
Intramolecular Photo-Oxidation as a Potential Source to Probe Biological Electron Damage: A Carboxylated Adenosine Analogue as Case Study


Abstract
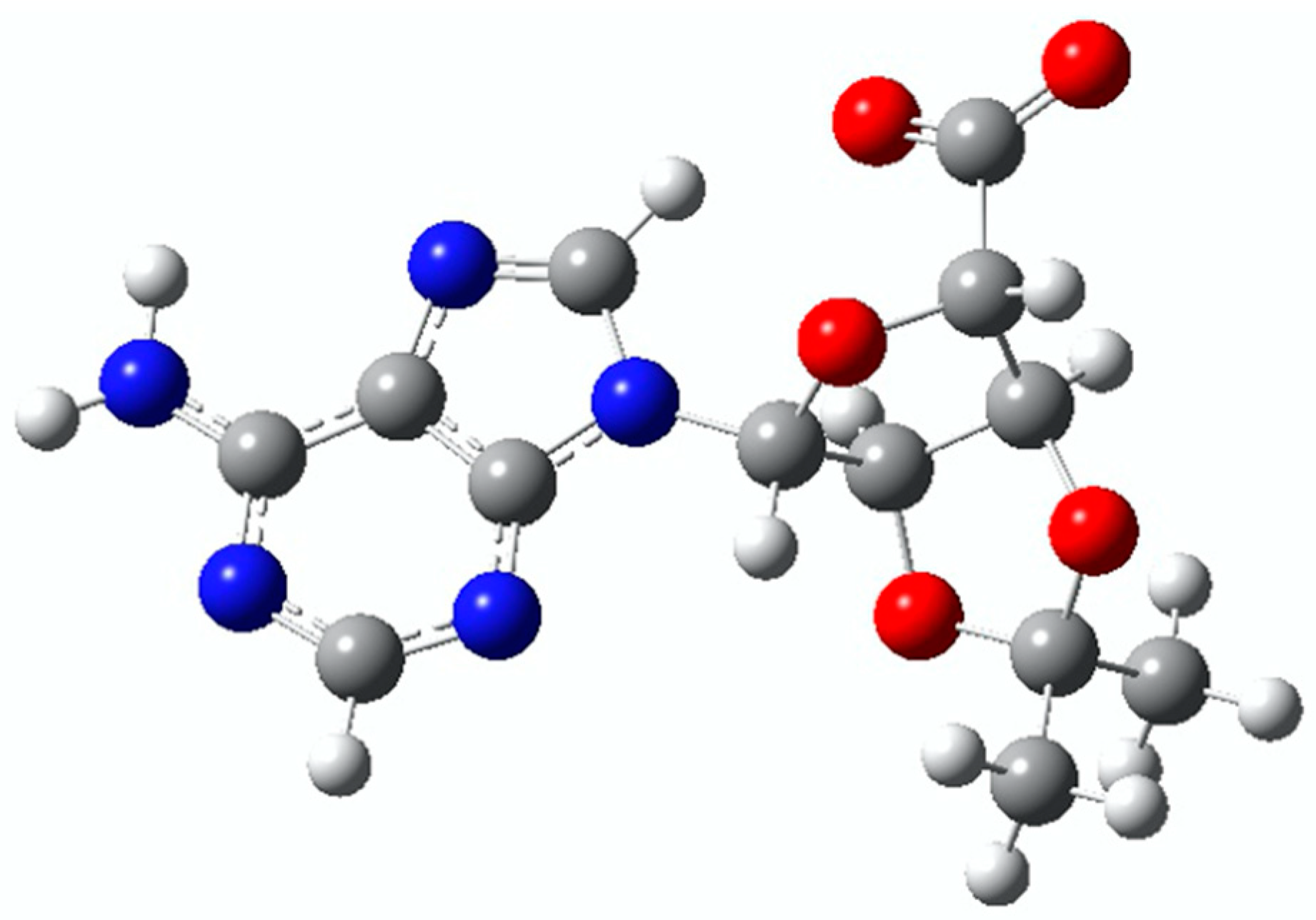
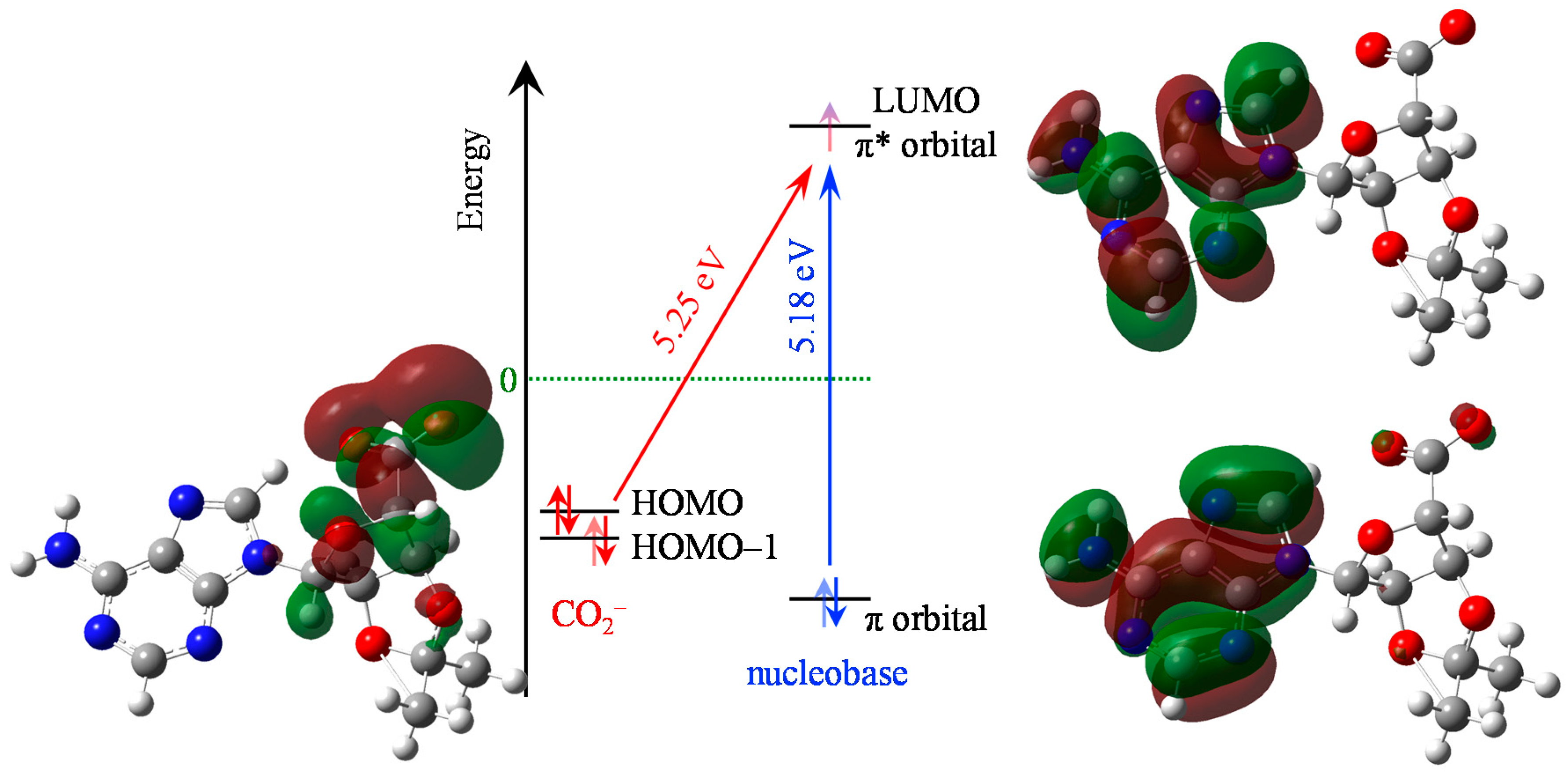
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alberts, B. Molecular Biology of the Cell, 5th ed.; Garland Science: New York, NY, USA, 2008. [Google Scholar]
- Steenken, S. Purine bases, nucleosides, and nucleotides: Aqueous solution redox chemistry and transformation reactions of their radical cations and e- and OH adducts. Chem. Rev. 1989, 89, 503–520. [Google Scholar] [CrossRef]
- Burrows, C.J.; Muller, J.G. Oxidative nucleobase modifications leading to strand scission. Chem. Rev. 1998, 98, 1109–1151. [Google Scholar] [CrossRef]
- Simons, J. How do low-energy (0.1–2 eV) electrons cause DNA-strand breaks? Acc. Chem. Res. 2006, 39, 772–779. [Google Scholar] [CrossRef]
- Kumar, A.; Becker, D.; Adhikary, A.; Sevilla, M.D. Reaction of Electrons with DNA: Radiation Damage to Radiosensitization. Int. J. Mol. Sci. 2019, 20, 3998. [Google Scholar] [CrossRef]
- Zheng, Y.; Cloutier, P.; Hunting, D.J.; Wagner, J.R.; Sanche, L. Glycosidic bond cleavage of thymidine by low-energy electrons. J. Am. Chem. Soc. 2004, 126, 1002–1003. [Google Scholar] [CrossRef]
- Aflatooni, K.; Gallup, G.A.; Burrow, P.D. Electron attachment energies of the DNA bases. J. Phys. Chem. A 1998, 102, 6205–6207. [Google Scholar] [CrossRef]
- Ptasińska, S.; Denifl, S.; Scheier, P.; Märk, T.D. Inelastic electron interaction (attachment/ionization) with deoxyribose. J. Chem. Phys. 2004, 120, 8505–8511. [Google Scholar] [CrossRef]
- Li, X.; Sevilla, M.D.; Sanche, L. Density functional theory studies of electron interaction with DNA: Can zero eV electrons induce strand breaks? J. Am. Chem. Soc. 2003, 125, 13668–13669. [Google Scholar] [CrossRef]
- Scheer, A.M.; Aflatooni, K.; Gallup, G.A.; Burrow, P.D. Bond Breaking and Temporary Anion States in Uracil and Halouracils: Implications for the DNA Bases. Phys. Rev. Lett. 2004, 92, 068102. [Google Scholar] [CrossRef]
