
Biosynthesis of Nature-Inspired Unnatural Cannabinoids

 , , , and
, , , and 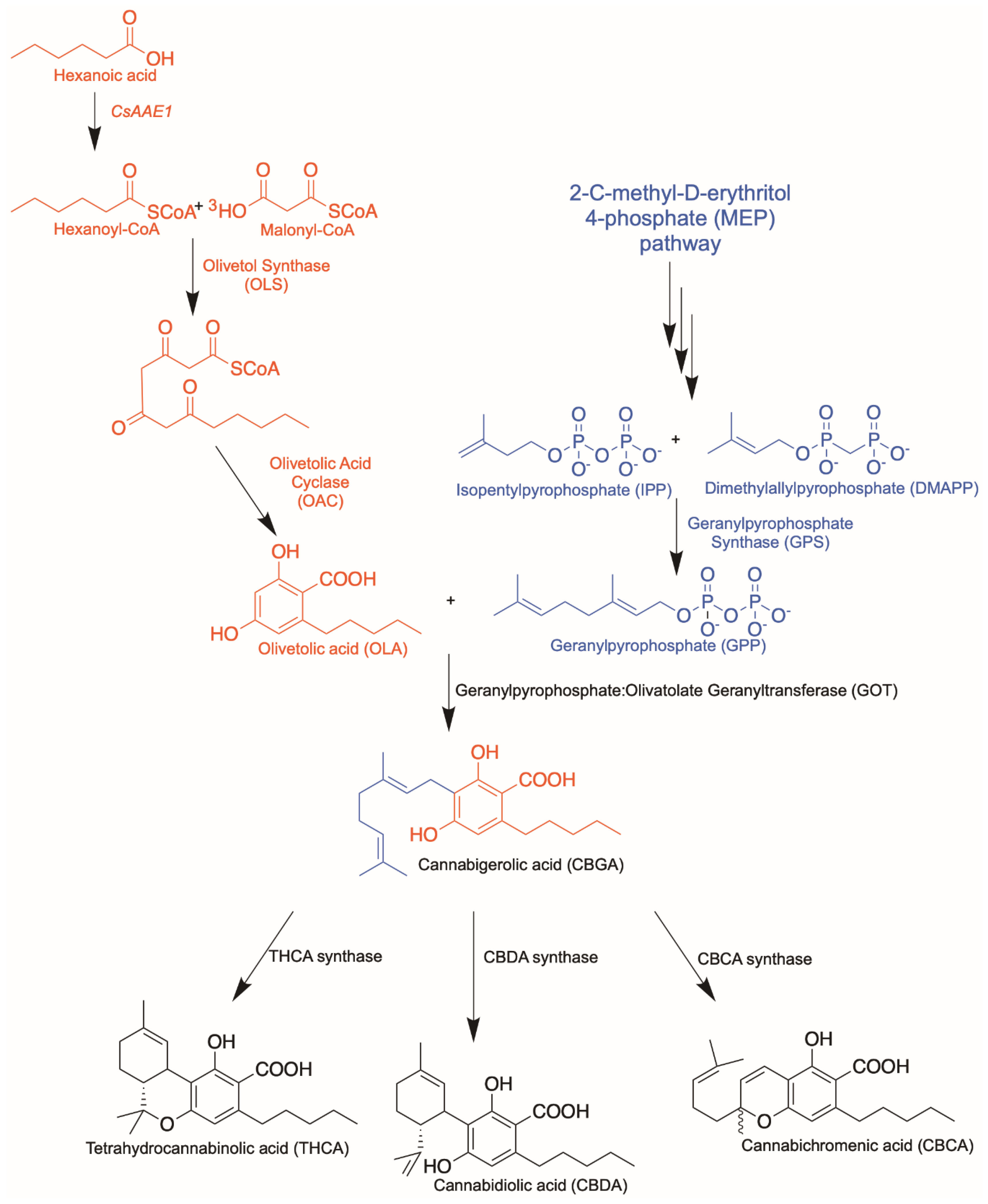{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
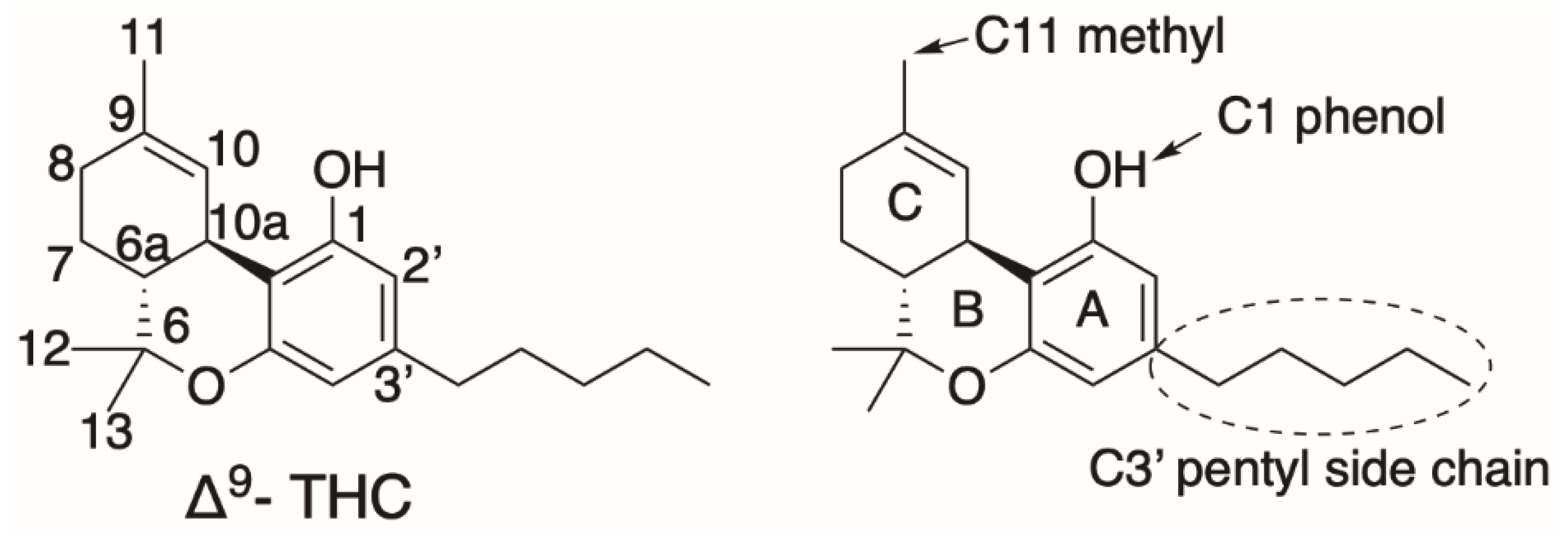
1. Cannabis sativa and Cannabinoids—An Introduction
Cannabinoid Biosynthetic Pathway
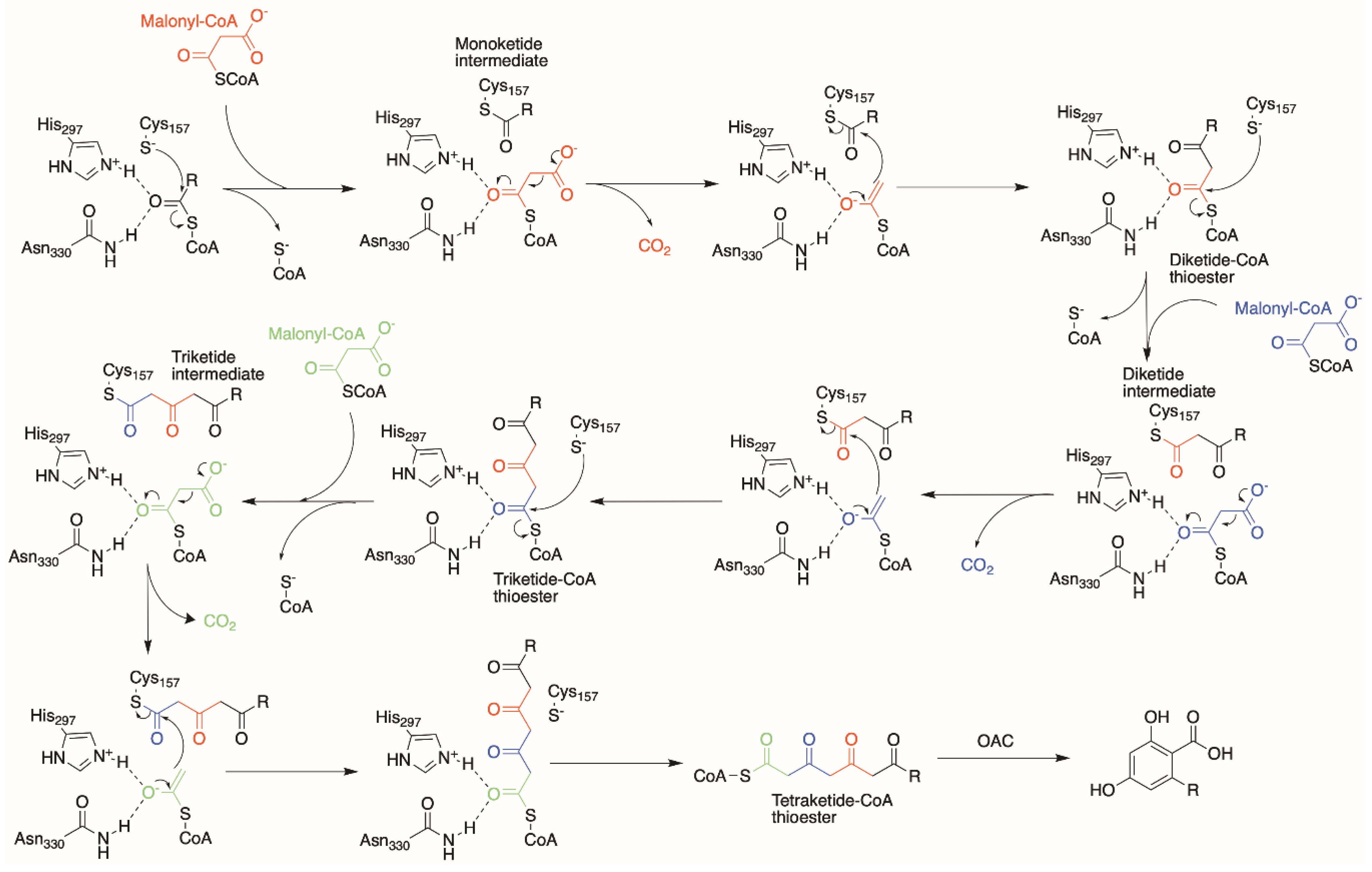
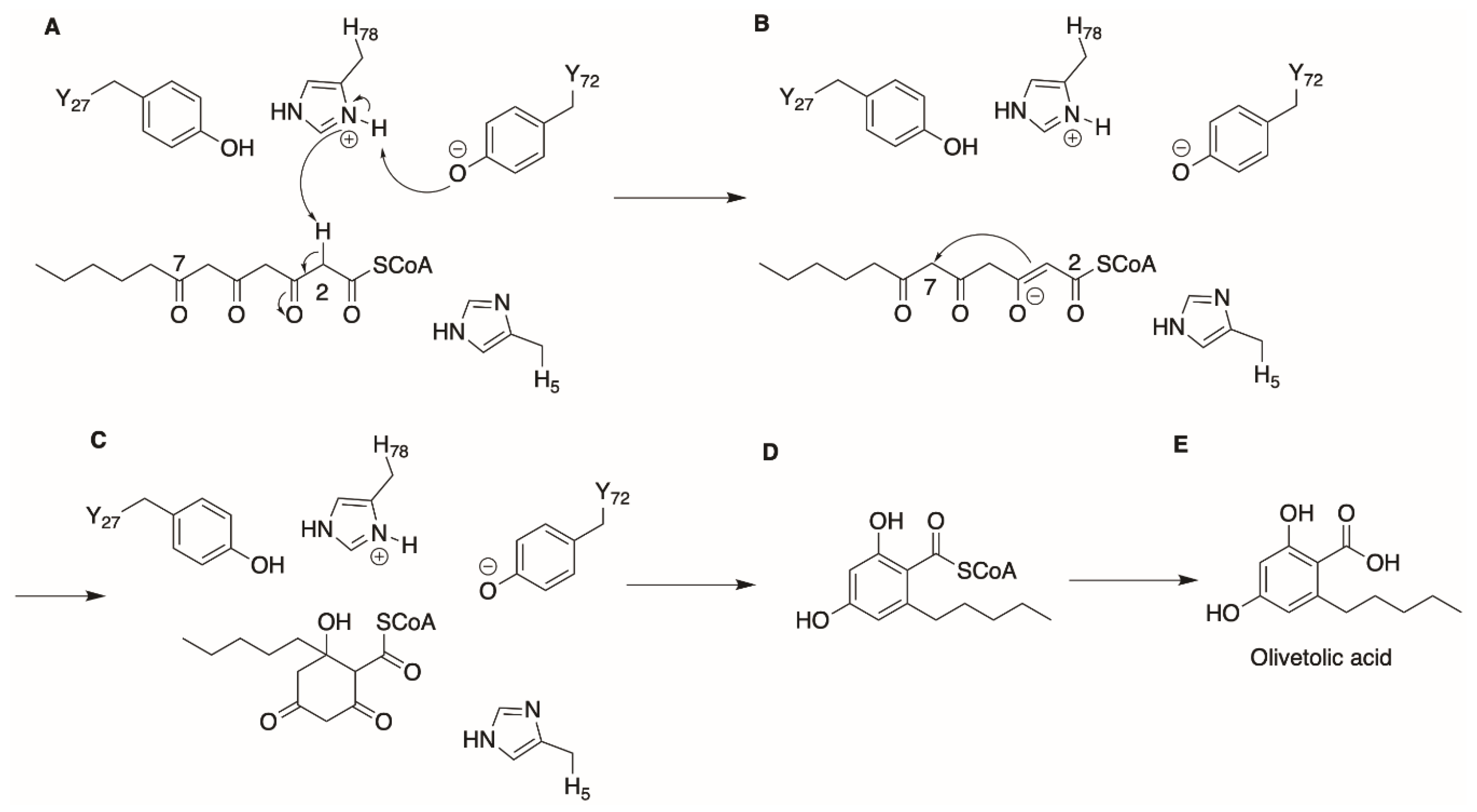
2. Olivetolic Acid Derivatives
2.1. Precursor-Directed Combinatorial Biosynthesis
2.2. Protein Engineering
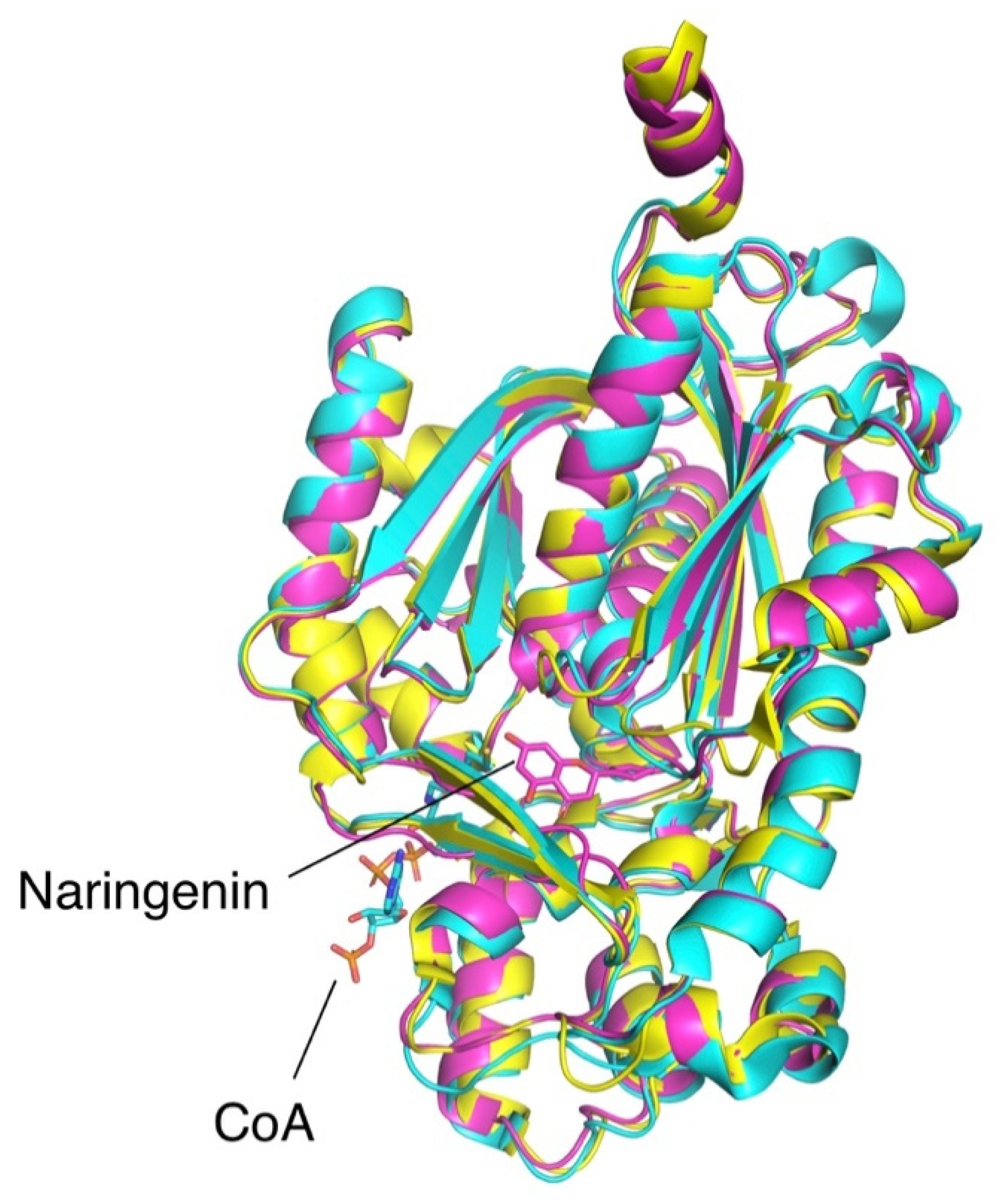
2.3. Orthologues of Interest
3. Aromatic Prenyltransferases
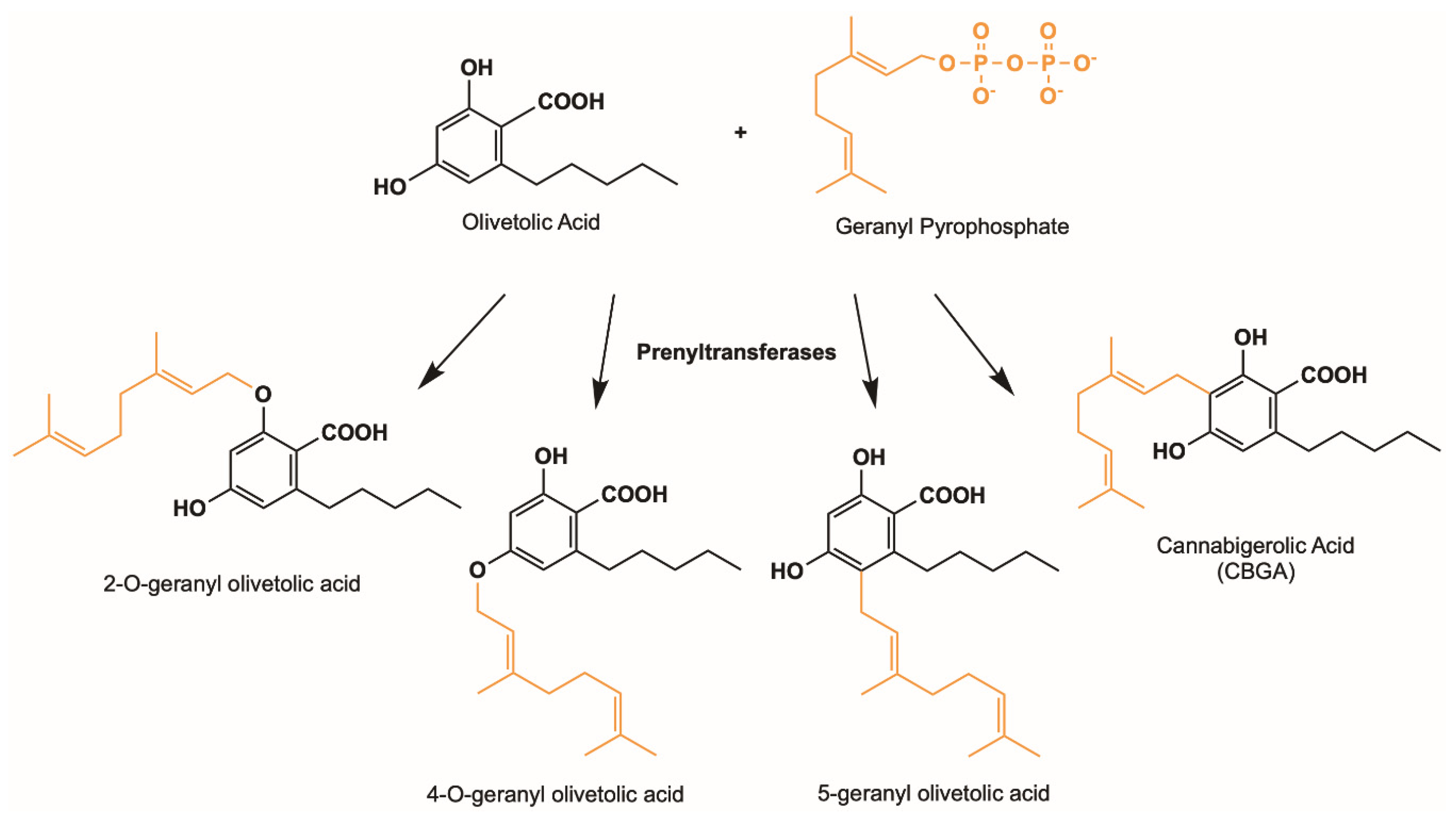
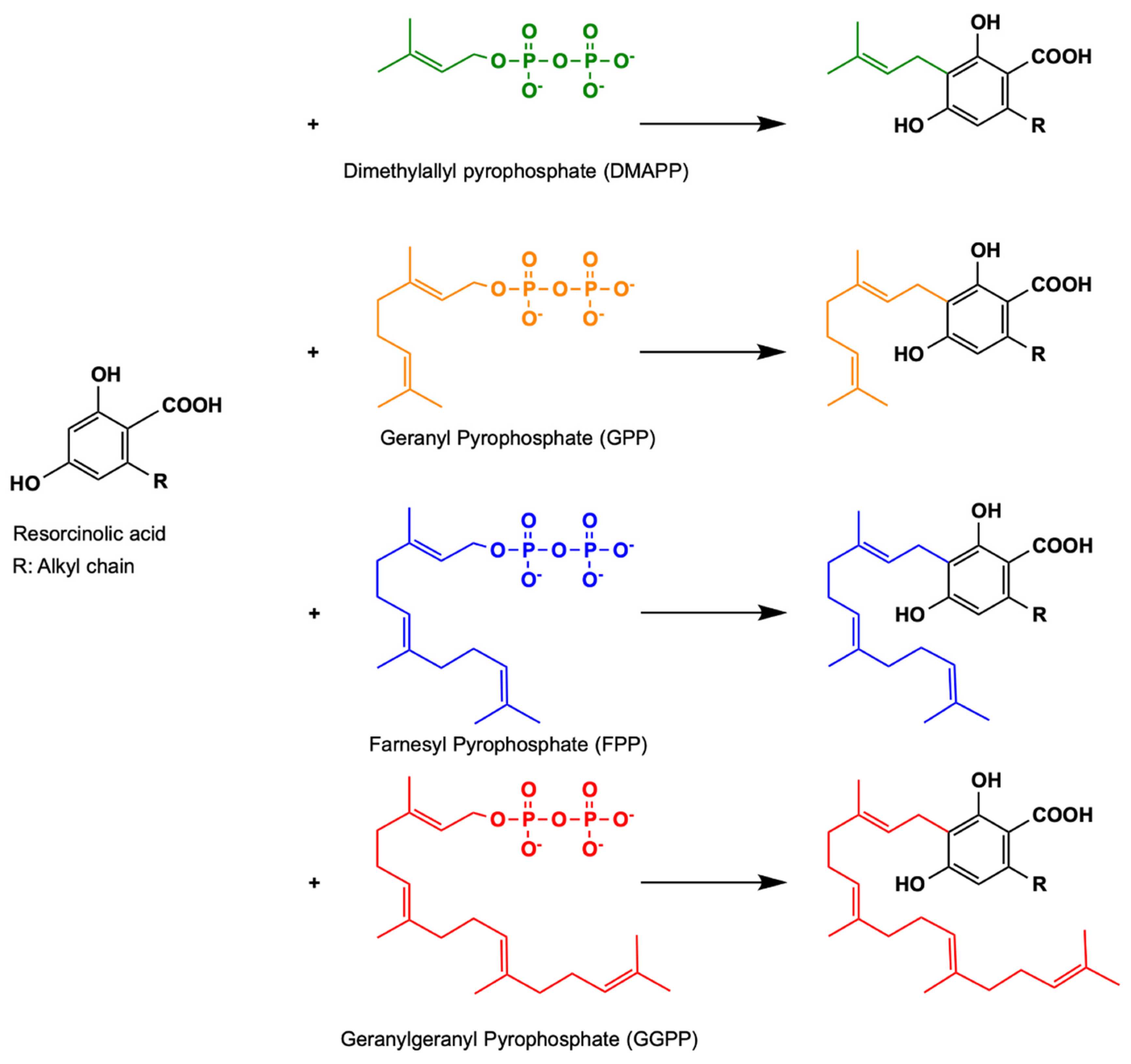
3.1. Plant Aromatic Prenyltransferases
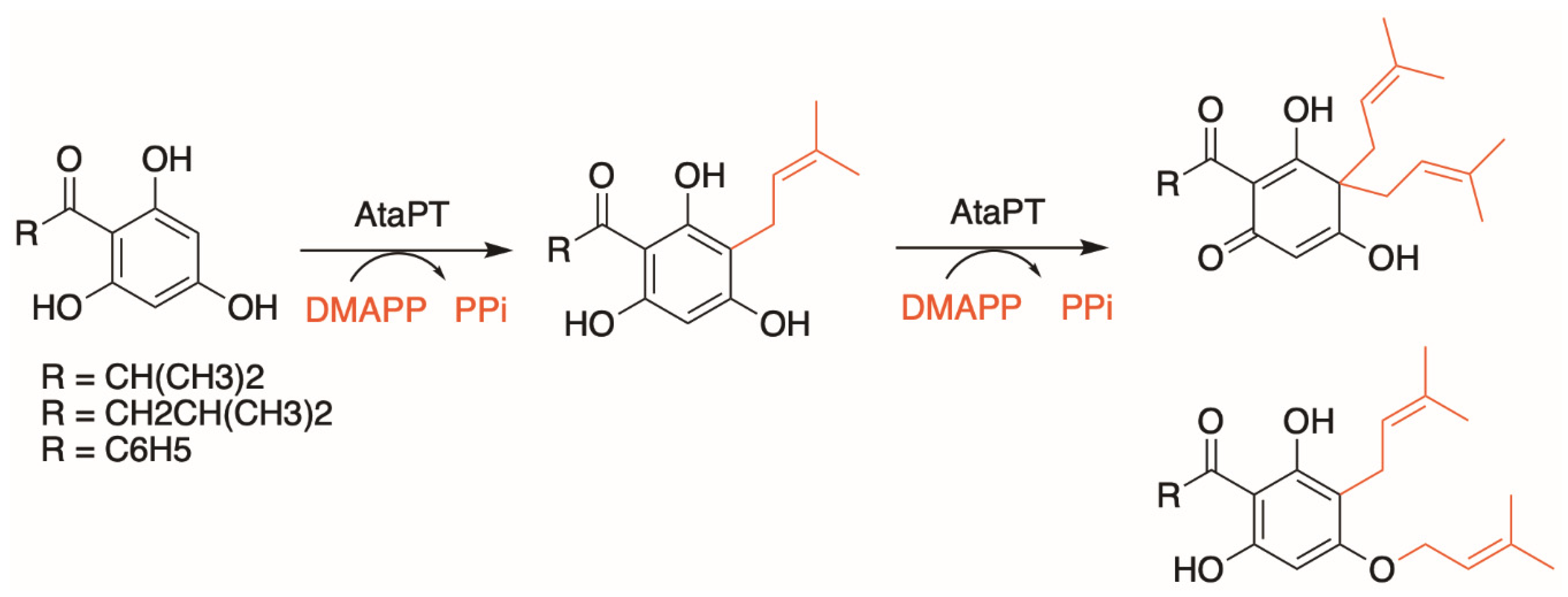
3.2. Alternative Soluble Prenyltransferases
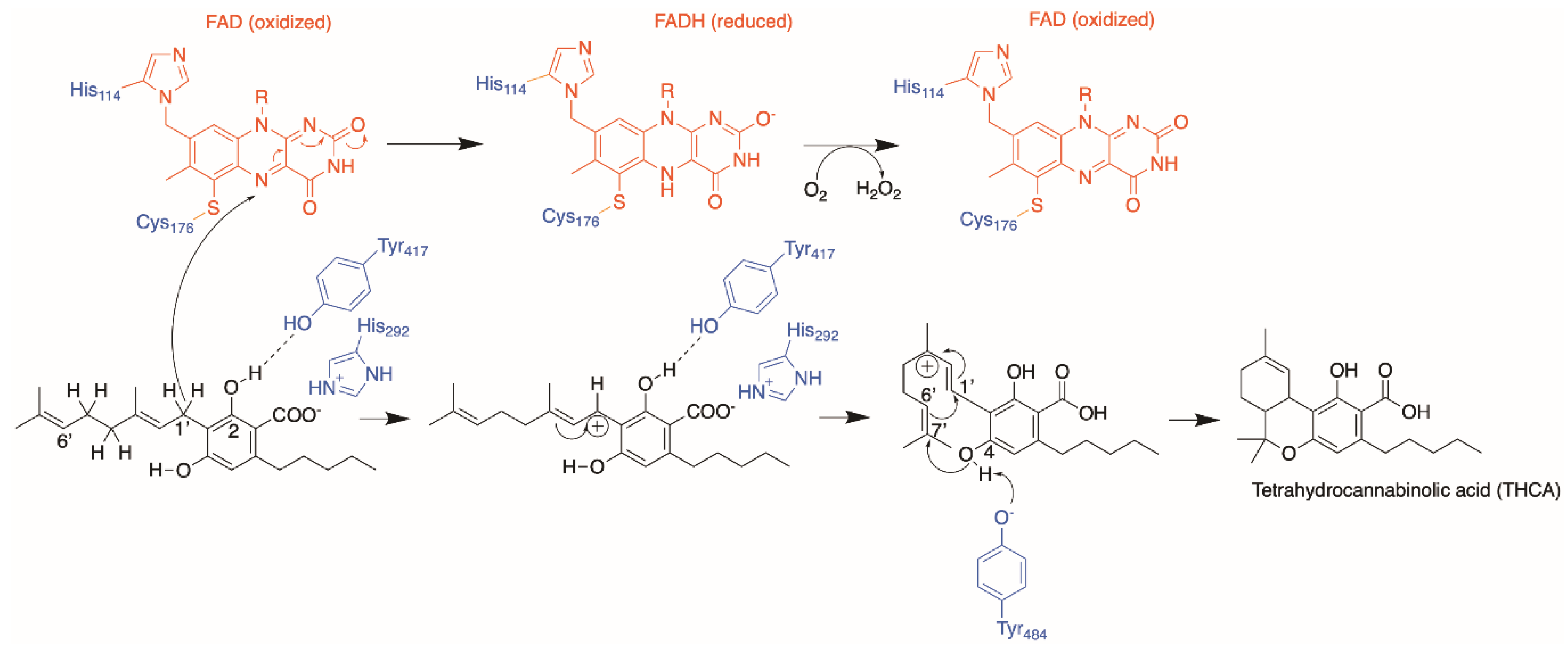
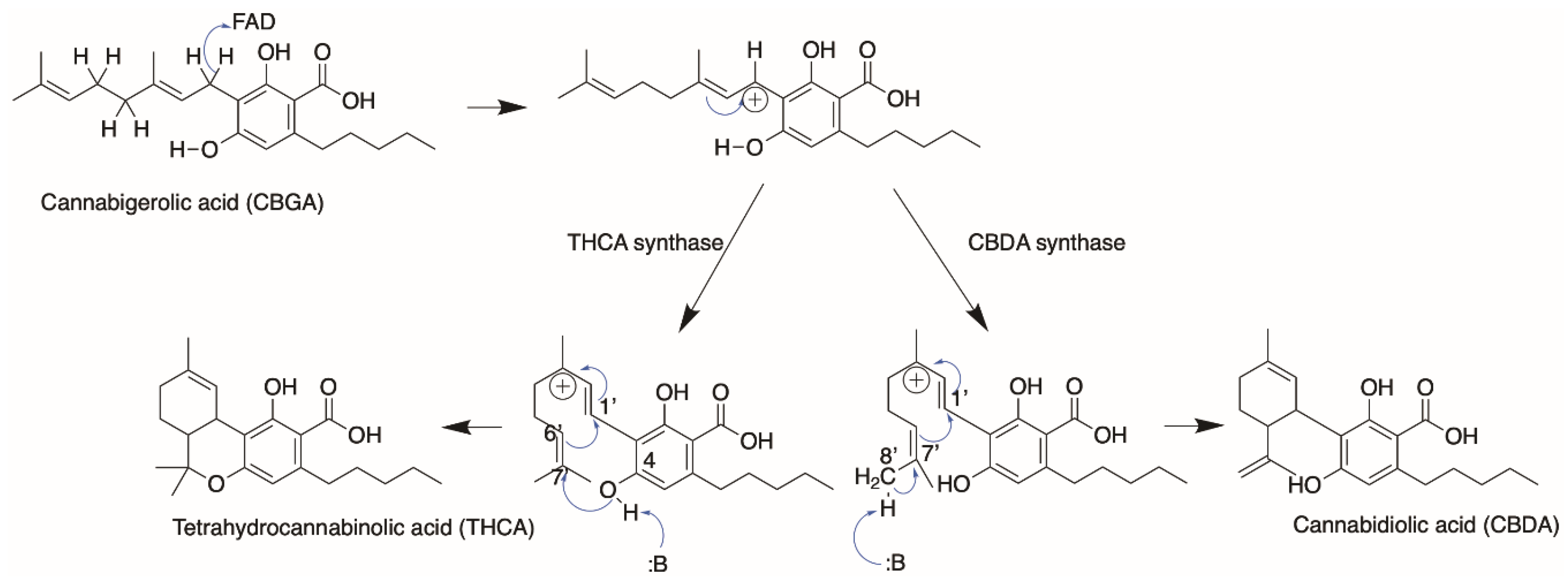
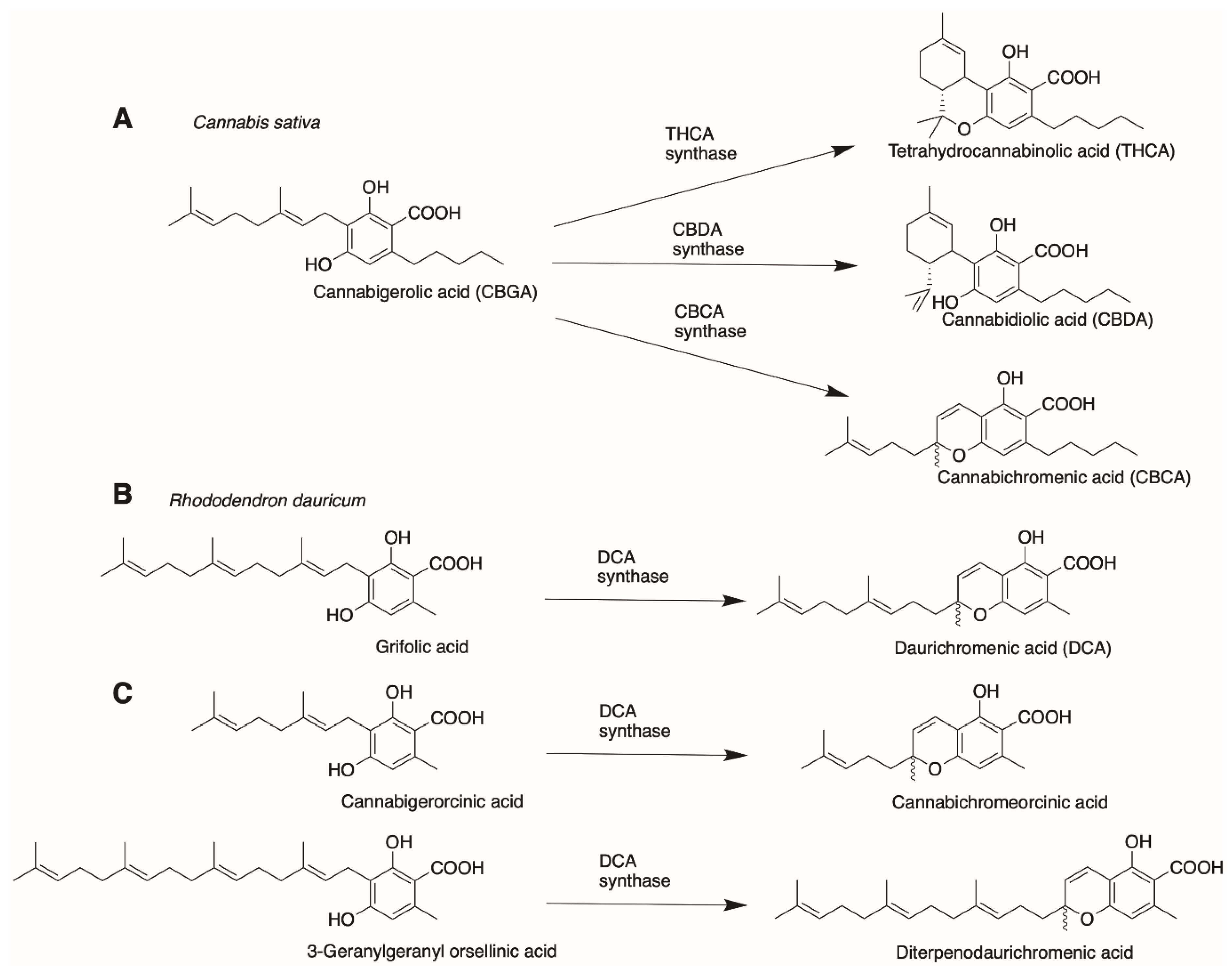
4. Cannabinoid Synthases
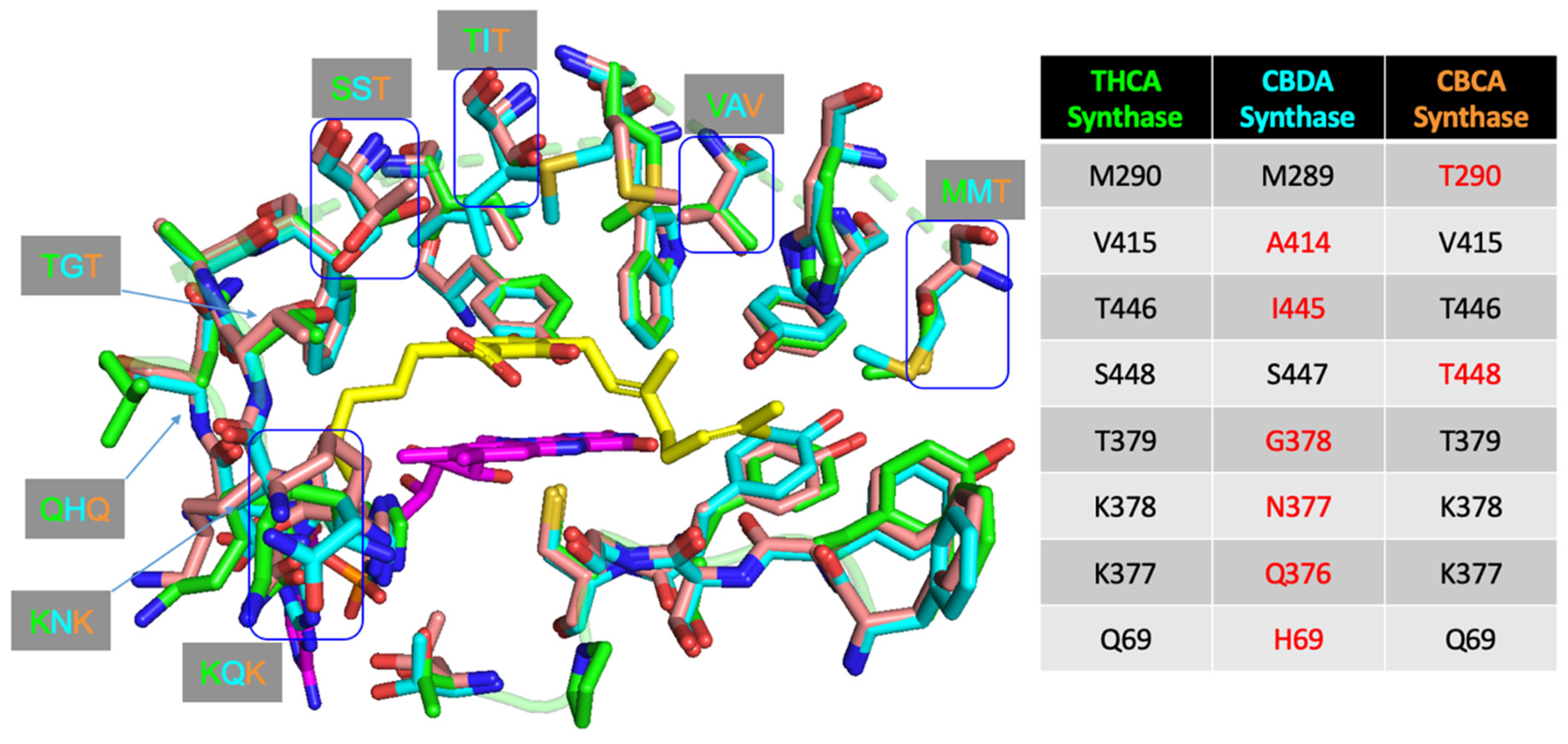
4.1. Protein Engineering
4.2. Orthologues of Interest
5. CB Receptors and Their Ligands
5.1. Classical Cannabinoid Receptors and Their Ligands
5.2. Orphan GPCRs and Other Cannabinoid-Related GPCRs
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Appendino, G.; Gibbons, S.; Giana, A.; Pagani, A.; Grassi, G.; Stavri, M.; Smith, E.; Rahman, M.M. Antibacterial cannabinoids from Cannabis sativa: A structure-activity study. J. Nat. Prod. 2008, 71, 1427–1430. [Google Scholar] [CrossRef]
- Pryce, G.; Riddall, D.R.; Selwood, D.L.; Giovannoni, G.; Baker, D. Neuroprotection in Experimental Autoimmune Encephalomyelitis and Progressive Multiple Sclerosis by Cannabis-Based Cannabinoids. J. Neuroimmune Pharmacol. 2015, 10, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Sanchez, C.; Guzman, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Gulck, T.; Moller, B.L. Phytocannabinoids: Origins and Biosynthesis. Trends Plant Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Galiègue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carrière, D.; Carayon, P.; Bouaboula, M.; Shire, D.; Le Fur, G.; Casellas, P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef]
- Ben-Shabat, S.; Fride, E.; Sheskin, T.; Tamiri, T.; Rhee, M.-H.; Vogel, Z.; Bisogno, T.; De Petrocellis, L.; Di Marzo, V.; Mechoulam, R. An entourage effect: Inactive endogenous fatty acid glycerol esters enhance 2-arachidonoyl-glycerol cannabinoid activity. Eur. J. Pharmacol. 1998, 353, 23–31. [Google Scholar] [CrossRef]
- Russo, E.B. The Case for the Entourage Effect and Conventional Breeding of Clinical Cannabis: No “Strain,” No Gain. Front. Plant Sci. 2019, 9. [Google Scholar] [CrossRef]
- Cogan, P.S. The ‘entourage effect’ or ‘hodge-podge hashish’: The questionable rebranding, marketing, and expectations of cannabis polypharmacy. Expert Rev. Clin. Pharmacol. 2020, 13, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Duggan, P.J. The Chemistry of Cannabis and Cannabinoids. Aust. J. Chem. 2021. [Google Scholar] [CrossRef]
- Anand, U.; Pacchetti, B.; Anand, P.; Sodergren, M.H. Cannabis-based medicines and pain: A review of potential synergistic and entourage effects. Pain Manag. 2021, 11, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Flachenecker, P.; Henze, T.; Zettl, U.K. Nabiximols (THC/CBD oromucosal spray, Sativex(R)) in clinical practice--results of a multicenter, non-interventional study (MOVE 2) in patients with multiple sclerosis spasticity. Eur. Neurol. 2014, 71, 271–279. [Google Scholar] [CrossRef]
- Chen, J.W.; Borgelt, L.M.; Blackmer, A.B. Cannabidiol: A New Hope for Patients With Dravet or Lennox-Gastaut Syndromes. Ann Pharmacol. 2019, 53, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Taura, F. Studies on tetrahydrocannabinolic acid synthase that produces the acidic precursor of tetrahydrocannabinol, the pharmacologically active cannabinoid in marijuana. Drug Discov. Ther. 2009, 3, 83–87. [Google Scholar]
