
A Concise Synthesis of a BODIPY-Labeled Tetrasaccharide Related to the Antitumor PI-88

 , , ,
, , ,  ,
,
Abstract
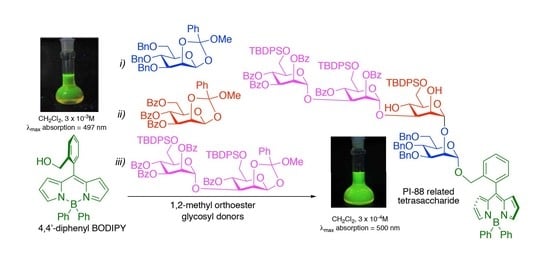
:
1. Introduction
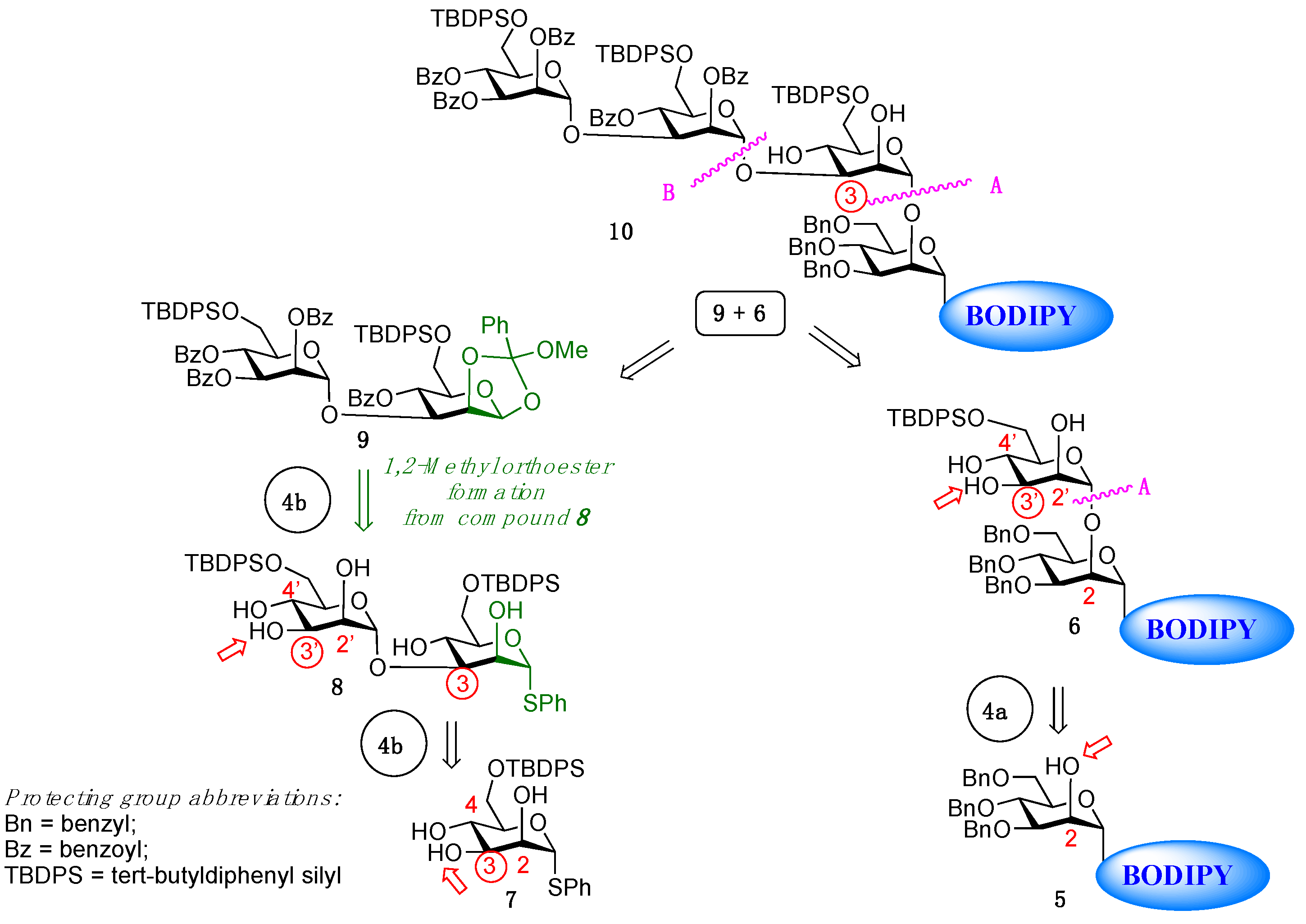
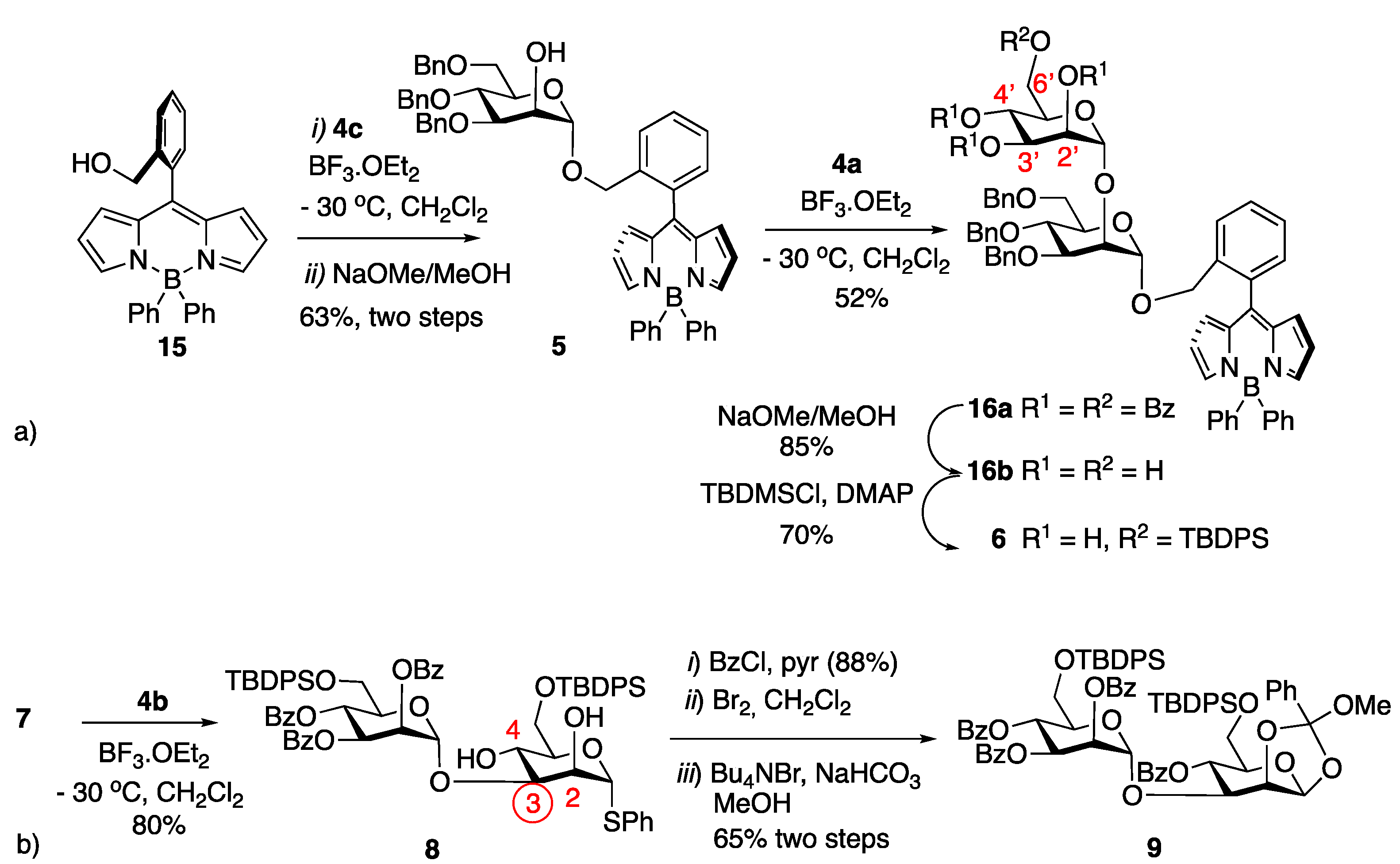
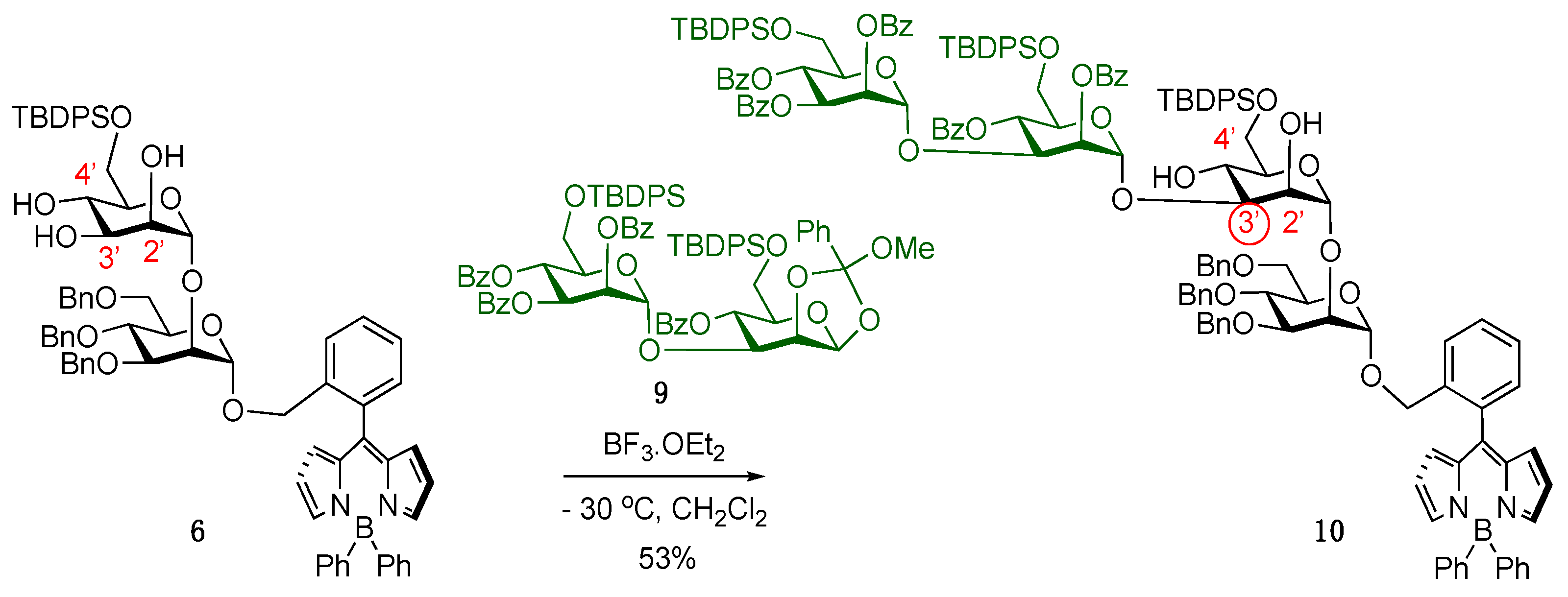
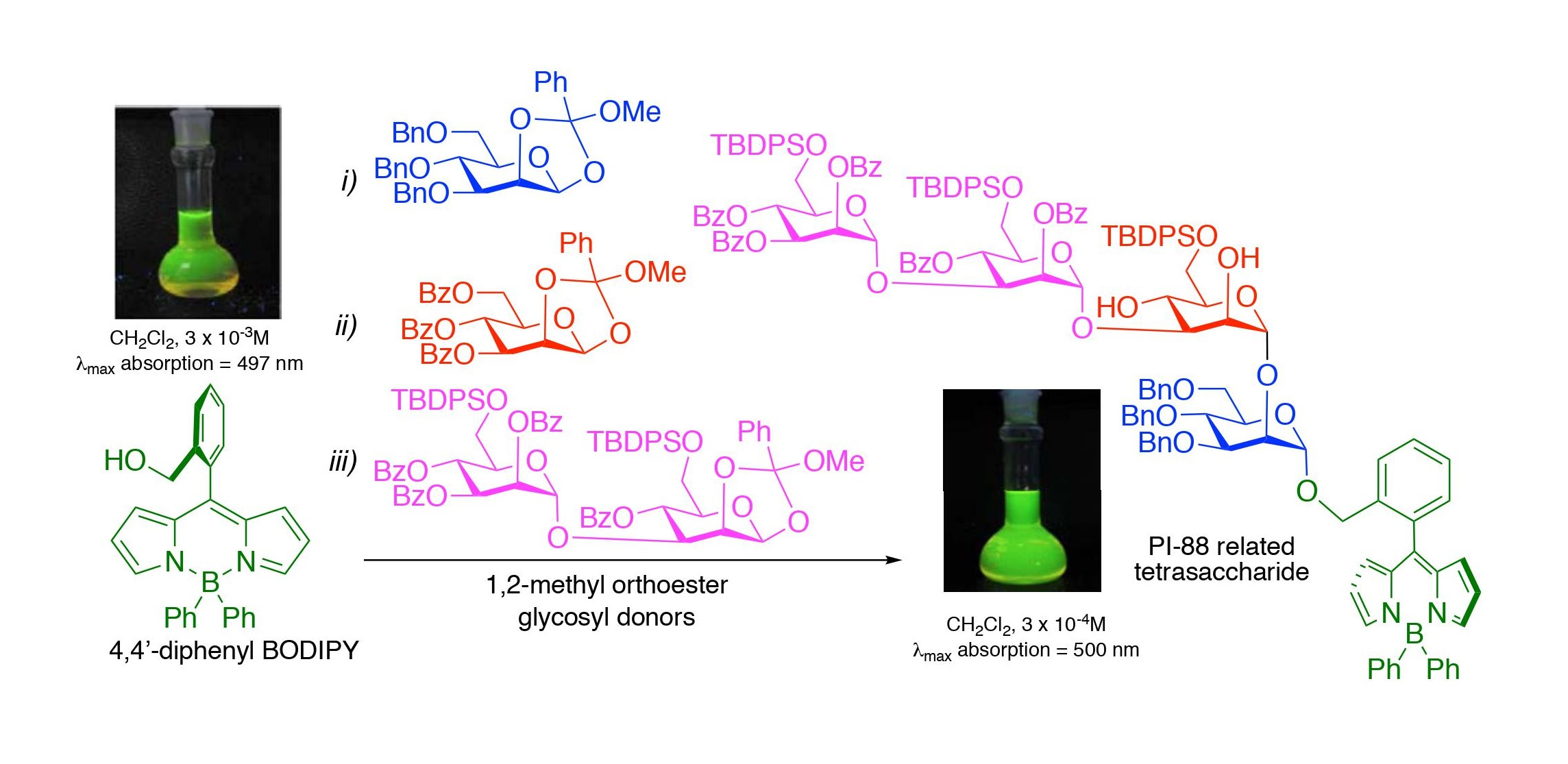
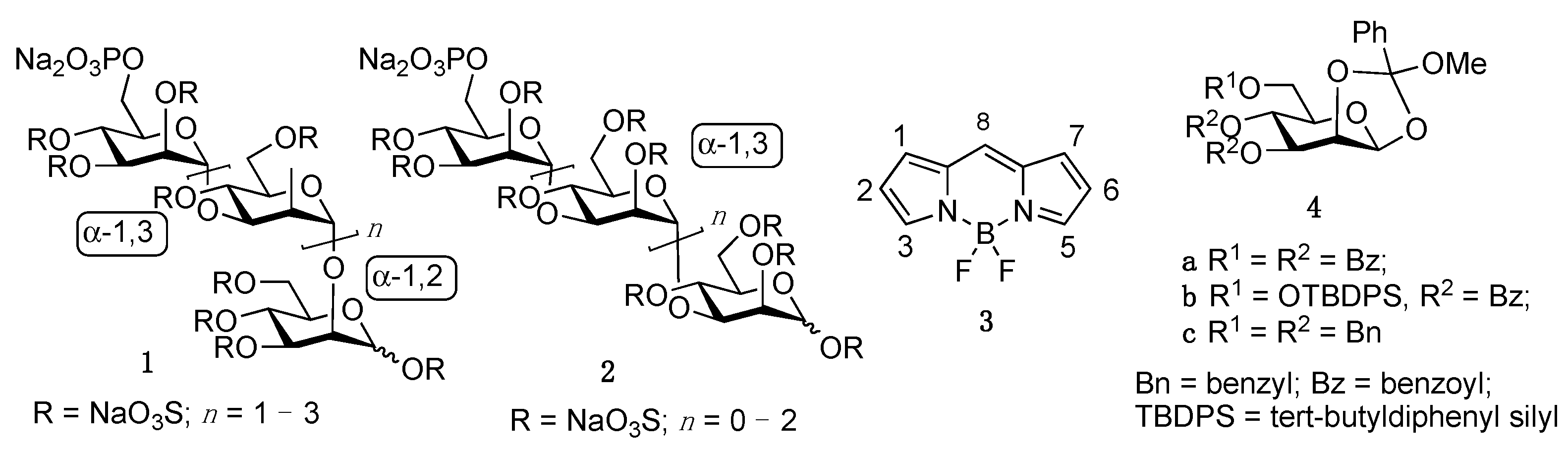
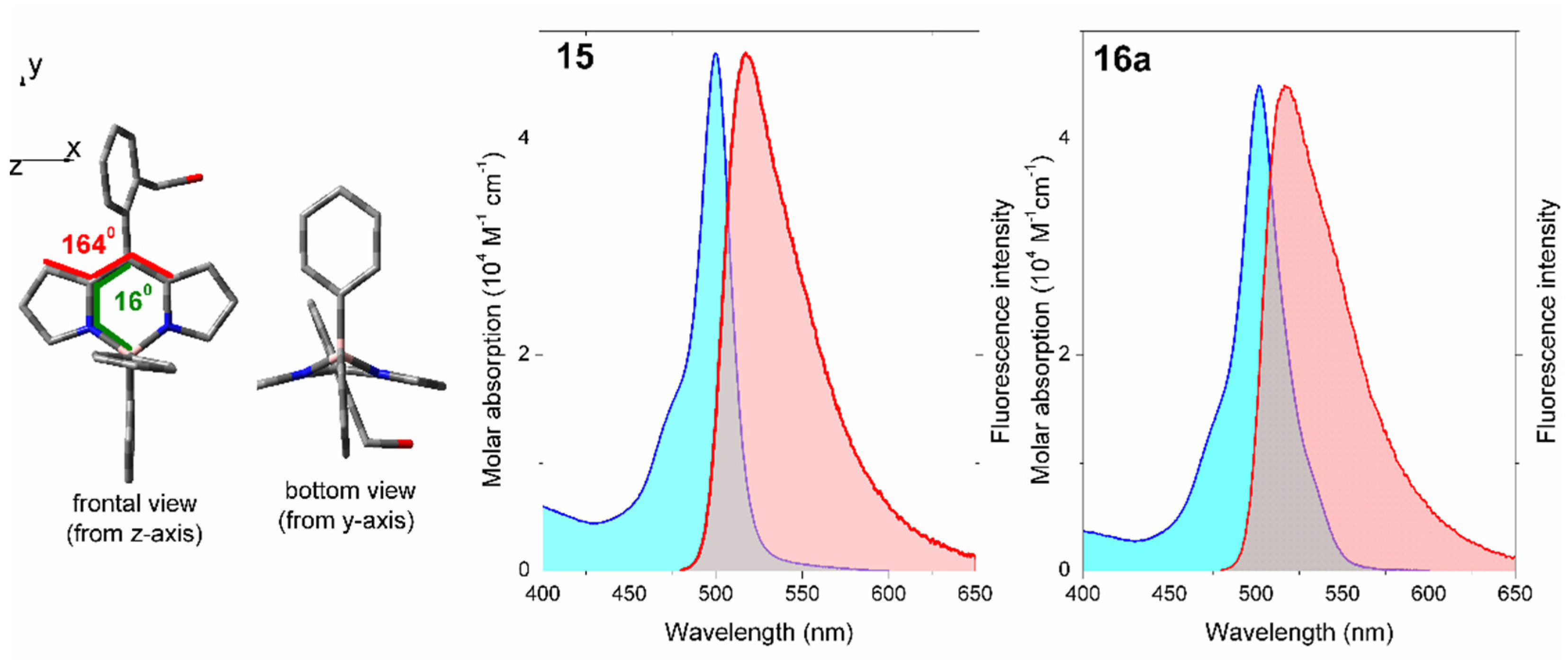
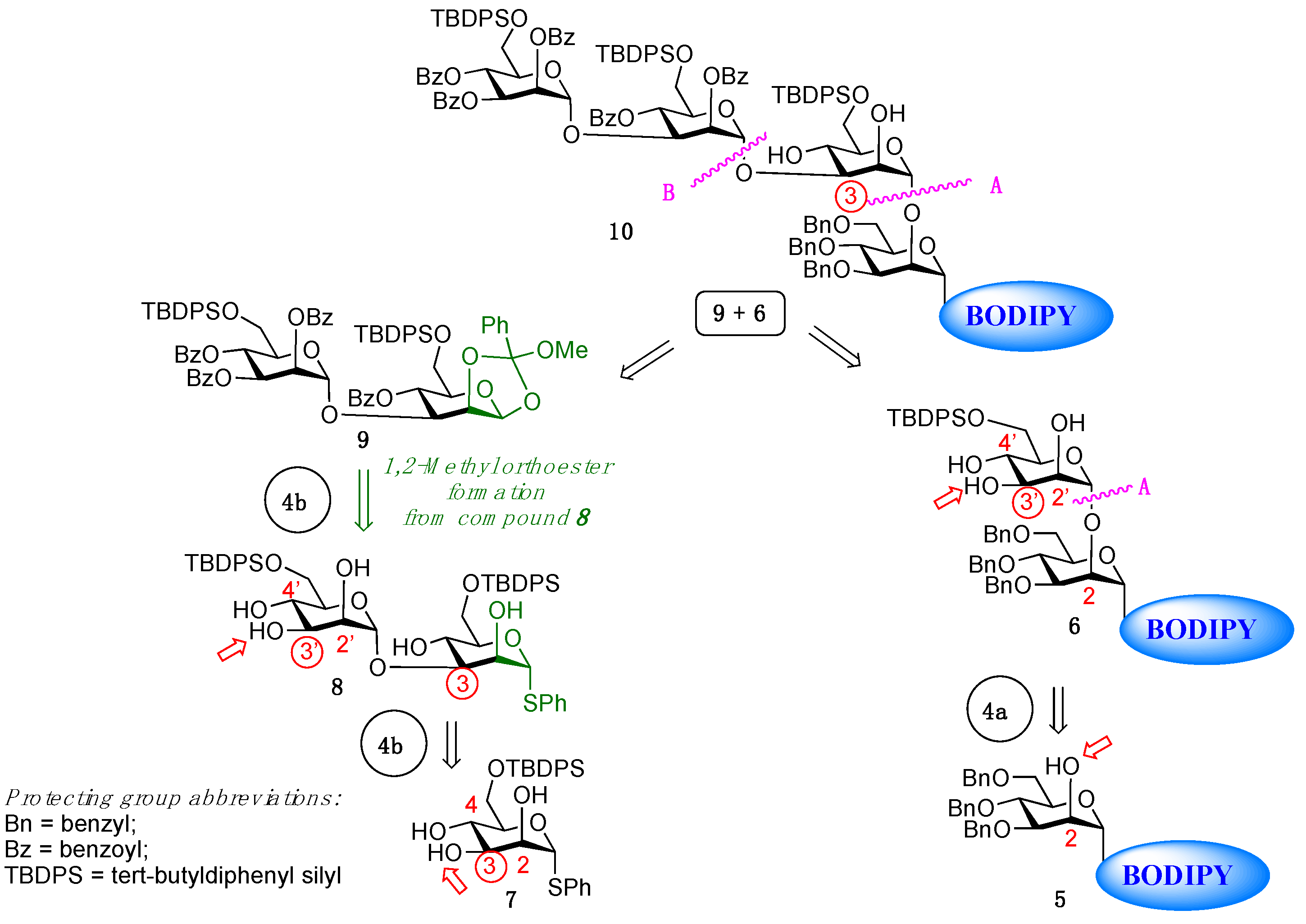
2. Results
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. General Procedures
4.2.1. General Procedure for Glycosylation. Procedure A
4.2.2. General Procedure for Debenzoylation. Procedure B
4.2.3. General Procedure for Silylation. Procedure C
4.2.4. General Procedure for Characterization of Polyol Derivatives as Peracetates. Procedure D
4.3. Synthesis
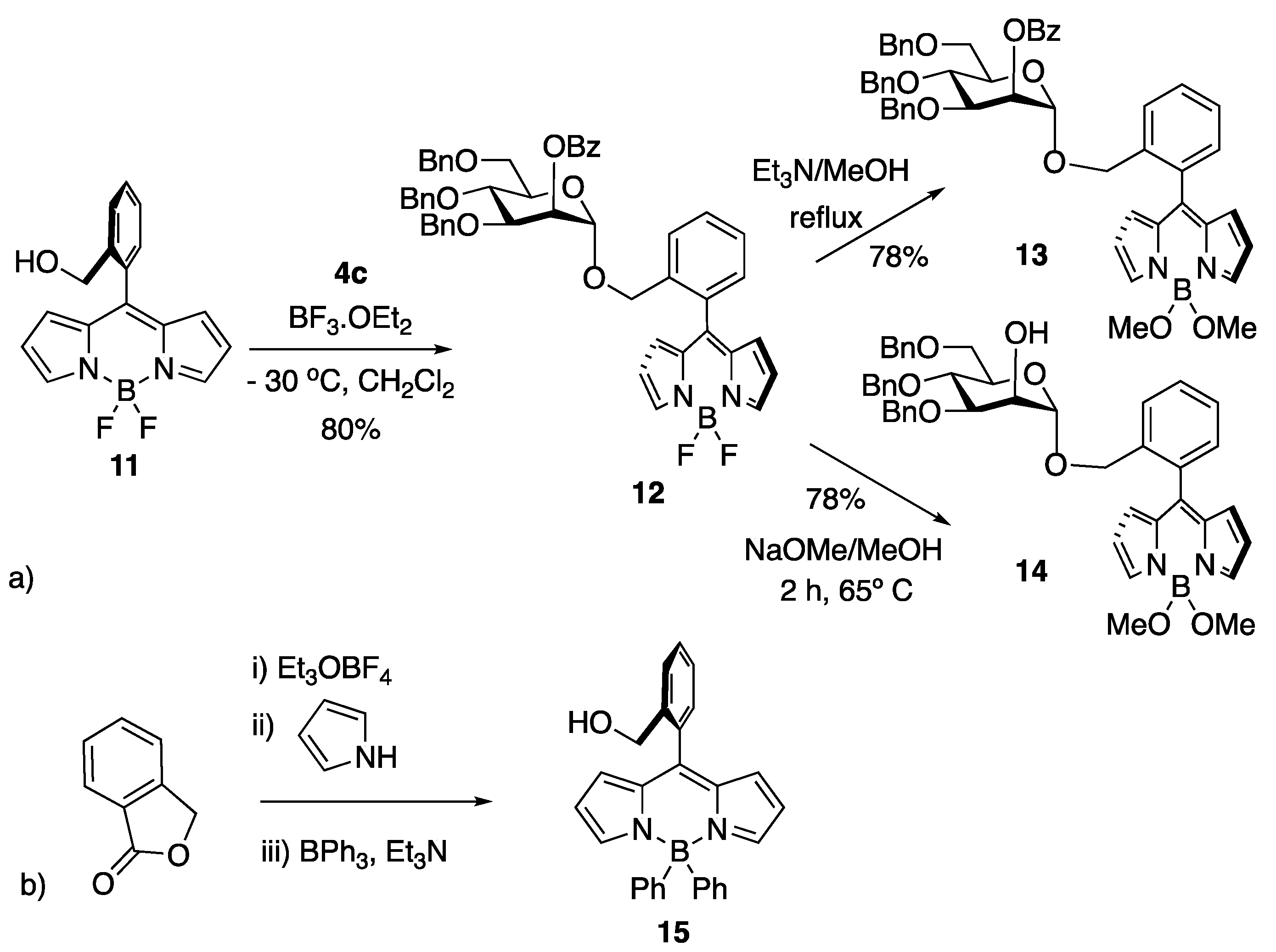
4.3.1. BF2-Bodipy 11
4.3.2. Mannopyranosyl BODIPY 12
4.3.3. Mannopyranosyl BODIPY 13
4.3.4. Mannopyranosyl BODIPY 14
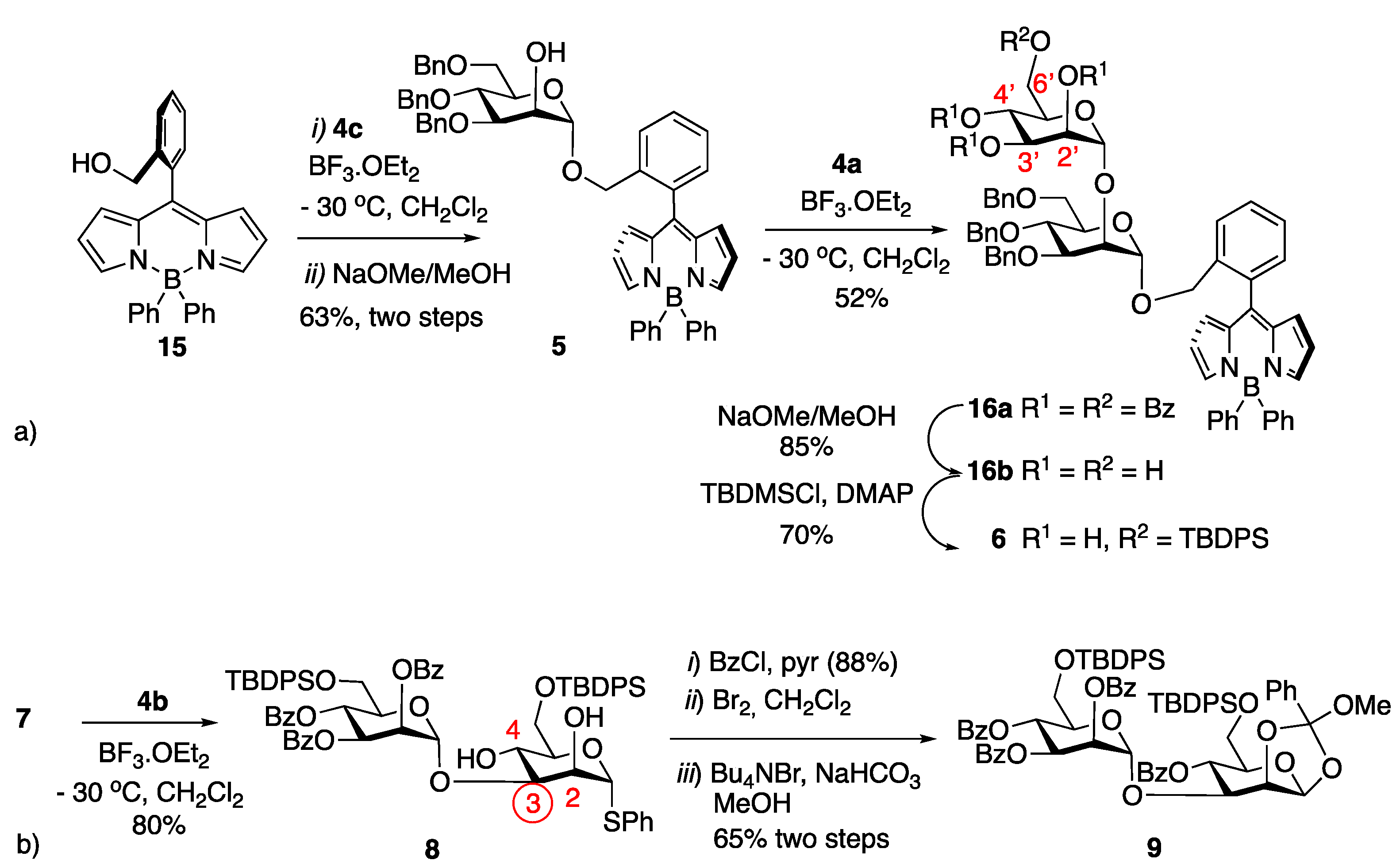
4.3.5. B(Ph)2-BODIPY 15
4.3.6. Mannopyranosyl BODIPY 5
4.3.7. BODIPY Disaccharide 16a
4.3.8. BODIPY-Disaccharide Tetraol 16b
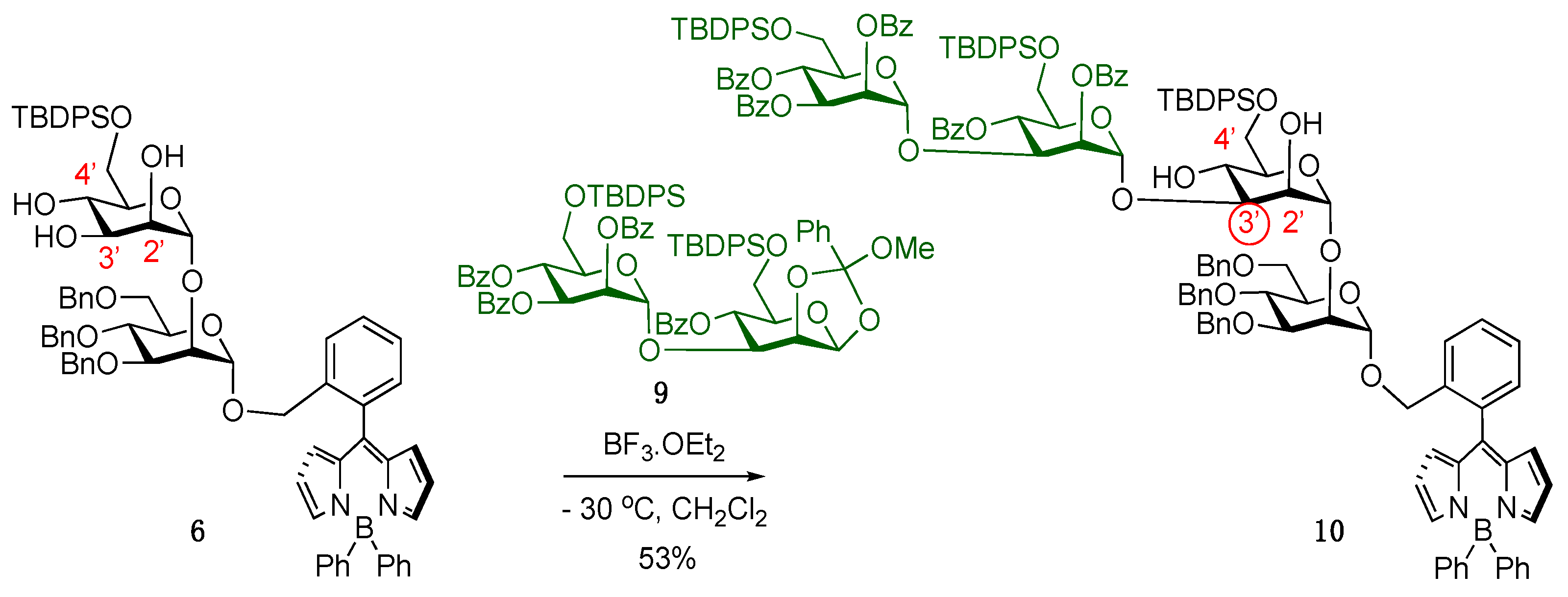
4.3.9. BODIPY-Disaccharide Silylated Triol 6
4.3.10. Thioglycosyl Disaccharide 8
4.3.11. 1,2-Methyl Orthoester Disaccharide 9
4.3.12. BODIPY-Tetrasaccharide 10
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ferro, V.; Hammond, E.; Fairweather, J.K. The development of inhibitors of heparanase, a key enzyme involved in tumour metastasis, angiogenesis and inflammation. Rev. Med. Chem. 2004, 4, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.; Bytheway, I.; Ferro, V. Heparanase