- Baccarelli, I.; Bald, I.; Gianturco, F.A.; Illenberger, E.; Kopyra, J. Electron-induced damage of DNA and its components: Experiments and theoretical models. Phys. Rep. 2011, 508, 1–44. [Google Scholar] [CrossRef]
- Sanche, L. Role of secondary low energy electrons in radiobiology and chemoradiation therapy of cancer. Chem. Phys. Lett. 2009, 474, 1–6. [Google Scholar] [CrossRef]
- Sanche, L. Low energy electron-driven damage in biomolecules. Eur. Phys. J. D 2005, 35, 367–390. [Google Scholar] [CrossRef]
- Boudaiffa, B.; Cloutier, P.; Hunting, D.; Huels, M.A.; Sanche, L. Resonant formation of DNA strand breaks by low-energy (3 to 20 eV) electrons. Science 2000, 287, 1658–1660. [Google Scholar]
- Burrow, P.D.; Gallup, G.A.; Modelli, A. Are There π* Shape Resonances in Electron Scattering from Phosphate Groups? J. Phys. Chem. A 2008, 112, 4106–4113. [Google Scholar] [CrossRef]
- Fennimore, M.A.; Matsika, S. Core-excited and shape resonances of uracil. Phys. Chem. Chem. Phys. 2016, 18, 30536–30545. [Google Scholar] [CrossRef]
- Abdoul-Carime, H.; Gohlke, S.; Illenberger, E. Site-specific dissociation of DNA bases by slow electrons at early stages of irradiation. Phys. Rev. Lett. 2004, 92, 168103. [Google Scholar] [CrossRef]
- Barrios, R.; Skurski, P.; Simons, J. Mechanism for damage to DNA by low-energy electrons. J. Phys. Chem. B 2002, 106, 7991–7994. [Google Scholar] [CrossRef]
- Berdys, J.; Anusiewicz, I.; Skurski, P.; Simons, J. Damage to Model DNA Fragments from Very Low-Energy (<1 eV) Electrons. J. Am. Chem. Soc. 2004, 126, 6441–6447. [Google Scholar] [CrossRef]
- Bhaskaran, R.; Sarma, M. The role of the shape resonance state in low energy electron induced single strand break in 2′-deoxycytidine-5′-monophosphate. Phys. Chem. Chem. Phys. 2015, 17, 15250–15257. [Google Scholar] [CrossRef] [PubMed]
- Khorsandgolchin, G.; Sanche, L.; Cloutier, P.; Wagner, J.R. Strand Breaks Induced by Very Low Energy Electrons: Product Analysis and Mechanistic Insight into the Reaction with TpT. J. Am. Chem. Soc. 2019, 141, 10315–10323. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Cloutier, P.; Hunting, D.; Sanche, L. Dissociative Electron Attachment to DNA. Phys. Rev. Lett. 2003, 90, 208102. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Burrow, P.D.; Cai, Z.L.; Cloutier, P.; Hunting, D.; Sanche, L. DNA strand breaks induced by 0-4 eV electrons: The role of shape resonances. Phys. Rev. Lett. 2004, 93, 068101. [Google Scholar] [CrossRef]
