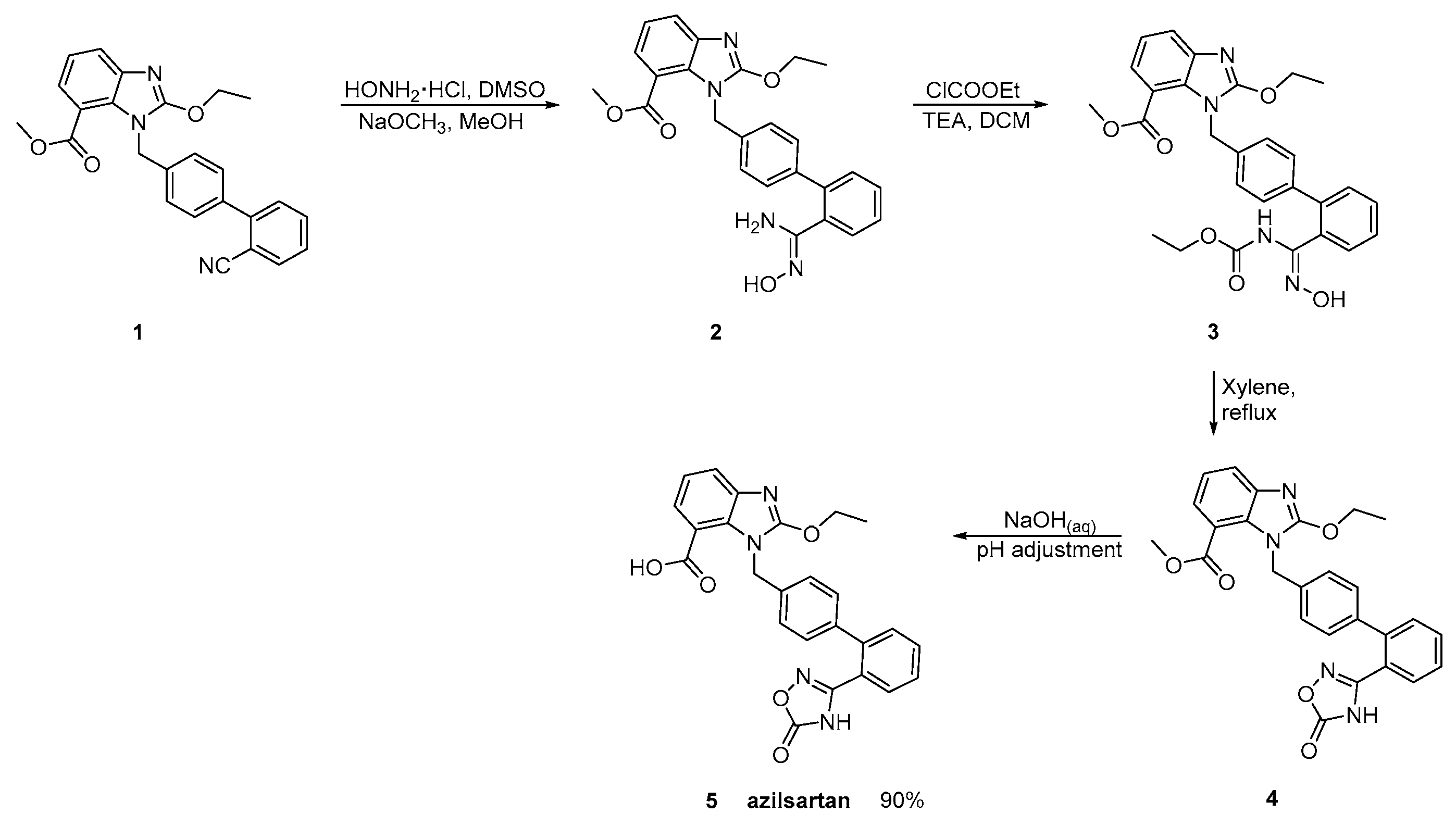
2.4. Synthetic Routes of Azilsartan


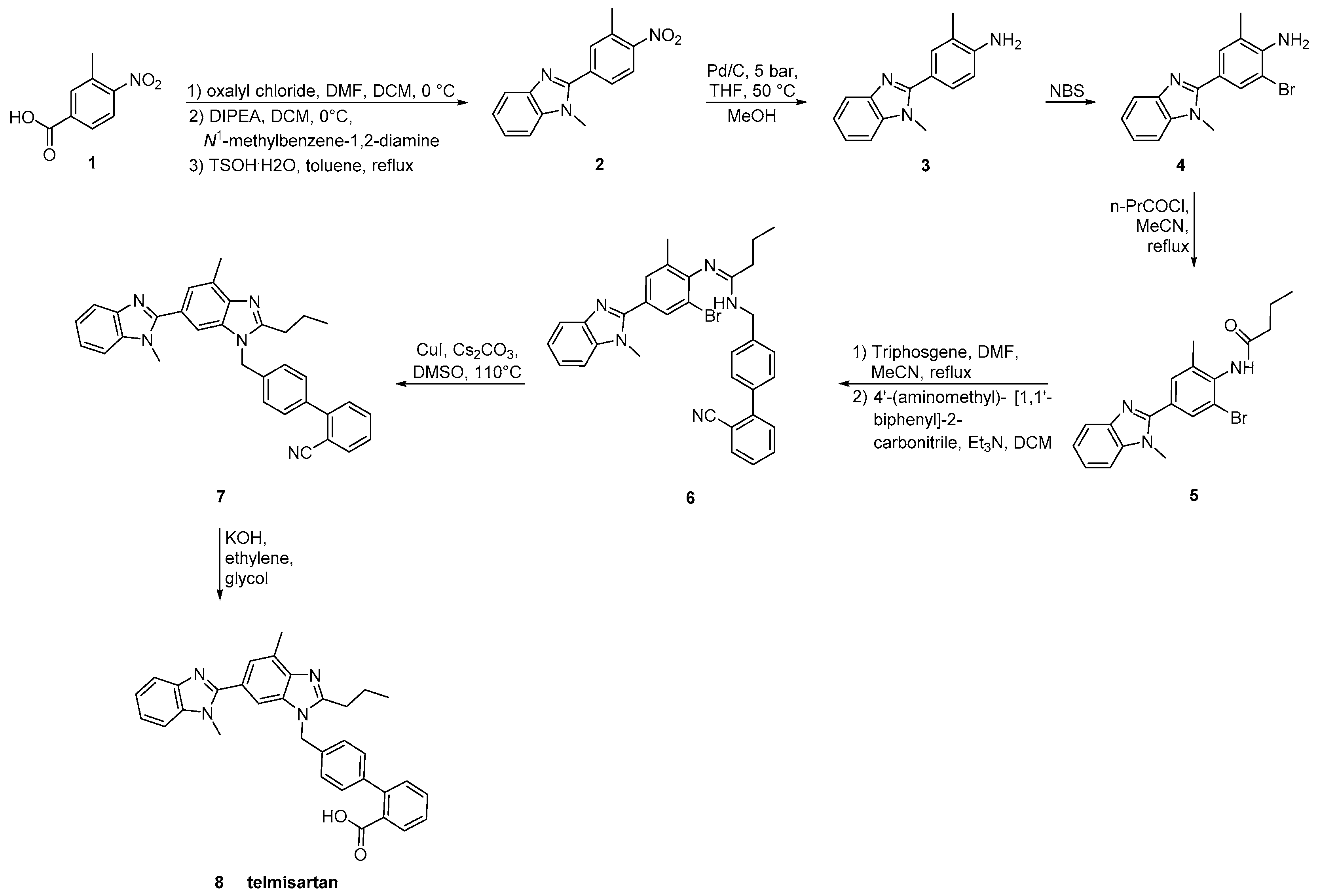
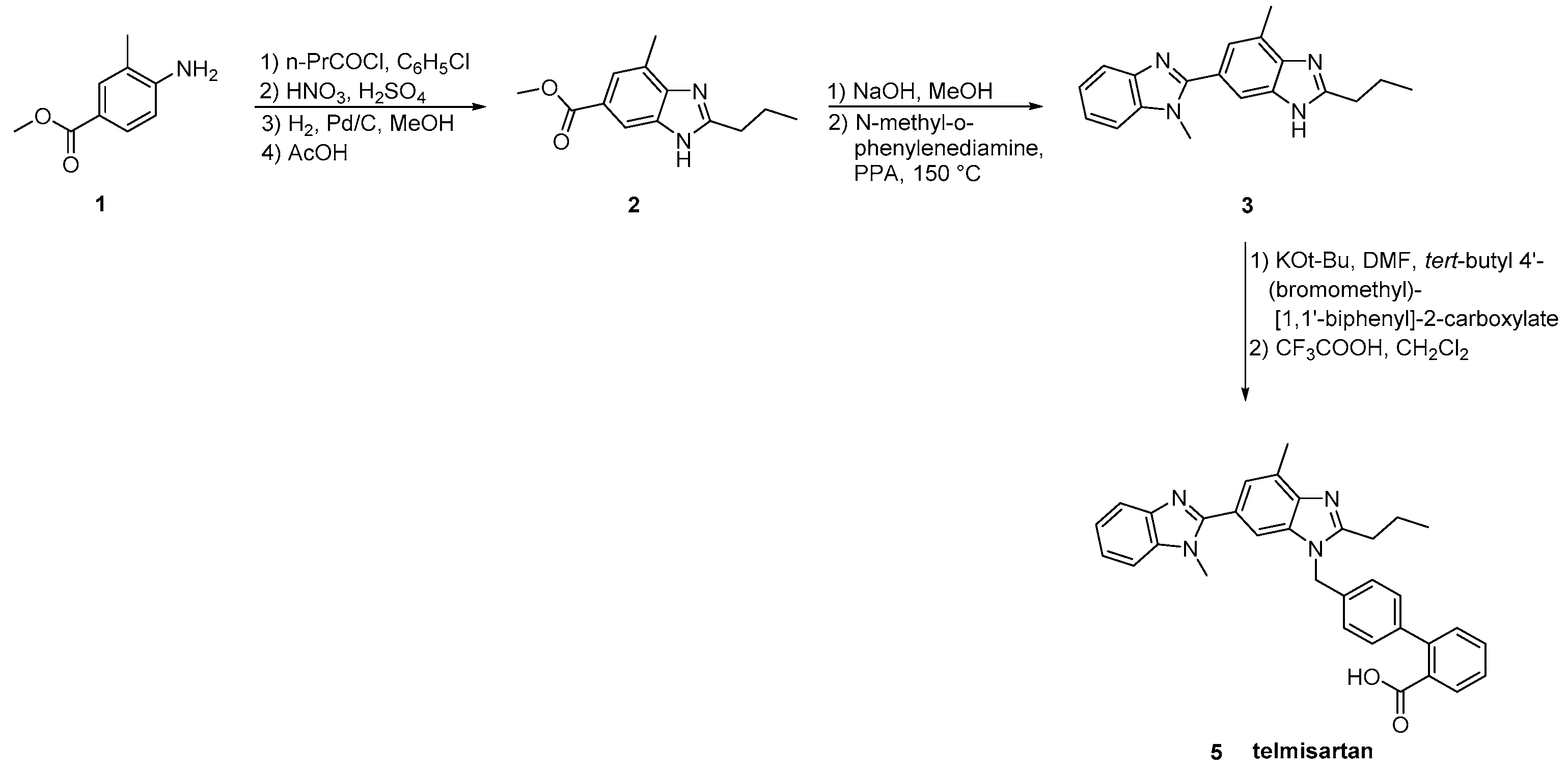
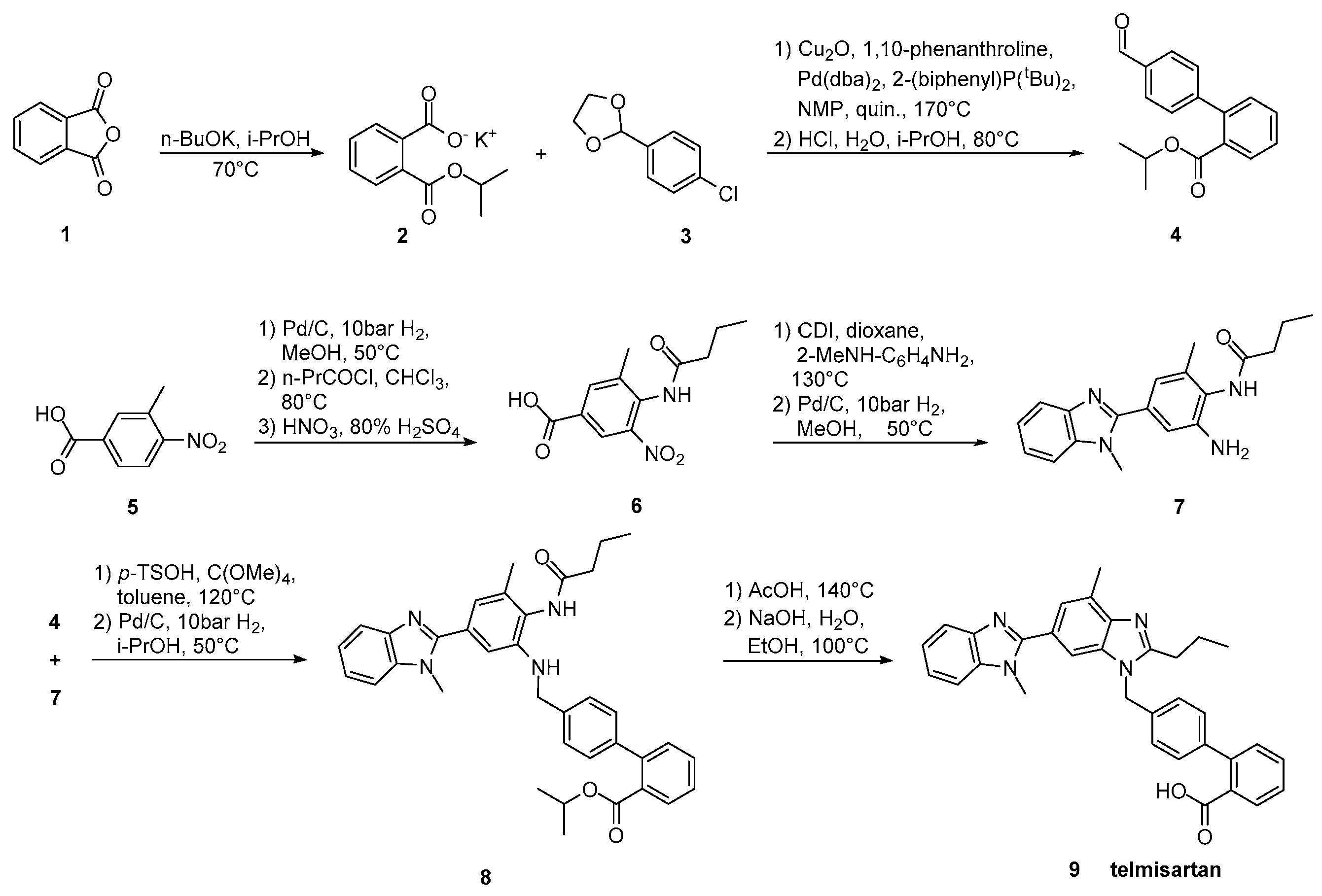
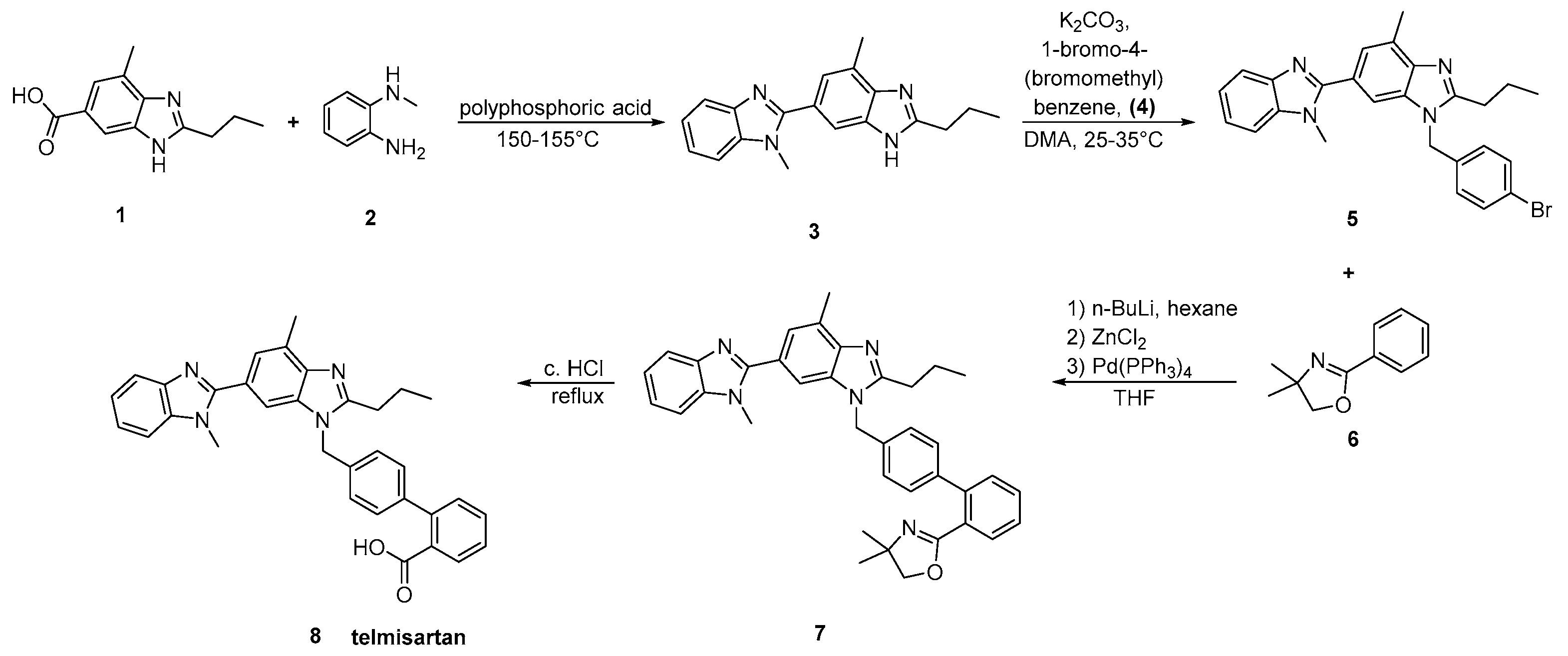
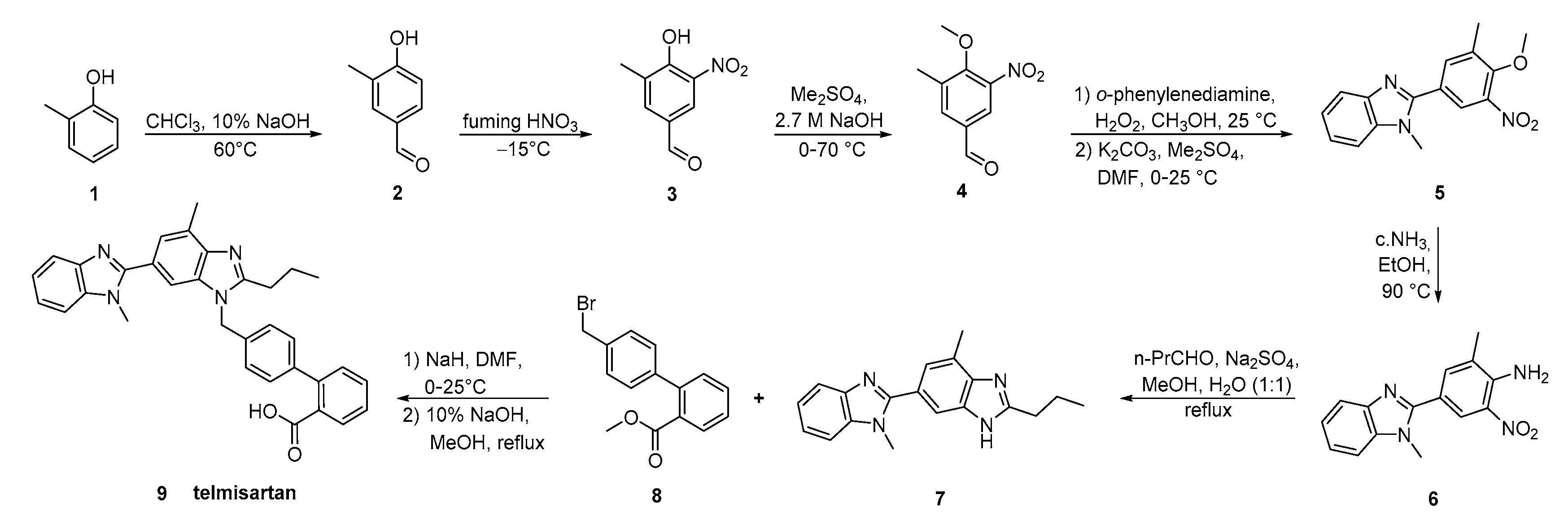
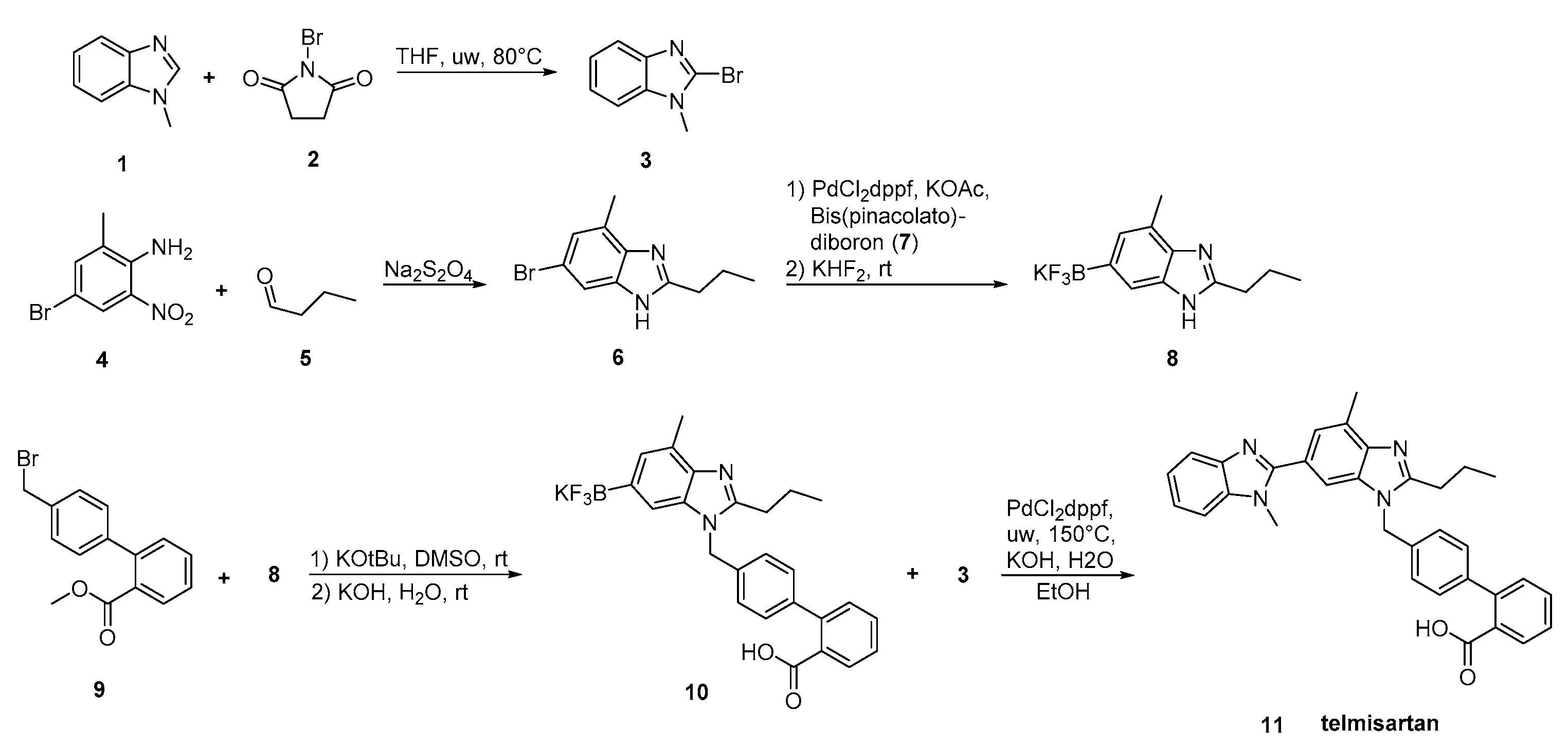
2.5. Synthetic Routes of Telmisartan





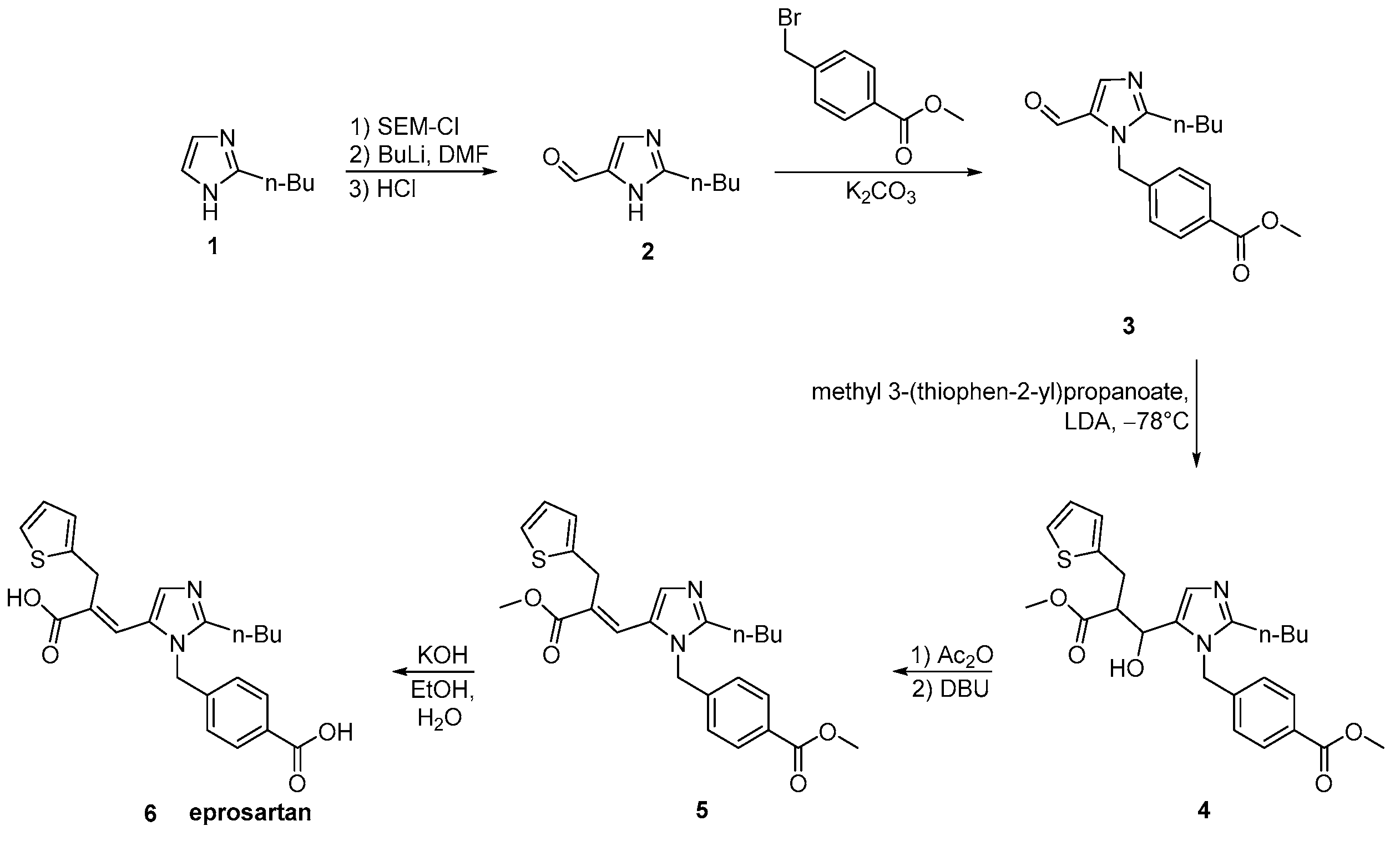
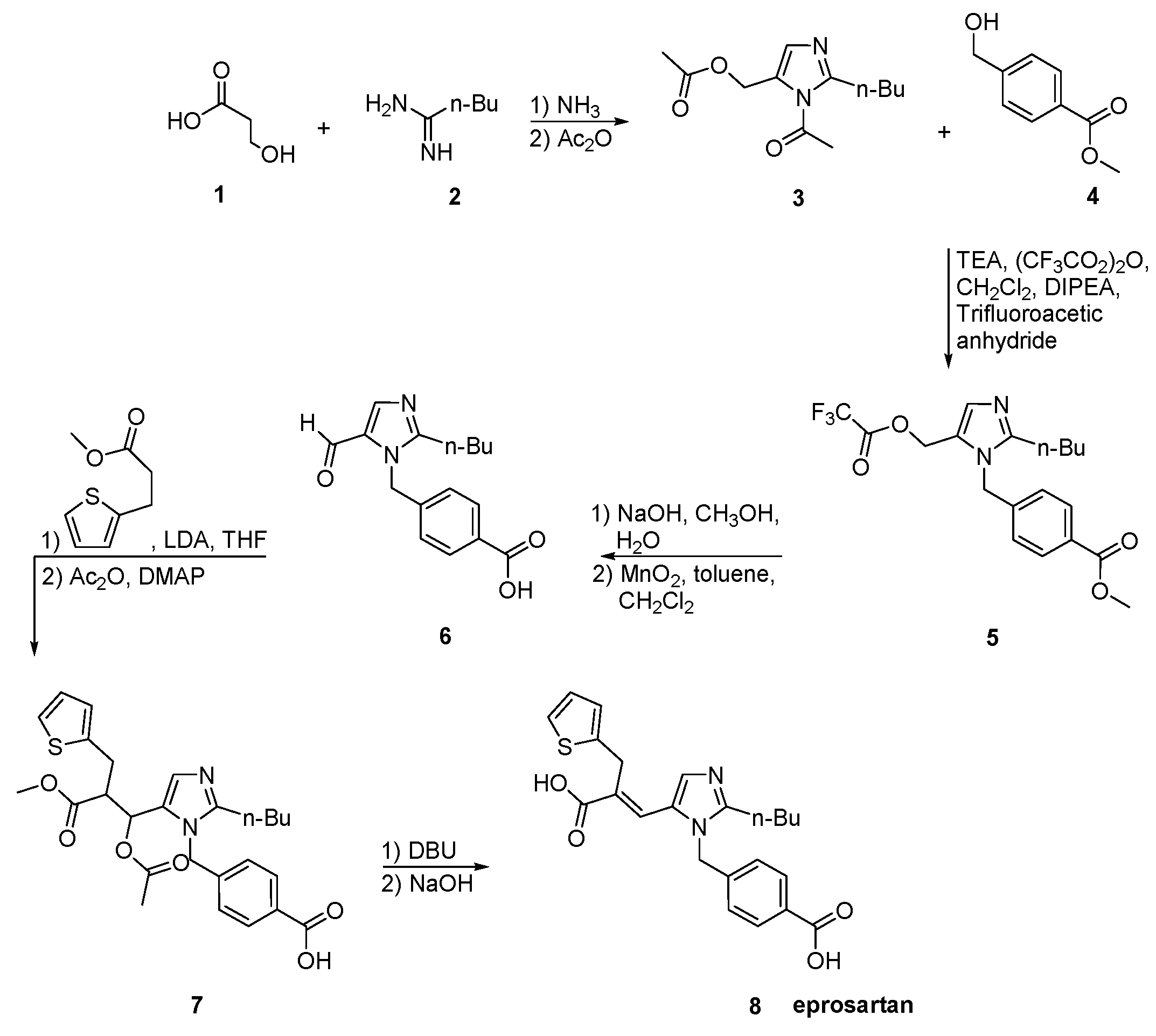
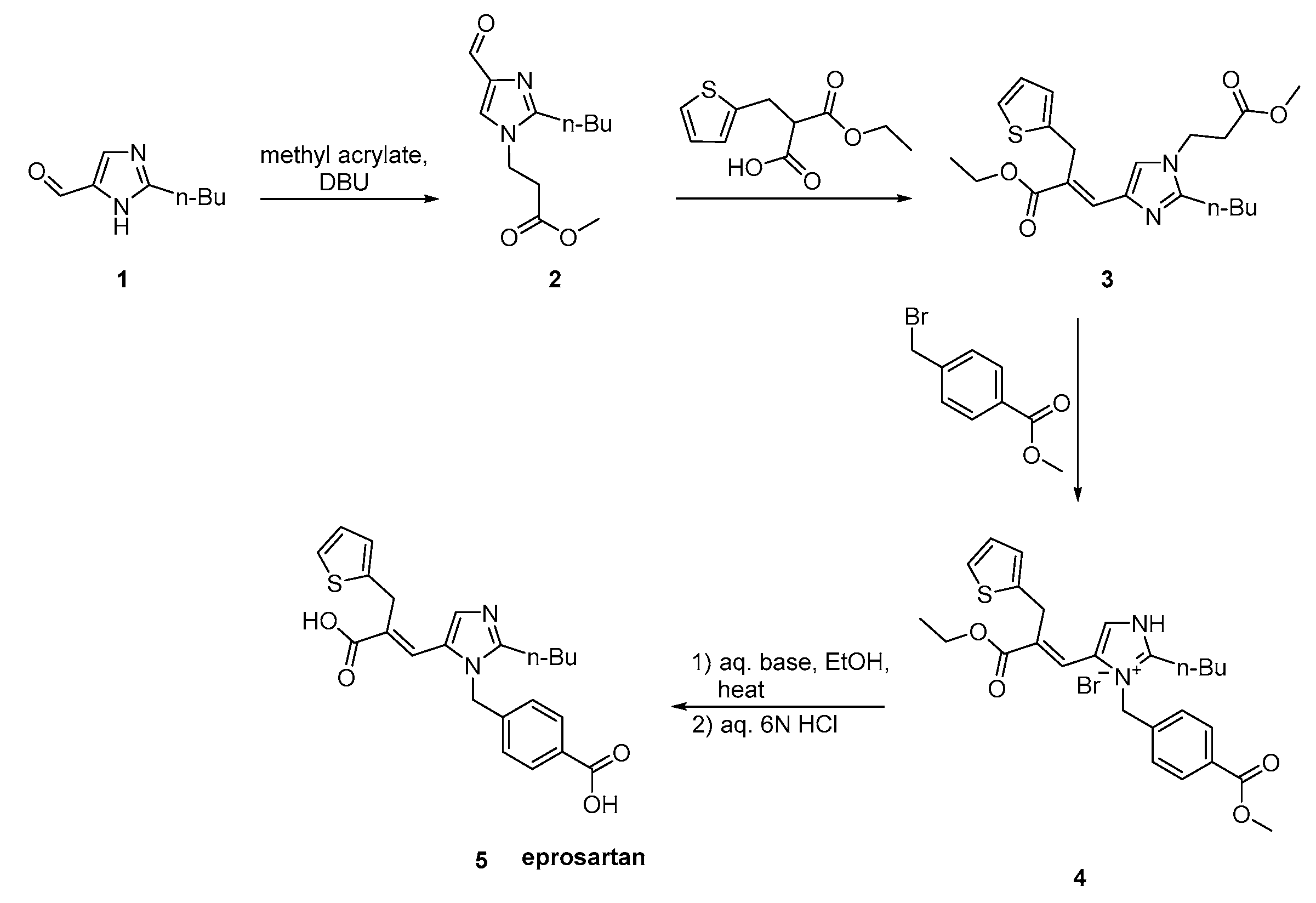
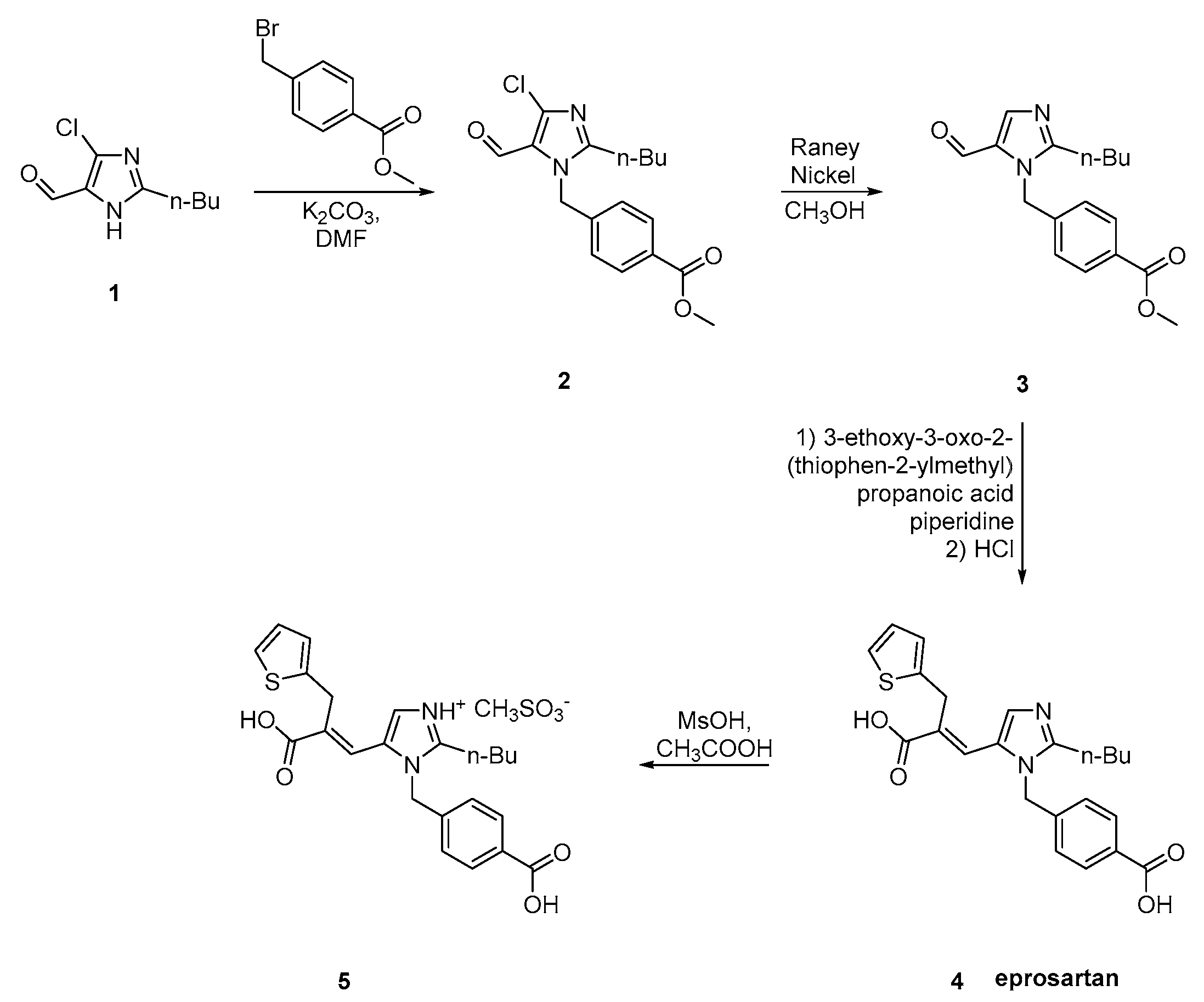
2.6. Synthetic Routes of Eprosartan



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
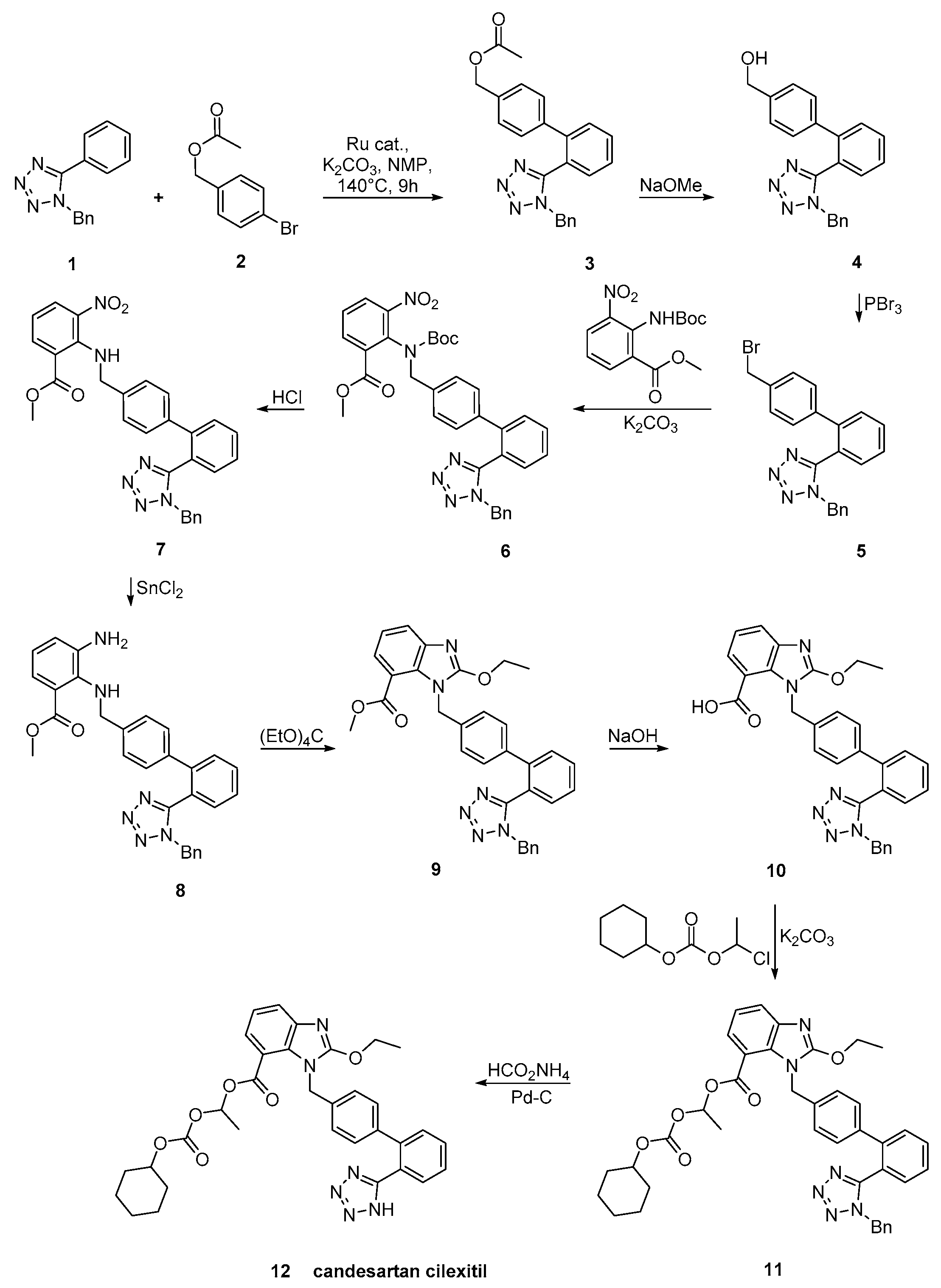
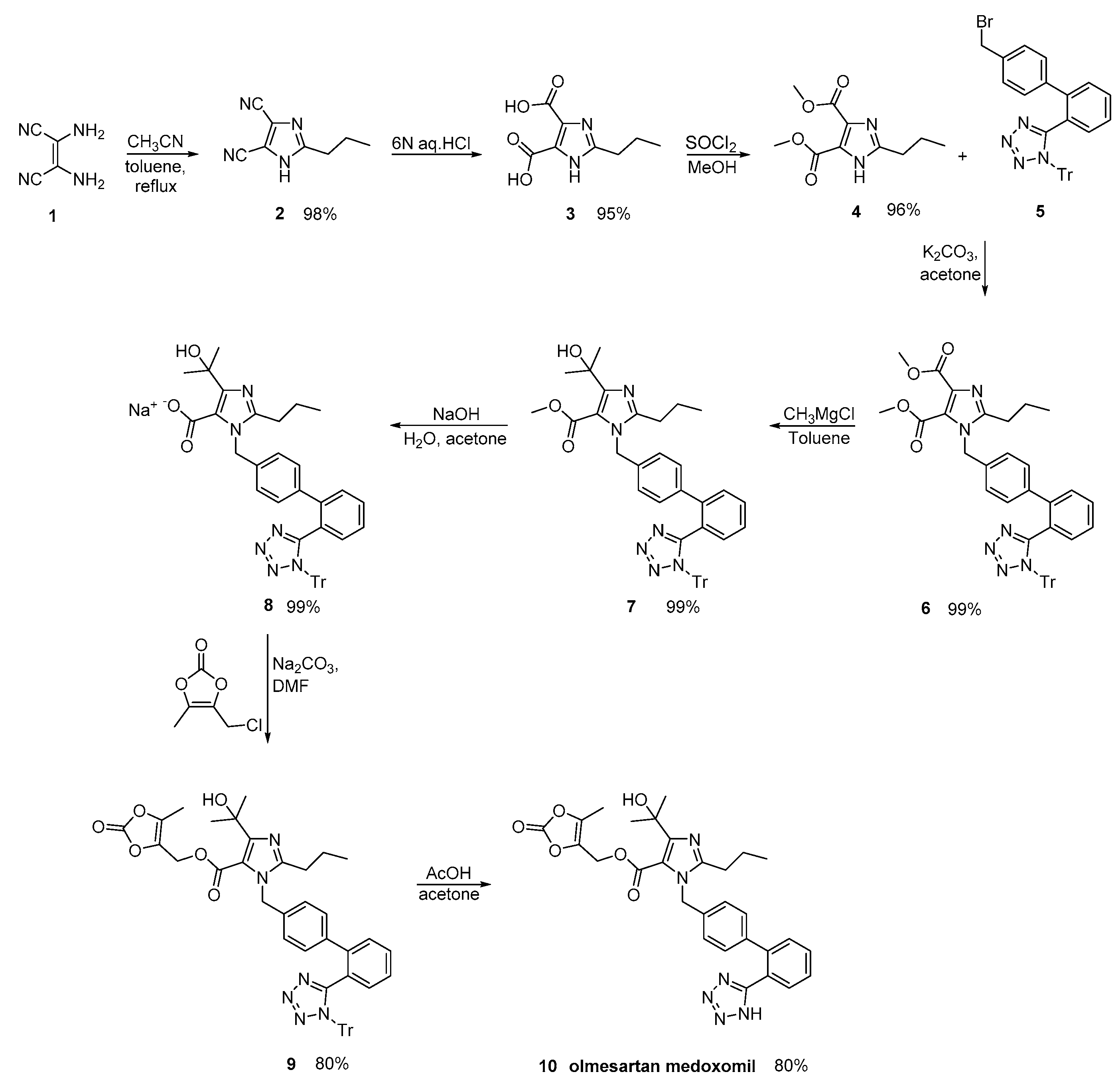
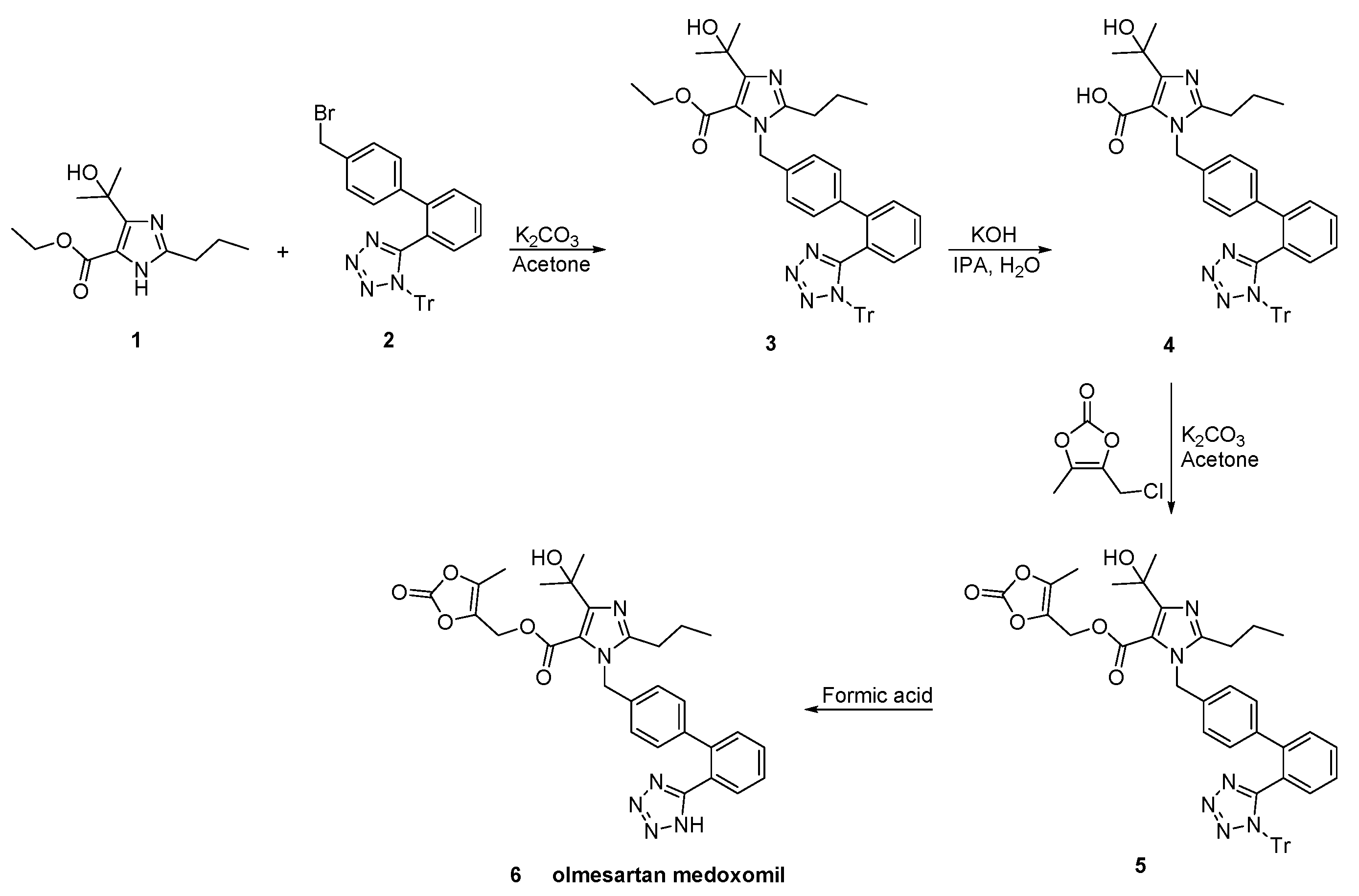
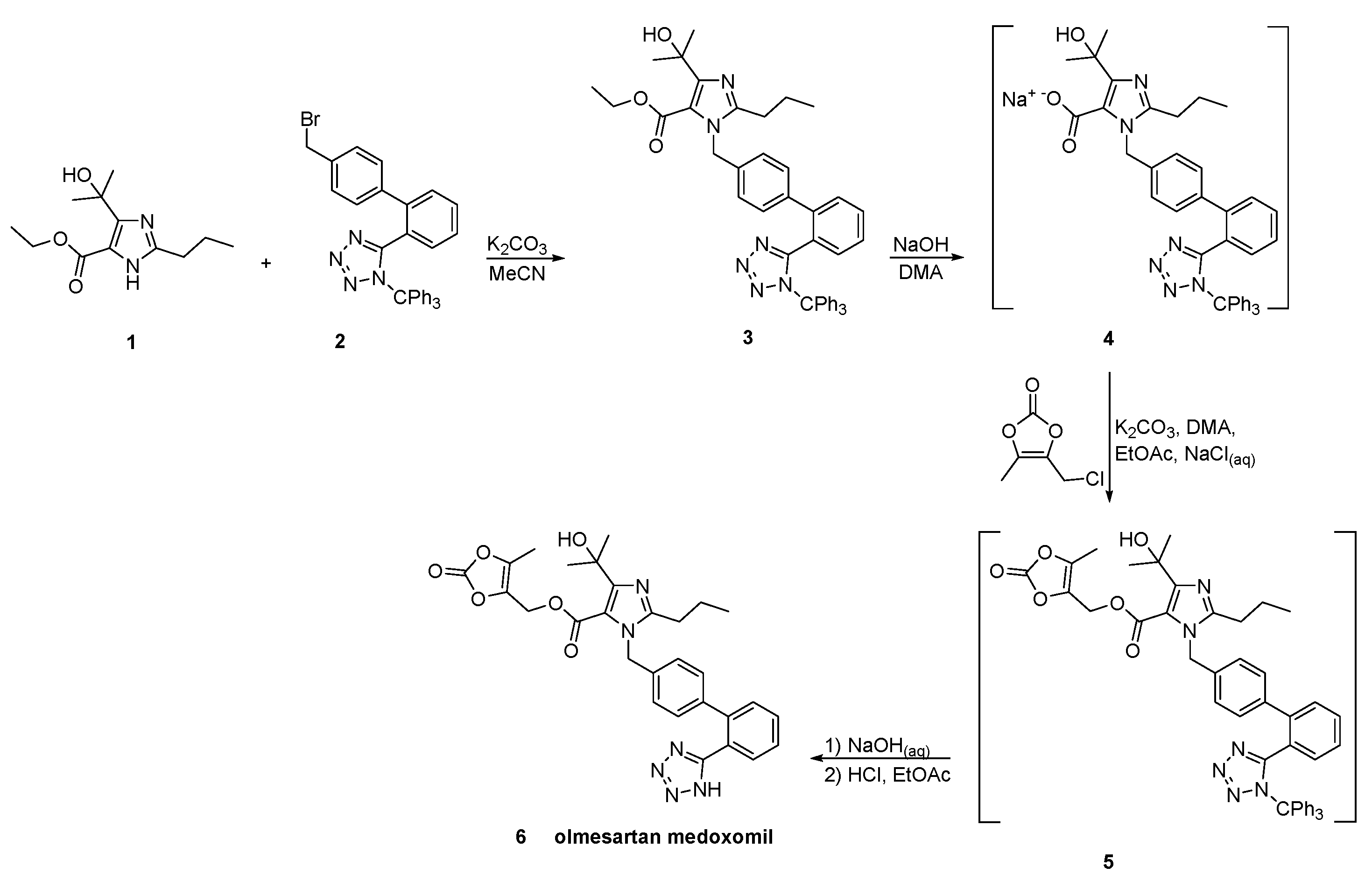
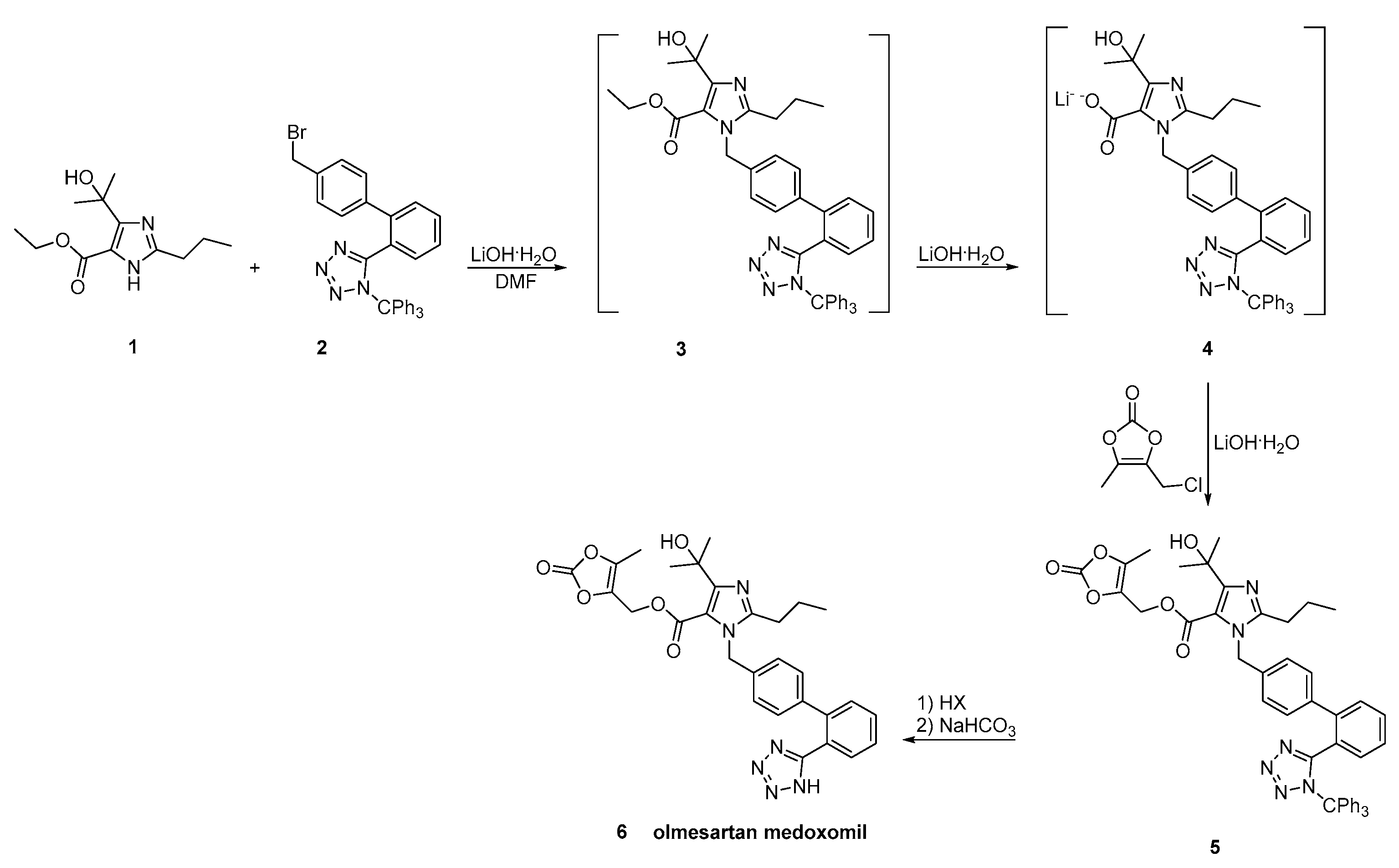
2.7. Synthetic Routes of Olmesartan Medoxomil & Candesartan Cilexetil





3. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higuchi, S.; Ohtsu, H.; Suzuki, H.; Shirai, H.; Frank, G.D.; Eguchi, S. Angiotensin II signal transduction through the AT1 receptor: Novel insights into mechanisms and pathophysiology. Clin. Sci. 2007, 112, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Duncia, J.V.; Carini, D.J.; Chiu, A.T.; Johnson, A.L.; Price, W.A.; Wong, P.C.; Wexler, R.R.; Timmermans, P.B.M. The discovery of DuP 753, a potent, orally active nonpeptide angiotensin II receptor antagonist. Med. Res. Rev. 1992, 12, 149–191. [Google Scholar] [CrossRef] [PubMed]