as a target for anticancer therapeutics: New developments and future prospects. In New Developments in Therapeutic Glycomics; Delehedde, M., Lortat-Jacob, H., Eds.; Research Signpost: Trivandrum, India, 2006; pp. 251–282. [Google Scholar]
- Miao, H.Q.; Liu, H.; Navarro, E.; Kussie, P.; Zhu, Z. Development of heparanase inhibitors for anti-cancer therapy. Curr. Med. Chem. 2006, 13, 2101–2111. [Google Scholar]
- McKenzie, E.A. Heparanase: A target for drug discovery in cancer and inflammation. Br. J. Pharmacol. 2007, 151, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Ma, S. Recent advances in the discovery of heparanase inhibitors as anticancer agents. Eur. J. Med. Chem. 2016, 121, 209–220. [Google Scholar] [CrossRef]
- Chhabra, M.; Ferro, V. The development of assays for heparanase enzymatic activity: Towards a gold standard. Molecules 2018, 23, 2971. [Google Scholar] [CrossRef] [Green Version]
- Chhabra, M.; Ferro, V. PI-88 and related heparan sulfate mimetics. In Heparanase: From Basic Research to Clinical Applications, 1st ed.; Vlodavsky, I., Sanderson, R.D., Ilan, N., Eds.; Springer-Nature: Cham, Switzerland, 2020; Chapter 19; pp. 473–492. [Google Scholar]
- Mechref, Y.; Novotny, M.V. Structural investigations of glycoconjugates at high sensitivity. Chem. Rev. 2002, 102, 321–369. [Google Scholar] [CrossRef]
- Xia, B.; Kawar, Z.S.; Ju, T.; Alvarez, R.A.; Sachdev, G.P.; Cummings, R.D. Versatile fluorescent derivatization of glycans for glycomic analysis. Nat. Methods 2005, 2, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Ogawa, M.; Alford, R.; Choyke, P.L.; Urano, Y. New strategies for fluorescent