- Zheng, Y.; Cloutier, P.; Hunting, D.J.; Wagner, J.R.; Sanche, L. Phosphodiester and N-glycosidic bond cleavage in DNA induced by 4-15 eV electrons. J. Chem. Phys. 2006, 124, 064710. [Google Scholar] [CrossRef]
- Li, Z.; Cloutier, P.; Sanche, L.; Wagner, J.R. Low-energy electron-induced DNA damage: Effect of base sequence in oligonucleotide trimers. J. Am. Chem. Soc. 2010, 132, 5422–5427. [Google Scholar] [CrossRef]
- Horke, D.A.; Li, Q.; Blancafort, L.; Verlet, J.R. Ultrafast above-threshold dynamics of the radical anion of a prototypical quinone electron-acceptor. Nat. Chem. 2013, 5, 711–717. [Google Scholar] [CrossRef] [PubMed]
- West, C.W.; Bull, J.N.; Antonkov, E.; Verlet, J.R. Anion resonances of para-benzoquinone probed by frequency-resolved photoelectron imaging. J. Phys. Chem. A 2014, 118, 11346–11354. [Google Scholar] [CrossRef]
- Dessent, C.E.H.; Kim, J.; Johnson, M.A. Photochemistry of Halide Ion−Molecule Clusters: Dipole-Bound Excited States and the Case for Asymmetric Solvation. Acc. Chem. Res. 1998, 31, 527–534. [Google Scholar] [CrossRef]
- Serxner, D.; Dessent, C.E.H.; Johnson, M.A. Precursor of the Iaq− charge-transfer-to-solvent (CTTS) band in I—(H2O)n clusters. J. Chem. Phys. 1996, 105, 7231–7234. [Google Scholar] [CrossRef]
- Talbot, J.J.; Yang, N.; Huang, M.; Duong, C.H.; McCoy, A.B.; Steele, R.P.; Johnson, M.A. Spectroscopic Signatures of Mode-Dependent Tunnel Splitting in the Iodide–Water Binary Complex. J. Phys. Chem. A 2020, 124, 2991–3001. [Google Scholar] [CrossRef]
- Dessent, C.E.H.; Bailey, C.G.; Johnson, M.A. Dipole-bound excited states of the I−CH3CN and I−(CH3CN)2 ion–molecule complexes: Evidence for asymmetric solvation. J. Chem. Phys. 1995, 103, 2006–2015. [Google Scholar] [CrossRef]
- Dessent, C.E.H.; Kim, J.; Johnson, M.A. Spectroscopic observation of vibrational Feshbach resonances in near-threshold photoexcitation of X−·CH3NO2 (X− = I− and Br−). Faraday Discuss. 2000, 115, 395–406. [Google Scholar] [CrossRef]