- Aizpurua-Olaizola, O.; Soydaner, U.; Ozturk, E.; Schibano, D.; Simsir, Y.; Navarro, P.; Etxebarria, N.; Usobiaga, A. Evolution of the Cannabinoid and Terpene Content during the Growth of Cannabis sativa Plants from Different Chemotypes. J. Nat. Prod. 2016, 79, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Mills, E. The carbon footprint of indoor Cannabis production. Energy Policy 2012, 46, 58–67. [Google Scholar] [CrossRef]
- Edwards, E.; Bunting, W.; Garcia, L. The War on Marijuana in Black and White; American Civil Liberties Union: New York, NY, USA, 2013. [Google Scholar]
- Tan, Z.; Clomburg, J.M.; Gonzalez, R. Synthetic Pathway for the Production of Olivetolic Acid in Escherichia coli. ACS Synth. Biol. 2018, 7, 1886–1896. [Google Scholar] [CrossRef]
- Zirpel, B.; Stehle, F.; Kayser, O. Production of Δ9-tetrahydrocannabinolic acid from cannabigerolic acid by whole cells of Pichia (Komagataella) pastoris expressing Δ9-tetrahydrocannabinolic acid synthase from Cannabis sativa L. Biotechnol. Lett. 2015, 37, 1869–1875. [Google Scholar] [CrossRef]
- Luo, X.; Reiter, M.A.; d’Espaux, L.; Wong, J.; Denby, C.M.; Lechner, A.; Zhang, Y.; Grzybowski, A.T.; Harth, S.; Lin, W.; et al. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature 2019, 567, 123–126. [Google Scholar] [CrossRef]
- Stout, J.M.; Boubakir, Z.; Ambrose, S.J.; Purves, R.W.; Page, J.E. The hexanoyl-CoA precursor for cannabinoid biosynthesis is formed by an acyl-activating enzyme in Cannabis sativa trichomes. Plant J. 2012, 71, 353–365. [Google Scholar] [CrossRef]
- Taura, F.; Tanaka, S.; Taguchi, C.; Fukamizu, T.; Tanaka, H.; Shoyama, Y.; Morimoto, S. Characterization of olivetol synthase, a polyketide synthase putatively involved in cannabinoid biosynthetic pathway. FEBS Lett. 2009, 583, 2061–2066. [Google Scholar] [CrossRef]
- Kearsey, L.J.; Prandi, N.; Karuppiah, V.; Yan, C.; Leys, D.; Toogood, H.; Takano, E.; Scrutton, N.S. Structure of the Cannabis sativa olivetol-producing enzyme reveals cyclization plasticity in type III polyketide synthases. FEBS J. 2020, 287, 1511–1524. [Google Scholar] [CrossRef]
- Gagne, S.J.; Stout, J.M.; Liu, E.; Boubakir, Z.; Clark, S.M.; Page, J.E. Identification of olivetolic acid cyclase from Cannabis sativa reveals a unique catalytic route to plant polyketides. Proc. Natl. Acad. Sci. USA 2012, 109, 12811–12816. [Google Scholar] [CrossRef] [PubMed]
- Fellermeier, M.; Zenk, M.H. Prenylation of olivetolate by a hemp transferase yields cannabigerolic acid, the precursor of tetrahydrocannabinol. FEBS Lett. 1998, 427, 283–285. [Google Scholar] [CrossRef]
- Sirikantaramas, S.; Morimoto, S.; Shoyama, Y.; Ishikawa, Y.; Wada, Y.; Shoyama, Y.; Taura, F. The gene controlling marijuana psychoactivity: Molecular cloning and heterologous expression of Delta1-tetrahydrocannabinolic acid synthase from Cannabis sativa L. J. Biol. Chem. 2004, 279, 39767–39774. [Google Scholar] [CrossRef] [PubMed]
- Taura, F.; Sirikantaramas, S.; Shoyama, Y.; Yoshikai, K.; Shoyama, Y.; Morimoto, S. Cannabidiolic-acid synthase, the chemotype-determining enzyme in the fiber-type Cannabis sativa. FEBS Lett. 2007, 581, 2929–2934. [Google Scholar] [CrossRef]
- Austin, M.B.; Bowman, M.E.; Ferrer, J.L.; Schroder, J.; Noel, J.P. An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chem. Biol. 2004, 11, 1179–1194. [Google Scholar] [CrossRef]
- Ferrer, J.L.; Jez, J.M.; Bowman, M.E.; Dixon, R.A.; Noel, J.P. Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat. Struct. Biol. 1999, 6, 775–784. [Google Scholar] [CrossRef]
- Go, M.K.; Wongsantichon, J.; Cheung, V.W.N.; Chow, J.Y.; Robinson, R.C.; Yew, W.S. Synthetic Polyketide Enzymology: Platform for Biosynthesis of Antimicrobial Polyketides. ACS Catal. 2015, 5, 4033–4042. [Google Scholar] [CrossRef]
- Torkkell, S.; Kunnari, T.; Palmu, K.; Hakala, J.; Mantsala, P.; Ylihonko, K. Identification of a cyclase gene dictating the C-9 stereochemistry of anthracyclines from Streptomyces nogalater. Antimicrob. Agents Chemother. 2000, 44, 396–399. [Google Scholar] [CrossRef]