probe design in medical diagnostic imaging. Chem. Rev. 2010, 110, 2620–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, T.; Nagano, T. Fluorescent probes for sensing and imaging. Nat. Methods 2011, 8, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Keithley, R.B.; Rosenthal, A.S.; Essaka, D.C.; Tanaka, H.; Yoshimura, Y.; Palcic, M.M.; Hindsgaul, O.; Dovichi, N.J. Capillary electrophoresis with three-color fluorescence detection for the analysis of glycosphingolipid metabolism. Analyst 2013, 138, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Louret, A.; Burgess, K. BODIPY Dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar]
- Ziessel, R.; Ulrich, G.; Harriman, A. The chemistry of BODIPY: A new El Dorado for fluorescence tools. New. J. Chem. 2007, 31, 496–501. [Google Scholar] [CrossRef]
- Ulrich, G.; Ziessel, R.; Harriman, A. The chemistry of fluorescent BODIPY dyes: Versatility unsurpassed. Angew. Chem. Int. Ed. 2008, 47, 1184–1201. [Google Scholar] [CrossRef] [PubMed]
- Boens, N.; Verbelen, B.; Dehaen, W. Postfunctionalization of the BODIPY core: Synthesis and spectroscopy. Eur. J. Org. Chem. 2015, 30, 6577–6595. [Google Scholar] [CrossRef] [Green Version]
- Bañuelos, J. BODIPY Dye, the most versatile fluorophore ever? Chem. Rec. 2016, 16, 335–348. [Google Scholar] [CrossRef]
- Boens, N.; Verbelen, B.; Ortiz, M.J.; Jiao, L.; Dehaen, W. Synthesis of BODIPY dyes through postfunctionalization of the boron dipyrromethene core. Coord. Chem. Rev. 2019, 399, 213024. [Google Scholar] [CrossRef]