- Dessent, C.E.H.; Bailey, C.G.; Johnson, M.A. Observation of the dipole-bound excited state of the I−⋅acetone ion-molecule complex. J. Chem. Phys. 1995, 102, 6335–6338. [Google Scholar] [CrossRef]
- Kunin, A.; Neumark, D.M. Time-resolved radiation chemistry: Femtosecond photoelectron spectroscopy of electron attachment and photodissociation dynamics in iodide-nucleobase clusters. Phys. Chem. Chem. Phys. 2019, 21, 7239–7255. [Google Scholar] [CrossRef] [PubMed]
- King, S.B.; Yandell, M.A.; Stephansen, A.B.; Neumark, D.M. Time-resolved radiation chemistry: Dynamics of electron attachment to uracil following UV excitation of iodide-uracil complexes. J. Chem. Phys. 2014, 141, 224310. [Google Scholar] [CrossRef] [PubMed]
- King, S.B.; Yandell, M.A.; Neumark, D.M. Time-resolved photoelectron imaging of the iodide–thymine and iodide–uracil binary cluster systems. Faraday Discuss. 2013, 163, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Yandell, M.A.; King, S.B.; Neumark, D.M. Time-Resolved Radiation Chemistry: Photoelectron Imaging of Transient Negative Ions of Nucleobases. J. Am. Chem. Soc. 2013, 135, 2128–2131. [Google Scholar] [CrossRef]
- King, S.B.; Stephansen, A.B.; Yokoi, Y.; Yandell, M.A.; Kunin, A.; Takayanagi, T.; Neumark, D.M. Electron accommodation dynamics in the DNA base thymine. J. Chem. Phys. 2015, 143, 024312. [Google Scholar] [CrossRef]
- Stephansen, A.B.; King, S.B.; Yokoi, Y.; Minoshima, Y.; Li, W.-L.; Kunin, A.; Takayanagi, T.; Neumark, D.M. Dynamics of dipole- and valence bound anions in iodide-adenine binary complexes: A time-resolved photoelectron imaging and quantum mechanical investigation. J. Chem. Phys. 2015, 143, 104308. [Google Scholar] [CrossRef]
- Rogers, J.P.; Anstoter, C.S.; Verlet, J.R.R. Ultrafast dynamics of low-energy electron attachment via a non-valence correlation-bound state. Nat. Chem. 2018, 10, 341–346. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.-B.; Vorpagel, E.R.; Wang, L.-S. Direct experimental observation of the low ionization potentials of guanine in free oligonucleotides by using photoelectron spectroscopy. Proc. Natl. Acad. Sci. USA 2004, 101, 17588–17592. [Google Scholar] [CrossRef]