- Carini, D.J.; Duncia, J.V.; Aldrich, P.E.; Chiu, A.T.; Johnson, A.L.; Pierce, M.E.; Price, W.A.; Santella, J.B.; Wells, G.J. Nonpeptide angiotensin II receptor antagonists: The discovery of a series of N-(biphenylylmethyl)imidazoles as potent, orally active antihypertensives. J. Med. Chem. 1991, 34, 2525–2547. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kishimoto, S.; Nishikawa, K. Hypotensive Imidazole-5-Acetic Acid Derivatives. U.S. Patent 4,355,040, 19 October 1989. [Google Scholar]
- Bhardwaj, G. How the antihypertensive losartan was discovered. Expert Opin. Drug Discov. 2006, 1, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Middlemiss, D.; Drew, G.M.; Ross, B.C.; Robertson, M.J.; Scopes, D.I.; Dowle, M.D.; Akers, J.; Cardwell, K.; Clark, K.L.; Coote, S.; et al. Bromobenzofurans: A new class of potent, non-peptide antagonists of angiotensin II. Bioorg. Med. Chem. Lett. 1991, 1, 711–716. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Mantlo, N.B.; Chakravarty, P.K.; Ondeyka, D.L.; Siegl, P.K.S.; Chang, R.S.; Lotti, V.J.; Faust, K.A.; Schorn, T.W.; Chen, T.B. Potent, orally active imidazo[4,5-b] pyridine-based angiotensin II receptor antagonists. J. Med. Chem. 1991, 34, 2919–2922. [Google Scholar] [CrossRef] [PubMed]
- Mavromoustakos, T.; Apostolopoulos, V.; Matsoukas, J. Antihypertensive drugs that act on renin-angiotensin system with emphasis in AT1 antagonists. Mini Rev. Med. Chem. 2001, 1, 207–217. [Google Scholar] [CrossRef]
- Wingler, L.M.; McMahon, C.; Staus, D.P.; Lefkowitz, R.J.; Kruse, A.C. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell 2019, 176, 479–490. [Google Scholar] [CrossRef]
- De, B.; Winn, M.; Zydowsky, T.M.; Kerkman, D.J.; DeBernardis, J.F.; Lee, J.; Buckner, S.; Warner, R.; Brune, M.; Hancock, A.; et al. Discovery of a novel class of orally active, non-peptide angiotensin II antagonists. J. Med. Chem. 1992, 35, 3714–3717. [Google Scholar] [CrossRef]