- Sultana, A.; Kallio, P.; Jansson, A.; Wang, J.S.; Niemi, J.; Mantsala, P.; Schneider, G. Structure of the polyketide cyclase SnoaL reveals a novel mechanism for enzymatic aldol condensation. EMBO J. 2004, 23, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Hutchinson, C.R. Deciphering the mechanism for the assembly of aromatic polyketides by a bacterial polyketide synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 6600–6604. [Google Scholar] [CrossRef]
- Ames, B.D.; Korman, T.P.; Zhang, W.; Smith, P.; Vu, T.; Tang, Y.; Tsai, S.C. Crystal structure and functional analysis of tetracenomycin ARO/CYC: Implications for cyclization specificity of aromatic polyketides. Proc. Natl. Acad. Sci. USA 2008, 105, 5349–5354. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Hutchinson, C.R. Tetracenomycin F2 cyclase: Intramolecular aldol condensation in the biosynthesis of tetracenomycin C in Streptomyces glaucescens. Biochemistry 1993, 32, 11149–11154. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.B.; Katayama, K.; Watanabe, K.; Hutchinson, C.R.; Rayment, I. Structural and functional analysis of tetracenomycin F2 cyclase from Streptomyces glaucescens. A type II polyketide cyclase. J. Biol. Chem. 2004, 279, 37956–37963. [Google Scholar] [CrossRef]
- Yang, X.; Matsui, T.; Kodama, T.; Mori, T.; Zhou, X.; Taura, F.; Noguchi, H.; Abe, I.; Morita, H. Structural basis for olivetolic acid formation by a polyketide cyclase from Cannabis sativa. FEBS J. 2016, 283, 1088–1106. [Google Scholar] [CrossRef]
- Gulck, T.; Booth, J.K.; Carvalho, A.; Khakimov, B.; Crocoll, C.; Motawia, M.S.; Moller, B.L.; Bohlmann, J.; Gallage, N.J. Synthetic Biology of Cannabinoids and Cannabinoid Glucosides in Nicotiana benthamiana and Saccharomyces cerevisiae. J. Nat. Prod. 2020, 83, 2877–2893. [Google Scholar] [CrossRef]
- Lim, Y.P.; Go, M.K.; Raida, M.; Inoue, T.; Wenk, M.R.; Keasling, J.D.; Chang, M.W.; Yew, W.S. Synthetic Enzymology and the Fountain of Youth: Repurposing Biology for Longevity. ACS Omega 2018, 3, 11050–11061. [Google Scholar] [CrossRef]
- Karchin, J.M.; Ha, J.-H.; Namitz, K.E.; Cosgrove, M.S.; Loh, S.N. Small Molecule-Induced Domain Swapping as a Mechanism for Controlling Protein Function and Assembly. Sci. Rep. 2017, 7, 44388. [Google Scholar] [CrossRef]
- Liu, Y.; Eisenberg, D. 3D domain swapping: As domains continue to swap. Protein Sci. 2002, 11, 1285–1299. [Google Scholar] [CrossRef]
- Barajas, J.F.; Blake-Hedges, J.M.; Bailey, C.B.; Curran, S.; Keasling, J.D. Engineered polyketides: Synergy between protein and host level engineering. Synth. Syst. Biotechnol. 2017, 2, 147–166. [Google Scholar] [CrossRef]
- Taura, F.; Iijima, M.; Yamanaka, E.; Takahashi, H.; Kenmoku, H.; Saeki, H.; Morimoto, S.; Asakawa, Y.; Kurosaki, F.; Morita, H. A Novel Class of Plant Type III Polyketide Synthase Involved in Orsellinic Acid Biosynthesis from Rhododendron dauricum. Front. Plant Sci. 2016, 7, 1452. [Google Scholar] [CrossRef] [PubMed]
- Austin, M.B.; Noel, J.P. The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 2003, 20, 79–110. [Google Scholar] [CrossRef]
- Shoyama, Y.; Tamada, T.; Kurihara, K.; Takeuchi, A.; Taura, F.; Arai, S.; Blaber, M.; Morimoto, S.; Kuroki, R. Structure and function of ∆1-tetrahydrocannabinolic acid (THCA) synthase, the enzyme controlling the psychoactivity of Cannabis sativa. J. Mol. Biol. 2012, 423, 96–105. [Google Scholar] [CrossRef]
- Gerlt, J.A.; Bouvier, J.T.; Davidson, D.B.; Imker, H.J.; Sadkhin, B.; Slater, D.R.; Whalen, K.L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 2015, 1854, 1019–1037. [Google Scholar] [CrossRef]