- Uriel, C.; Permingeat, C.; Ventura, J.; Avellanal-Zaballa, E.; Bañuelos, J.; Garcia-Moreno, I.; Gómez, A.M.; López, J.C. BODIPYs as chemically stable fluorescent tags for synthetic glycosylation strategies towards fluorescently labeled saccharides. Chem. Eur. J. 2020, 26, 5388–5399. [Google Scholar] [CrossRef]
- Ekblad, M.; Adamiak, B.; Bergstrom, T.; Johnstone, K.D.; Karoli, T.; Liu, L.; Ferro, V.; Trybala, E. A highly lipophilic sulfated tetrasaccharide glycoside related to muparfostat (PI-88) exhibits virucidal activity against herpes simplex virus. Antivir. Res. 2010, 86, 196–203. [Google Scholar] [CrossRef]
- Handley, P.N.; Carroll, A.; Ferro, V. New structural insights into the oligosaccharide phosphate fraction of Pichia (Hansenula) holstii NRRL Y2448 phosphomannan. Carbohydr. Res. 2017, 446–447, 68–75. [Google Scholar] [CrossRef]
- Gu, G.; Wei, G.; Du, Y. Synthesis of a 6V-sulfated mannopentasaccharide analogue related to PI-88. Carbohydr. Res. 2004, 339, 1155–1162. [Google Scholar] [CrossRef]
- Namme, R.; Mitsugi, T.; Takahashi, H.; Ikegami, S. Synthesis of PI-88 analogue using novel O-glycosidation of exo-methylenesugars. Tetrahedron Lett. 2005, 46, 3033–3036. [Google Scholar] [CrossRef]
- Fairweather, J.K.; Hammond, E.; Johnstone, K.D.; Ferro, V. Synthesis and heparanase inhibitory activity of sulfated mannooligosaccharides related to the antiangiogenic agent PI-88. Bioorg. Med. Chem. 2008, 16, 699–709. [Google Scholar] [CrossRef]
- Valerio, S.; Pastore, A.; Adinolfi, M.; Iadonisi, A. Sequential one-pot glycosidations catalytically promoted: Unprecedented strategy in oligosaccharide synthesis for the straightforward assemblage of the antitumor PI-88 pentasaccharide. J. Org. Chem. 2008, 73, 4496–4503. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Johnstone, K.D.; Fairweather, J.K.; Dredge, K.; Ferro, V. An improved synthetic route to the potent angiogenesis inhibitor benzyl Manα(1→3)-Manα(1→3)-Manα(1→3)-Manα(1→2)-Man hexadecasulfate. Aust. J. Chem. 2009, 62, 546–552. [Google Scholar] [CrossRef] [Green Version]
- Pastore, A.; Adinolfi, M.; Iadonisi, A.; Valerio, S. One-pot catalytic glycosidation/Fmoc removal−An iterable sequence for straightforward assembly of oligosaccharides related to HIC gp120. Eur. J. Org. Chem. 2010, 4, 711–718. [Google Scholar] [CrossRef]
- Mong, K.-K.T.; Shiau, K.-S.; Lin, Y.H.; Cheng, K.-C.; Lin, C.-H. A concise synthesis of single components of partially sulfated oligomannans. Org. Biomol. Chem. 2015, 13, 11550–11560. [Google Scholar] [CrossRef]
- Kochetkov, N.K.; Khorlin, A.J.; Bochkov, A.F. New synthesis of glycosides. Tetrahedron Lett. 1964, 5, 289–293. [Google Scholar] [CrossRef]
- Kochetkov, N.K.; Khorlin, A.J.; Bochkov, A.F. A new method of glycosylation. Tetrahedron 1967, 23, 693–707. [Google Scholar] [CrossRef]
- Uriel, C.; Ventura, J.; Gómez, A.M.; López, J.C.; Fraser-Reid, B. Methyl 1,2-orthoesters as useful glycosyl donors in glycosylation reactions: A comparison with n-pent-4-enyl 1,2 orthoesters. Eur. J. Org. Chem. 2012, 16, 3122–3131. [Google Scholar] [CrossRef]
- Uriel, C.; Ventura, J.; Gómez, A.M.; López, J.C.; Fraser-Reid, B. Unexpected stereocontrolled access to 1α,1’β-disaccharides from methyl 1,2-ortho esters. J. Org. Chem. 2012, 77, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Uriel, C.; Rijo, P.; Fernandes, A.S.; Gómez, A.M.; Fraser-Reid, B.; López, J.C. Methyl 1,2-orthoesters in acid-washed molecular sieves mediated glycosylations. Chem. Select 2016, 1, 6011–6015. [Google Scholar] [CrossRef]