- Lin, H.S.; Rampersaud, A.A.; Zimmerman, K.; Steinberg, M.I.; Boyd, D.B. Nonpeptide angiotensin II receptor antagonists: Synthetic and computational chemistry of N-[[4-[2-(2H-tetrazol-5-yl)-1-cycloalken-1-yl] phenyl]methyl]imidazole derivatives and their in vitro activity. J. Med. Chem. 1992, 35, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, R.H.; Allott, C.P.; Dennis, M.; Girdwood, J.A.; Kenny, P.W.; Major, J.S.; Oldham, A.A.; Ratcliffe, A.H.; Rivett, J.E. New nonpeptide angiotensin II receptor antagonists. 3. Synthesis, biological properties, and structure-activity relationships of 2-alkyl-4-(biphenylylmethoxy) pyridine derivatives. J. Med. Chem. 1993, 36, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Ries, U.J.; Mihm, G.; Narr, B.; Hasselbach, K.M.; Wittneben, H.; Entzeroth, M.; van Meel, J.C.A.; Wienen, W.; Hauel, N.H. 6-Substituted benzimidazoles as new nonpeptide angiotensin II receptor antagonists: Synthesis, biological activity, and structure-activity relationships. J. Med. Chem. 1993, 36, 4040–4051. [Google Scholar] [CrossRef] [PubMed]
- Kellici, T.; Tzakos, A.; Mavromoustakos, T. rational drug design and synthesis of molecules targeting the angiotensin II type 1 and type 2 receptors. Molecules 2015, 20, 3868–3897. [Google Scholar] [CrossRef]
- Mavromoustakos, T.; Agelis, G.; Durdagi, S. AT1 antagonists: A patent review (2008–2012). Expert Opin. Ther. Pat. 2013, 23, 1483–1494. [Google Scholar] [CrossRef]
- Mavromoustakos, T.; Zoumpoulakis, P.; Kyrikou, I.; Zoga, A.; Siapi, E.; Zervou, M.; Daliani, I.; Dimitriou, D.; Pitsas, A.; Kamoutsis, C.; et al. Efforts to understand the molecular basis of hypertension through drug: Membrane interactions. Curr. Top. Med. Chem. 2004, 4, 445–459. [Google Scholar] [CrossRef]