- Cornilescu, G.; Cornilescu, C.C.; Zhao, Q.; Frederick, R.O.; Peterson, F.C.; Thao, S.; Markley, J.L. Solution structure of a homodimeric hypothetical protein, At5g22580, a structural genomics target from Arabidopsis thaliana. J. Biomol. NMR 2004, 29, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Dgany, O.; Gonzalez, A.; Sofer, O.; Wang, W.; Zolotnitsky, G.; Wolf, A.; Shoham, Y.; Altman, A.; Wolf, S.G.; Shoseyov, O.; et al. The structural basis of the thermostability of SP1, a novel plant (Populus tremula) boiling stable protein. J. Biol. Chem. 2004, 279, 51516–51523. [Google Scholar] [CrossRef]
- Koehl, P. Relaxed specificity in aromatic prenyltransferases. Nat. Chem. Biol. 2005, 1, 71–72. [Google Scholar] [CrossRef]
- Palsuledesai, C.C.; Distefano, M.D. Protein prenylation: Enzymes, therapeutics, and biotechnology applications. ACS Chem. Biol. 2015, 10, 51–62. [Google Scholar] [CrossRef]
- Kumano, T.; Richard, S.B.; Noel, J.P.; Nishiyama, M.; Kuzuyama, T. Chemoenzymatic syntheses of prenylated aromatic small molecules using Streptomyces prenyltransferases with relaxed substrate specificities. Bioorg. Med. Chem. 2008, 16, 8117–8126. [Google Scholar] [CrossRef]
- Zirpel, B.; Degenhardt, F.; Martin, C.; Kayser, O.; Stehle, F. Engineering yeasts as platform organisms for cannabinoid biosynthesis. J. Biotechnol. 2017, 259, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Page, J.E.; Boubakir, Z. Aromatic Prenyltransferase from Cannabis. U.S. Patent US8884100B2, 11 November 2014. [Google Scholar]
- De Bruijn, W.J.C.; Levisson, M.; Beekwilder, J.; van Berkel, W.J.H.; Vincken, J.P. Plant Aromatic Prenyltransferases: Tools for Microbial Cell Factories. Trends Biotechnol. 2020, 38, 917–934. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jiang, W.; Xia, Y.; Wang, X.; Shen, G.; Pang, Y. Genistein-Specific G6DT Gene for the Inducible Production of Wighteone in Lotus japonicus. Plant Cell Physiol. 2018, 59, 128–141. [Google Scholar] [CrossRef]
- Yazaki, K.; Kunihisa, M.; Fujisaki, T.; Sato, F. Geranyl diphosphate:4-hydroxybenzoate geranyltransferase from Lithospermum erythrorhizon. Cloning and characterization of a ket enzyme in shikonin biosynthesis. J. Biol. Chem. 2002, 277, 6240–6246. [Google Scholar] [CrossRef]
- Saeki, H.; Hara, R.; Takahashi, H.; Iijima, M.; Munakata, R.; Kenmoku, H.; Fuku, K.; Sekihara, A.; Yasuno, Y.; Shinada, T.; et al. An Aromatic Farnesyltransferase Functions in Biosynthesis of the Anti-HIV Meroterpenoid Daurichromenic Acid. Plant Physiol. 2018, 178, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Kuzuyama, T.; Noel, J.P.; Richard, S.B. Structural basis for the promiscuous biosynthetic prenylation of aromatic natural products. Nature 2005, 435, 983–987. [Google Scholar] [CrossRef]
- Metzger, U.; Keller, S.; Stevenson, C.E.; Heide, L.; Lawson, D.M. Structure and mechanism of the magnesium-independent aromatic prenyltransferase CloQ from the clorobiocin biosynthetic pathway. J. Mol. Biol. 2010, 404, 611–626. [Google Scholar] [CrossRef]
- Metzger, U.; Schall, C.; Zocher, G.; Unsold, I.; Stec, E.; Li, S.M.; Heide, L.; Stehle, T. The structure of dimethylallyl tryptophan synthase reveals a common architecture of aromatic prenyltransferases in fungi and bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 14309–14314. [Google Scholar] [CrossRef]
- Valliere, M.A.; Korman, T.P.; Woodall, N.B.; Khitrov, G.A.; Taylor, R.E.; Baker, D.; Bowie, J.U. A cell-free platform for the prenylation of natural products and application to cannabinoid production. Nat. Commun. 2019, 10, 565. [Google Scholar] [CrossRef]
- Qian, S.; Clomburg, J.M.; Gonzalez, R. Engineering Escherichia coli as a platform for the in vivo synthesis of prenylated aromatics. Biotechnol. Bioeng. 2019, 116, 1116–1127. [Google Scholar] [CrossRef]
- Mori, T. Enzymatic studies on aromatic prenyltransferases. J. Nat. Med. 2020, 74, 501–512. [Google Scholar] [CrossRef]
- Li, S.M. Evolution of aromatic prenyltransferases in the biosynthesis of indole derivatives. Phytochemistry 2009, 70, 1746–1757. [Google Scholar] [CrossRef]