- Chatterley, A.S.; Johns, A.S.; Stavros, V.G.; Verlet, J.R.R. Base-Specific Ionization of Deprotonated Nucleotides by Resonance Enhanced Two-Photon Detachment. J. Phys. Chem. A 2013, 117, 5299–5305. [Google Scholar] [CrossRef]
- Epp, J.B.; Widlanski, T.S. Facile Preparation of Nucleoside-5‘-carboxylic Acids. J. Org. Chem. 1999, 64, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Chatterley, A.S.; West, C.W.; Roberts, G.M.; Stavros, V.G.; Verlet, J.R.R. Mapping the Ultrafast Dynamics of Adenine onto Its Nucleotide and Oligonucleotides by Time-Resolved Photoelectron Imaging. J. Phys. Chem. Lett. 2014, 5, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Stavros, V.G.; Verlet, J.R.R. Gas-Phase Femtosecond Particle Spectroscopy: A Bottom-Up Approach to Nucleotide Dynamics. Annu. Rev. Phys. Chem. 2016, 67, 211–232. [Google Scholar] [CrossRef]
- Castellani, M.E.; Avagliano, D.; Verlet, J.R.R. Ultrafast Dynamics of the Isolated Adenosine-5′-triphosphate Dianion Probed by Time-Resolved Photoelectron Imaging. J. Phys. Chem. A 2021. [Google Scholar] [CrossRef]
- Castellani, M.E.; Avagliano, D.; González, L.; Verlet, J.R.R. Site-Specific Photo-oxidation of the Isolated Adenosine-5′-triphosphate Dianion Determined by Photoelectron Imaging. J. Phys. Chem. Lett. 2020, 11, 8195–8201. [Google Scholar] [CrossRef]
- Chatterley, A.S.; West, C.W.; Stavros, V.G.; Verlet, J.R.R. Time-resolved photoelectron imaging of the isolated deprotonated nucleotides. Chem. Sci. 2014, 5, 3963–3975. [Google Scholar] [CrossRef]
- Vysotskii, Y.B. Effect of chemical substitution on the ionization potentials and electron affinity of systems with conjugated bonds. Theor. Exp. Chem. 1982, 17, 363–371. [Google Scholar] [CrossRef]
- Tully, J.C.; Preston, R.K. Trajectory Surface Hopping Approach to Nonadiabatic Molecular Collisions: The Reaction of H+ with D2. J. Chem. Phys. 1971, 55, 562–572. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S.; Tully, J.C. Proton transfer in solution: Molecular dynamics with quantum transitions. J. Chem. Phys. 1994, 101, 4657–4667. [Google Scholar] [CrossRef]