- Lu, J.; Fraser-Reid, B. The antituberculosis, antitumor, multibranched dodecafuranoarabinan of Mycobacterium species has been assembled from a single n-pentenylfuranoside source. Chem. Commun. 2005, 7, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakash, K.N.; Lu, J.; Fraser-Reid, B. Synthesis of a lipomannan component of the cell-wall complex of Mycobacterium tuberculosis is based on Paulsen’s concept of donor/acceptor “match”. Angew. Chem. Int. Ed. 2005, 44, 5894–5898. [Google Scholar] [CrossRef]
- Fraser-Reid, B.; Loópez, J.C. Orthoesters and Related Derivatives. In Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Demchenko, A.V., Ed.; Wiley-VCH: New York, NY, USA, 2008; Chapter 5.1. [Google Scholar]
- Roacho, R.I.; Metta-Magaña, A.J.; Peña-Cabrera, E.; Pannell, K.H. Synthesis, structural characterization, and spectroscopic properties of the ortho, meta and para isomers of 8-(HOCH2-C6H4)-BODIPY and 8-(MeOC6H4)-BODIPY. J. Phys. Org. Chem. 2013, 26, 345–351. [Google Scholar] [CrossRef]
- Del Rio, M.; Lobo, F.; Lopez, J.C.; Oliden, A.; Bañuelos, J.; Lopez-Arbeloa, I.; Garcia-Moreno, I.; Gomez, A.M. One-pot synthesis of rotationally restricted, conjugatable, BODIPY derivatives from phthalides. J. Org. Chem. 2017, 82, 1240–1247. [Google Scholar] [CrossRef]
- Bodio, E.; Goze, C. Investigation of B-F substitution of BODIPY and aza-BODIPY dyes: Development of B-O and B-C BODIPYs. Dyes Pigment. 2019, 160, 700–710. [Google Scholar] [CrossRef]