- Tanner, M.E. Mechanistic studies on the indole prenyltransferases. Nat. Prod. Rep. 2015, 32, 88–101. [Google Scholar] [CrossRef]
- Bonitz, T.; Alva, V.; Saleh, O.; Lupas, A.N.; Heide, L. Evolutionary relationships of microbial aromatic prenyltransferases. PLoS ONE 2011, 6, e27336. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Gao, B.; Liu, X.; Ruan, F.; Zhang, Y.; Lou, J.; Feng, K.; Wunsch, C.; Li, S.M.; Dai, J.; et al. Molecular insights into the enzyme promiscuity of an aromatic prenyltransferase. Nat. Chem. Biol. 2017, 13, 226–234. [Google Scholar] [CrossRef]
- Zhou, K.; Wunsch, C.; Dai, J.; Li, S.M. gem-Diprenylation of Acylphloroglucinols by a Fungal Prenyltransferase of the Dimethylallyltryptophan Synthase Superfamily. Org. Lett. 2017, 19, 388–391. [Google Scholar] [CrossRef]
- Yu, X.; Zocher, G.; Xie, X.; Liebhold, M.; Schutz, S.; Stehle, T.; Li, S.M. Catalytic mechanism of stereospecific formation of cis-configured prenylated pyrroloindoline diketopiperazines by indole prenyltransferases. Chem. Biol. 2013, 20, 1492–1501. [Google Scholar] [CrossRef]
- Shoyama, Y.; Takeuchi, A.; Taura, F.; Tamada, T.; Adachi, M.; Kuroki, R.; Morimoto, S. Crystallization of Delta1-tetrahydrocannabinolic acid (THCA) synthase from Cannabis sativa. Acta Cryst. Sect. F Struct. Biol. Cryst. Commun. 2005, 61, 799–801. [Google Scholar] [CrossRef]
- Zirpel, B.; Kayser, O.; Stehle, F. Elucidation of structure-function relationship of THCA and CBDA synthase from Cannabis sativaL. J. Biotechnol. 2018, 284, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Komatsu, K.; Taura, F.; Shoyama, Y. Enzymological Evidence for Cannabichromenic Acid Biosynthesis. J. Nat. Prod. 1997, 60, 854–857. [Google Scholar] [CrossRef]
- Morimoto, S.; Komatsu, K.; Taura, F.; Shoyama, Y. Purification and characterization of cannabichromenic acid synthase from Cannabis sativa. Phytochemistry 1998, 49, 1525–1529. [Google Scholar] [CrossRef]
- Onofri, C.; de Meijer, E.P.M.; Mandolino, G. Sequence heterogeneity of cannabidiolic- and tetrahydrocannabinolic acid-synthase in Cannabis sativa L. and its relationship with chemical phenotype. Phytochemistry 2015, 116, 57–68. [Google Scholar] [CrossRef]
- Bow, E.W.; Rimoldi, J.M. The Structure-Function Relationships of Classical Cannabinoids: CB1/CB2 Modulation. Perspect. Med. Chem. 2016, 8, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Kashiwada, Y.; Yamazaki, K.; Ikeshiro, Y.; Yamagishi, T.; Fujioka, T.; Mihashi, K.; Mizuki, K.; Cosentino, L.M.; Fowke, K.; Morris-Natschke, S.L.; et al. Isolation of rhododaurichromanic acid B and the anti-HIV principles rhododaurichromanic acid A and rhododaurichromenic acid from Rhododendron dauricum. Tetrahedron 2001, 57, 1559–1563. [Google Scholar] [CrossRef]
- Hanus, L.O.; Meyer, S.M.; Munoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. [Google Scholar] [CrossRef]
- Citti, C.; Linciano, P.; Forni, F.; Vandelli, M.A.; Gigli, G.; Lagana, A.; Cannazza, G. Analysis of impurities of cannabidiol from hemp. Isolation, characterization and synthesis of cannabidibutol, the novel cannabidiol butyl analog. J. Pharmacol. Biomed. Anal. 2019, 175, 112752. [Google Scholar] [CrossRef] [PubMed]
- Linciano, P.; Citti, C.; Russo, F.; Tolomeo, F.; Lagana, A.; Capriotti, A.L.; Luongo, L.; Iannotta, M.; Belardo, C.; Maione, S.; et al. Identification of a new cannabidiol n-hexyl homolog in a medicinal cannabis variety with an antinociceptive activity in mice: Cannabidihexol. Sci. Rep. 2020, 10, 22019. [Google Scholar] [CrossRef]
- Citti, C.; Linciano, P.; Russo, F.; Luongo, L.; Iannotta, M.; Maione, S.; Lagana, A.; Capriotti, A.L.; Forni, F.; Vandelli, M.A.; et al. A novel phytocannabinoid isolated from Cannabis sativa L. with an in vivo cannabimimetic activity higher than Delta(9)-tetrahydrocannabinol: Delta(9)-Tetrahydrocannabiphorol. Sci. Rep. 2019, 9, 20335. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Munakata, R.; Takahashi, H.; Kenmoku, H.; Nakagawa, R.; Kodama, T.; Asakawa, Y.; Abe, I.; Yazaki, K.; Kurosaki, F.; et al. Identification and Characterization of Daurichromenic Acid Synthase Active in Anti-HIV Biosynthesis. Plant Physiol. 2017, 174, 2213–2230. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Harrison, T.J.; Wilson, P.D. A modular and concise total synthesis of (+/−)-daurichromenic acid and analogues. J. Org. Chem. 2004, 69, 3782–3786. [Google Scholar] [CrossRef]