- Mavromoustakos, T.; Zervou, M.; Zoumpoulakis, P.; Kyrikou, I.; Benetis, N.; Polevaya, L.; Roumelioti, P.; Giatas, N.; Zoga, A.; Minakakis, P. Conformation and bioactivity. design and discovery of novel antihypertensive drugs. Curr. Top. Med. Chem. 2004, 4, 385–401. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural basis for ligand recognition and functional selectivity at angiotensin receptor. J. Biol. Chem. 2015, 290, 29127–29139. [Google Scholar] [CrossRef]
- Wingler, L.M.; Skiba, M.A.; McMahon, C.; Staus, D.P.; Kleinhenz, A.L.W.; Suomivuori, C.-M.; Latorraca, N.R.; Dror, R.O.; Lefkowitz, R.J.; Kruse, A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 2020, 367, 888–892. [Google Scholar] [CrossRef]
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A rational approach in drug design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef]
- Thornber, C.W. Isosterism and molecular modification in drug design. Chem. Soc. Rev. 1979, 8, 563. [Google Scholar] [CrossRef]
- Lassalas, P.; Gay, B.; Lasfargeas, C.; James, M.J.; Tran, V.; Vijayendran, K.G.; Brunden, K.R.; Kozlowski, M.C.; Thomas, C.J.; Smith, A.B.; et al. Structure property relationships of carboxylic acid isosteres. J. Med. Chem. 2016, 59, 3183–3203. [Google Scholar] [CrossRef]
- Ballatore, C.; Huryn, D.M.; Smith, A.B. Carboxylic acid (bio) isosteres in drug design. ChemMedChem 2013, 8, 385–395. [Google Scholar] [CrossRef]
- Kurtz, T.W.; Kajiya, T. Differential pharmacology and benefit/risk of azilsartan compared to other sartans. Vasc. Health Risk Manag. 2012, 133. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shuangxia, F.; Zheng, G.; Yelv, T.; Hui, L.; Guofang, J. An efficient and green synthetic route to losartan. J. Chem. Res. 2015, 39, 451–454. [Google Scholar] [CrossRef]