- Yang, L.; Simionescu, R.; Lough, A.; Yan, H. Some observations relating to the stability of the BODIPY fluorophore under acidic and basic conditions. Dyes Pigment. 2011, 91, 264–267. [Google Scholar] [CrossRef]
- Nguyen, A.L.; Fronczek, F.R.; Smith, K.M.; Vicente, M.G.H. Synthesis of 4,4’-functionalized BODIPYs from dipyrrins. Tetrahedron Lett. 2015, 56, 6348–6351. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.C.; Agocs, A.; Uriel, C.; Gomez, A.M.; Fraser-Reid, B. Iterative, orthogonal strategy for oligosaccharide synthesis based on the regioselective glycosylation of polyol acceptors with partially unprotected n-pentenyl-orthoesters: Further evidence for reciprocal donor acceptor selectivity. Chem. Commun. 2005, 40, 5088–5090. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Zhao, J.; Shao, H. A facile method for the preparation of sugar orthoesters promoted by anhydrous sodium bicarbonate. Can. J. Chem. 2009, 87, 1733–1737. [Google Scholar] [CrossRef] [Green Version]
- Vidadala, S.R.; Thadke, S.A.; Hotha, S. Orthogonal activation of propargyl and n-pentenyl glycosides and 1,2-orthoesters. J. Org. Chem. 2009, 74, 9233–9236. [Google Scholar] [CrossRef] [PubMed]
- Duran-Sampedro, G.; Esnal, I.; Agarrabeitia, A.R.; Bañuelos-Prieto, J.; Cerdan, L.; Garcia-Moreno, I.; Costela, A.; Lopez-Arbeloa, I.; Ortiz, M.-J. First highly efficient and photostable E and C derivatives of 4,4,-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) as dye lasers in the liquid phase, thin films, and solid-state rods. Chem. Eur. J. 2014, 20, 2646–2653. [Google Scholar] [CrossRef] [PubMed]