- Kang, Y.; Mei, Y.; Du, Y.; Jin, Z. Total synthesis of the highly potent anti-HIV natural product daurichromenic acid along with its two chromane derivatives, rhododaurichromanic acids A and B. Org. Lett. 2003, 5, 4481–4484. [Google Scholar] [CrossRef]
- Lee, Y.R.; Wang, X. A short synthetic route to biologically active (+/−)-daurichromenic acid as highly potent anti-HIV agent. Org. Biomol. Chem. 2005, 3, 3955–3957. [Google Scholar] [CrossRef]
- Shoyama, Y.; Hirano, H.; Nishioka, I. Biosynthesis of propyl cannabinoid acid and its biosynthetic relationship with pentyl and methyl cannabinoid acids. Phytochemistry 1984, 23, 1909–1912. [Google Scholar] [CrossRef]
- Leferink, N.G.; Heuts, D.P.; Fraaije, M.W.; van Berkel, W.J. The growing VAO flavoprotein family. Arch. Biochem. Biophys. 2008, 474, 292–301. [Google Scholar] [CrossRef]
- Winkler, A.; Lyskowski, A.; Riedl, S.; Puhl, M.; Kutchan, T.M.; Macheroux, P.; Gruber, K. A concerted mechanism for berberine bridge enzyme. Nat. Chem. Biol. 2008, 4, 739–741. [Google Scholar] [CrossRef]
- Custers, J.H.; Harrison, S.J.; Sela-Buurlage, M.B.; van Deventer, E.; Lageweg, W.; Howe, P.W.; van der Meijs, P.J.; Ponstein, A.S.; Simons, B.H.; Melchers, L.S.; et al. Isolation and characterisation of a class of carbohydrate oxidases from higher plants, with a role in active defence. Plant J. 2004, 39, 147–160. [Google Scholar] [CrossRef]
- Eichhorn Bilodeau, S.; Wu, B.S.; Rufyikiri, A.S.; MacPherson, S.; Lefsrud, M. An Update on Plant Photobiology and Implications for Cannabis Production. Front. Plant Sci. 2019, 10, 296. [Google Scholar] [CrossRef]
- Go, M.K.; Lim, K.J.H.; Yew, W.S. Cannabinoid Biosynthesis using Noncanonical Cannabinoid Synthases. bioRxiv 2020. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Pertwee, R.G. Targeting the endocannabinoid system with cannabinoid receptor agonists: Pharmacological strategies and therapeutic possibilities. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3353–3363. [Google Scholar] [CrossRef]
- Cabral, G.A.; Raborn, E.S.; Griffin, L.; Dennis, J.; Marciano-Cabral, F. CB2 receptors in the brain: Role in central immune function. Br. J. Pharmacol. 2008, 153, 240–251. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936. [Google Scholar] [CrossRef]
- Stempel, A.V.; Stumpf, A.; Zhang, H.Y.; Ozdogan, T.; Pannasch, U.; Theis, A.K.; Otte, D.M.; Wojtalla, A.; Racz, I.; Ponomarenko, A.; et al. Cannabinoid Type 2 Receptors Mediate a Cell Type-Specific Plasticity in the Hippocampus. Neuron 2016, 90, 795–809. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef]
- Atwood, B.K.; Mackie, K. CB2: A cannabinoid receptor with an identity crisis. Br. J. Pharmacol. 2010, 160, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e714. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef]
- Shao, Z.; Yan, W.; Chapman, K.; Ramesh, K.; Ferrell, A.J.; Yin, J.; Wang, X.; Xu, Q.; Rosenbaum, D.M. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 2019, 15, 1199–1205. [Google Scholar] [CrossRef]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Locke, K.; et al. Crystal Structure of the Human Cannabinoid Receptor CB2. Cell 2019, 176, 459–467.e413. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Johnston, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e618. [Google Scholar] [CrossRef]