- Reddy, V. Process for the Preparation of Losartan. Patent Application Publication No. 2010/0222597 A1, 2 September 2010. [Google Scholar]
- Ghosh, S.; Kumar, A.S.; Soundararajan, R.; Mehta, G.N. improved synthesis of valsartan via nucleophilic aromatic substitution on aryloxazoline. Synth. Commun. 2009, 39, 3880–3887. [Google Scholar] [CrossRef]
- Zhang, C.; Zheng, G.; Fang, L.; Li, Y. Efficient synthesis of valsartan, a nonpeptide angiotensin II receptor–antagonist. Synlett 2006, 2006, 0475–0477. [Google Scholar] [CrossRef]
- Harel, Z.; Saba, K.; Rukhman, T.C. Process for the Preparation of Valsartan. Patent Application Publication No. US 2005/0010053 A1, 13 January 2005. U.S. provisional Appl. Ser. 2003. [Google Scholar]
- Kankan, R.N.; Rao, D.R.; Pathi, S.L.; Puppala, R. Process for Preparing Valsartan. Patent Application Publication No. US 2011/0105763 A1, 5 May 2011. [Google Scholar]
- Rao, K.; Dandala, R.; Handa, V.; Rao, I.; Rani, A.; Shivashankar, S.; Naidu, A. A novel approach for the conversion of primary amides into tetrazoles by using tributyltin chloride and sodium azide in the presence of DMF. Synlett 2007, 2007, 1289–1293. [Google Scholar] [CrossRef]
- Prasada Rao, K.V.V.; Dandala, R.; Handa, V.K.; Rao, I.V.S.; Rani, A.; Naidu, A. Improved methods for the synthesis of irbesartan, an antihypertensive active pharmaceutical ingredient. Synth. Commun. 2007, 37, 2897–2905. [Google Scholar] [CrossRef]
- Kumar, M.R.; Park, K.; Lee, S. Synthesis of amido-N-imidazolium salts and their applications as ligands in Suzuki-Miyaura reactions: Coupling of hetero-aromatic halides and the synthesis of milrinone and irbesartan. Adv. Synth. Catal. 2010, 352, 3255–3266. [Google Scholar] [CrossRef]