- Patel, I.; Woodcock, J.; Beams, R.; Stranick, S.J.; Niewendaal, R.; Gilman, J.W.; Mulenos, M.R.; Sayes, C.M.; Salari, M.; DeLoid, G.; et al. Fluorescently labeled cellulose nanofibers for environmental health and safety studies. Nanomaterials 2021, 11, 1015. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λab (nm) | εmax | λfl (nm) | ∅ | <τ> (ns) | λla (nm) | Eff (%) |
|---|---|---|---|---|---|---|---|
| 11 | 499.5 | 6.4 | 513.0 | 0.74 | 6.45 | 540 | 60 |
| 15 | 497.0 | 4.8 | 511.5 | 0.22 | 2.83 | 533 | 18 |
| 16a | 500.0 | 4.5 | 516.0 | 0.31 | 3.19 | 545 | 38 |
| 10 | 500.5 | 4.1 | 515.0 | 0.31 | 3.57 | 547 | 36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ventura, J.; Uriel, C.; Gomez, A.M.; Avellanal-Zaballa, E.; Bañuelos, J.; García-Moreno, I.; Lopez, J.C. A Concise Synthesis of a BODIPY-Labeled Tetrasaccharide Related to the Antitumor PI-88. Molecules 2021, 26, 2909. https://doi.org/10.3390/molecules26102909
Ventura J, Uriel C, Gomez AM, Avellanal-Zaballa E, Bañuelos J, García-Moreno I, Lopez JC. A Concise Synthesis of a BODIPY-Labeled Tetrasaccharide Related to the Antitumor PI-88. Molecules. 2021; 26(10):2909. https://doi.org/10.3390/molecules26102909
Chicago/Turabian StyleVentura, Juan, Clara Uriel, Ana M. Gomez, Edurne Avellanal-Zaballa, Jorge Bañuelos, Inmaculada García-Moreno, and Jose Cristobal Lopez. 2021. "A Concise Synthesis of a BODIPY-Labeled Tetrasaccharide Related to the Antitumor PI-88" Molecules 26, no. 10: 2909. https://doi.org/10.3390/molecules26102909
APA StyleVentura, J., Uriel, C., Gomez, A. M., Avellanal-Zaballa, E., Bañuelos, J., García-Moreno, I., & Lopez, J. C. (2021). A Concise Synthesis of a BODIPY-Labeled Tetrasaccharide Related to the Antitumor PI-88. Molecules, 26(10), 2909. https://doi.org/10.3390/molecules26102909