- Krishna Kumar, K.; Shalev-Benami, M.; Robertson, M.J.; Hu, H.; Banister, S.D.; Hollingsworth, S.A.; Latorraca, N.R.; Kato, H.E.; Hilger, D.; Maeda, S.; et al. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 2019, 176, 448–458.e412. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Zhuang, Y.; Xu, T.H.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L.; Meng, X.; Xue, Y.; Wang, J.; et al. Cryo-EM Structure of the Human Cannabinoid Receptor CB2-Gi Signaling Complex. Cell 2020, 180, 645–654.e613. [Google Scholar] [CrossRef]
- Reggio, P.H. Endocannabinoid binding to the cannabinoid receptors: What is known and what remains unknown. Curr. Med. Chem. 2010, 17, 1468–1486. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Peters, M.; Murillo-Rodriguez, E.; Hanus, L.O. Cannabidiol--recent advances. Chem. Biodivers. 2007, 4, 1678–1692. [Google Scholar] [CrossRef]
- Franco, V.; Bialer, M.; Perucca, E. Cannabidiol in the treatment of epilepsy: Current evidence and perspectives for further research. Neuropharmacology 2021, 185, 108442. [Google Scholar] [CrossRef]
- McPartland, J.M.; Glass, M.; Pertwee, R.G. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: Interspecies differences. Br. J. Pharmacol. 2007, 152, 583–593. [Google Scholar] [CrossRef]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef]
- Sabatucci, A.; Tortolani, D.; Dainese, E.; Maccarrone, M. In silico mapping of allosteric ligand binding sites in type-1 cannabinoid receptor. Biotechnol. Appl. Biochem. 2018, 65, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Tham, M.; Yilmaz, O.; Alaverdashvili, M.; Kelly, M.E.M.; Denovan-Wright, E.M.; Laprairie, R.B. Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol-dimethylheptyl at the type 1 and type 2 cannabinoid receptors. Br. J. Pharmacol. 2019, 176, 1455–1469. [Google Scholar] [CrossRef]
- Chung, H.; Fierro, A.; Pessoa-Mahana, C.D. Cannabidiol binding and negative allosteric modulation at the cannabinoid type 1 receptor in the presence of delta-9-tetrahydrocannabinol: An In Silico study. PLoS ONE 2019, 14, e0220025. [Google Scholar] [CrossRef]
- Shahbazi, F.; Grandi, V.; Banerjee, A.; Trant, J.F. Cannabinoids and Cannabinoid Receptors: The Story so Far. iScience 2020, 23, 101301. [Google Scholar] [CrossRef]
- Morales, P.; Reggio, P.H. An Update on Non-CB1, Non-CB2 Cannabinoid Related G-Protein-Coupled Receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef]
- Laun, A.S.; Song, Z.H. GPR3 and GPR6, novel molecular targets for cannabidiol. Biochem. Biophys. Res. Commun. 2017, 490, 17–21. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB(1) and CB(2). Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed]
- Balenga, N.A.; Henstridge, C.M.; Kargl, J.; Waldhoer, M. Pharmacology, signaling and physiological relevance of the G protein-coupled receptor 55. Adv. Pharmacol. 2011, 62, 251–277. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Balenga, N.A.; Ford, L.A.; Ross, R.A.; Waldhoer, M.; Irving, A.J. The GPR55 ligand L-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB J. 2009, 23, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, PsiGPR53 and GPR55: GPR55 is extensively expressed in human brain. Brain Res. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef]
- Kihara, Y.; Maceyka, M.; Spiegel, S.; Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 2014, 171, 3575–3594. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjogren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.Y.; Lu, H.C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Nakajima, K.; Yamashita, A.; Kishimoto, S.; Sugiura, T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun. 2007, 362, 928–934. [Google Scholar] [CrossRef]
- Yin, H.; Chu, A.; Li, W.; Wang, B.; Shelton, F.; Otero, F.; Nguyen, D.G.; Caldwell, J.S.; Chen, Y.A. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. J. Biol. Chem. 2009, 284, 12328–12338. [Google Scholar] [CrossRef]