- Chapala, V.L.; Katari, N.K.; Malavattu, G.P.; Marisetti, V.M.; Jonnalagadda, S.B. An improved preparation of azilsartan. Org. Prep. Proced. Int. 2020, 52, 550–555. [Google Scholar] [CrossRef]
- Raghava Reddy, A.V.; Garaga, S.; Takshinamoorthy, C.; Gupta, B.; Naidu, A. Improved synthesis of azilsartan: Development and control of process related impurities. Indo Am. J. Pharm. Res. 2015, 5, 2208–2216. [Google Scholar]
- FDA: Another Blood Pressure Drug, Irbesartan, Recalled over Cancer-Causing Impurity. Available online: https://eu.usatoday.com/story/news/health/2019/01/18/prinston-pharmaceuticals-recall-irbesartan-blood-pressure-medicines-fda-valsartan-losartan/2618695002/ (accessed on 20 January 2021).
- European Medicines Agency. Referral under Article 31 of Directive 2001/83/EC Angiotensin-II-Receptor Antagonists (Sartans) Containing a Tetrazole Group; Assessment Report; European Medicines Agency: Amsterdam, The Netherlands, 2019; Volume 31.
- Zhang, J.; Li, R.; Zhu, F.; Sun, C.; Shen, J. An improved synthesis of telmisartan via the copper-catalyzed cyclization of o -haloarylamidines. RSC Adv. 2020, 10, 13717–13721. [Google Scholar] [CrossRef]
- Goossen, L.J.; Knauber, T. Concise synthesis of telmisartan via decarboxylative cross-coupling. J. Org. Chem. 2008, 73, 8631–8634. [Google Scholar] [CrossRef]
- Wang, P.; Zheng, G.; Wang, Y.; Wang, X.; Wei, H.; Xiang, W. Highly practical and cost-efficient synthesis of telmisartan: An antihypertensive drug. Tetrahedron 2012, 68, 2509–2512. [Google Scholar] [CrossRef]