- Gantz, I.; Muraoka, A.; Yang, Y.K.; Samuelson, L.C.; Zimmerman, E.M.; Cook, H.; Yamada, T. Cloning and chromosomal localization of a gene (GPR18) encoding a novel seven transmembrane receptor highly expressed in spleen and testis. Genomics 1997, 42, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Hasegawa, H.; Inoue, A.; Muraoka, M.; Miyazaki, T.; Oka, K.; Yasukawa, M. Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem. Biophys. Res. Commun. 2006, 347, 827–832. [Google Scholar] [CrossRef]
- McHugh, D.; Hu, S.S.; Rimmerman, N.; Juknat, A.; Vogel, Z.; Walker, J.M.; Bradshaw, H.B. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Lu, V.B.; Puhl, H.L., III; Ikeda, S.R. N-Arachidonyl glycine does not activate G protein-coupled receptor 18 signaling via canonical pathways. Mol. Pharmacol. 2013, 83, 267–282. [Google Scholar] [CrossRef]
- Rajaraman, G.; Simcocks, A.; Hryciw, D.H.; Hutchinson, D.S.; McAinch, A.J. G protein coupled receptor 18: A potential role for endocannabinoid signaling in metabolic dysfunction. Mol. Nutr. Food Res. 2016, 60, 92–102. [Google Scholar] [CrossRef]
- McHugh, D. GPR18 in microglia: Implications for the CNS and endocannabinoid system signalling. Br. J. Pharmacol. 2012, 167, 1575–1582. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Felder, C.C. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: Evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 1997, 17, 5327–5333. [Google Scholar] [CrossRef] [PubMed]
- Przybyla, J.A.; Watts, V.J. Ligand-induced regulation and localization of cannabinoid CB1 and dopamine D2L receptor heterodimers. J. Pharmacol. Exp. Ther. 2010, 332, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Coke, C.J.; Scarlett, K.A.; Chetram, M.A.; Jones, K.J.; Sandifer, B.J.; Davis, A.S.; Marcus, A.I.; Hinton, C.V. Simultaneous Activation of Induced Heterodimerization between CXCR4 Chemokine Receptor and Cannabinoid Receptor 2 (CB2) Reveals a Mechanism for Regulation of Tumor Progression. J. Biol. Chem. 2016, 291, 9991–10005. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pinilla, E.; Reyes-Resina, I.; Onatibia-Astibia, A.; Zamarbide, M.; Ricobaraza, A.; Navarro, G.; Moreno, E.; Dopeso-Reyes, I.G.; Sierra, S.; Rico, A.J.; et al. CB1 and GPR55 receptors are co-expressed and form heteromers in rat and monkey striatum. Exp. Neurol. 2014, 261, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; Beukers, M.W.; Mulder-Krieger, T.; Ijzerman, A.P. The endocannabinoid 2-arachidonylglycerol is a negative allosteric modulator of the human A3 adenosine receptor. Biochem. Pharmacol. 2010, 79, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Kathmann, M.; Flau, K.; Redmer, A.; Trankle, C.; Schlicker, E. Cannabidiol is an allosteric modulator at mu- and delta-opioid receptors. Naunyn Schmiedebergs Arch. Pharmacol. 2006, 372, 354–361. [Google Scholar] [CrossRef]
- Cascio, M.G.; Gauson, L.A.; Stevenson, L.A.; Ross, R.A.; Pertwee, R.G. Evidence that the plant cannabinoid cannabigerol is a highly potent alpha2-adrenoceptor agonist and moderately potent 5HT1A receptor antagonist. Br. J. Pharmacol. 2010, 159, 129–141. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, version 2.0; Schrodinger, LLC: New York, NY, USA, 2015.















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, K.J.H.; Lim, Y.P.; Hartono, Y.D.; Go, M.K.; Fan, H.; Yew, W.S. Biosynthesis of Nature-Inspired Unnatural Cannabinoids. Molecules 2021, 26, 2914. https://doi.org/10.3390/molecules26102914
Lim KJH, Lim YP, Hartono YD, Go MK, Fan H, Yew WS. Biosynthesis of Nature-Inspired Unnatural Cannabinoids. Molecules. 2021; 26(10):2914. https://doi.org/10.3390/molecules26102914
Chicago/Turabian StyleLim, Kevin J. H., Yan Ping Lim, Yossa D. Hartono, Maybelle K. Go, Hao Fan, and Wen Shan Yew. 2021. "Biosynthesis of Nature-Inspired Unnatural Cannabinoids" Molecules 26, no. 10: 2914. https://doi.org/10.3390/molecules26102914
APA StyleLim, K. J. H., Lim, Y. P., Hartono, Y. D., Go, M. K., Fan, H., & Yew, W. S. (2021). Biosynthesis of Nature-Inspired Unnatural Cannabinoids. Molecules, 26(10), 2914. https://doi.org/10.3390/molecules26102914