- Martin, A.D.; Siamaki, A.R.; Belecki, K.; Gupton, B.F. A convergent approach to the total synthesis of telmisartan via a Suzuki cross-coupling reaction between two functionalized benzimidazoles. J. Org. Chem. 2015, 80, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.S.; Ghosh, S.; Mehta, G.N. A modification to the synthesis of Telmisartan: An antihypertensive drug. J. Chem. Res. 2010, 34, 95–97. [Google Scholar] [CrossRef]
- Matsuoka, R.T.; Peng Liu, P. Process for Preparing Eprosartan Using Regioselective Protection of 24-Disubstituted-Midazole Intermediates. U.S. Patent 6,458,963 B1, 1 October 2002. [Google Scholar]
- Ramakrishnan, A.; Upadhyay, U.; Bhawsar, S. Improved Process for Manufacturing Anhydrous (e)-3-[2-Butyl-i-((4-carboxyphenyl) methyl)-ih-imidazole-s-yl]-(thiophen-z-ylmethyl)prop-2-enoic Acid Methane Sulfonate. W.O Patent Number 2009/084028 A2, 9 July 2009. [Google Scholar]
- Weinstock, J.; Keenan, R.M.; Samanen, J.; Hempel, J.; Finkelstein, J.A.; Franz, R.G.; Gaitanopoulos, D.E.; Girard, G.R.; Gleason, J.G. 1-(Carboxybenzyl) imidazole-5-acrylic acids: Potent and selective angiotensin II receptor antagonists. J. Med. Chem. 1991, 34, 1514–1517. [Google Scholar] [CrossRef]
- Keenan, R.M.; Weinstock, J.; Finkelstein, J.A.; Franz, R.G.; Gaitanopoulos, D.E.; Girard, G.R.; Hill, D.T.; Morgan, T.M.; Samanen, J.M. Potent nonpeptide angiotensin II receptor antagonists. 2. 1-(Carboxybenzyl) imidazole-5-acrylic acids. J. Med. Chem. 1993, 36, 1880–1892. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.A.; Keenan, R.M.; Weinstock, J. Imidazolyl-Alkenoic Acids Useful as Angiotensin II Receptor Antagonists. U.S. Patent 5,185,351, 9 February 1993. [Google Scholar]
- Seki, M. An efficient c–h arylation of a 5-phenyl-1h-tetrazole derivative: A practical synthesis of an angiotensin II receptor blocker. Synthesis 2012, 44, 3231–3237. [Google Scholar] [CrossRef]
- Babu, K.S.; Reddy, M.S.; Tagore, A.R.; Reddy, G.S.; Sebastian, S.; Varma, M.S.; Venkateswarlu, G.; Bhattacharya, A.; Reddy, P.P.; Anand, R.V. Efficient synthesis of olmesartan medoxomil, an antihypertensive drug. Synth. Commun. 2008, 39, 291–298. [Google Scholar] [CrossRef]
- Venkanna, G.; Madhusudhan, G.; Mukkanti, K.; Sankar, A.; Sampath Kumar, Y.; Venakata Narayana, G. Synthesis and characterization of process-related impurities of antihypertensive drug olmesartan medoxomil. J. Chem. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Casar, T. Process for the Preparation of Olmesartan Medoxoml. Patent Application Publication No. US 2011/0224271 A1, 15 September 2011. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgiou, N.; Gkalpinos, V.K.; Katsakos, S.D.; Vassiliou, S.; Tzakos, A.G.; Mavromoustakos, T. Rational Design and Synthesis of AT1R Antagonists. Molecules 2021, 26, 2927. https://doi.org/10.3390/molecules26102927
Georgiou N, Gkalpinos VK, Katsakos SD, Vassiliou S, Tzakos AG, Mavromoustakos T. Rational Design and Synthesis of AT1R Antagonists. Molecules. 2021; 26(10):2927. https://doi.org/10.3390/molecules26102927
Chicago/Turabian StyleGeorgiou, Nikitas, Vasileios K. Gkalpinos, Spyridon D. Katsakos, Stamatia Vassiliou, Andreas G. Tzakos, and Thomas Mavromoustakos. 2021. "Rational Design and Synthesis of AT1R Antagonists" Molecules 26, no. 10: 2927. https://doi.org/10.3390/molecules26102927
APA StyleGeorgiou, N., Gkalpinos, V. K., Katsakos, S. D., Vassiliou, S., Tzakos, A. G., & Mavromoustakos, T. (2021). Rational Design and Synthesis of AT1R Antagonists. Molecules, 26(10), 2927. https://doi.org/10.3390/molecules26102927