- Mitrić, R.; Petersen, J.; Bonačić-Koutecký, V. Laser-field-induced surface-hopping method for the simulation and control of ultrafast photodynamics. Phys. Rev. A 2009, 79, 053416. [Google Scholar] [CrossRef]
- Mavri, J. Molecular Dynamics with Nonadiabatic Transitions: A Comparison of Methods. Mol. Simul. 2000, 23, 389–411. [Google Scholar] [CrossRef]
- Lecointre, J.; Roberts, G.M.; Horke, D.A.; Verlet, J.R.R. Ultrafast Relaxation Dynamics Observed Through Time-Resolved Photoelectron Angular Distributions. J. Phys. Chem. A 2010, 114, 11216–11224. [Google Scholar] [CrossRef]
- Stanley, L.H.; Anstöter, C.S.; Verlet, J.R.R. Resonances of the anthracenyl anion probed by frequency-resolved photoelectron imaging of collision-induced dissociated anthracene carboxylic acid. Chem. Sci. 2017, 8, 3054–3061. [Google Scholar] [CrossRef]
- Wiley, W.C.; McLaren, I.H. Time-of-Flight Mass Spectrometer with Improved Resolution. Rev. Sci. Instrum. 1955, 26, 1150–1157. [Google Scholar] [CrossRef]
- Horke, D.A.; Roberts, G.M.; Lecointre, J.; Verlet, J.R.R. Velocity-map imaging at low extraction fields. Rev. Sci. Instrum. 2012, 83, 063101. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.M.; Nixon, J.L.; Lecointre, J.; Wrede, E.; Verlet, J.R.R. Toward real-time charged-particle image reconstruction using polar onion-peeling. Rev. Sci. Instrum. 2009, 80, 053104. [Google Scholar] [CrossRef]
- Fingerman, S. CRC Handbook of Chemistry and Physics: A Ready-Reference Book of Chemical and Physical Data, 87th ed.; Sci-Tech News; CRC Press: Boca Raton, FL, USA, 2007; Volume 61, p. 38. [Google Scholar]
- Horke, D.A. Femtosecond Photoelectron Imaging of Anions; Durham University: Durham, UK, 2012. [Google Scholar]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castellani, M.E.; Verlet, J.R.R. Intramolecular Photo-Oxidation as a Potential Source to Probe Biological Electron Damage: A Carboxylated Adenosine Analogue as Case Study. Molecules 2021, 26, 2877. https://doi.org/10.3390/molecules26102877
Castellani ME, Verlet JRR. Intramolecular Photo-Oxidation as a Potential Source to Probe Biological Electron Damage: A Carboxylated Adenosine Analogue as Case Study. Molecules. 2021; 26(10):2877. https://doi.org/10.3390/molecules26102877
Chicago/Turabian StyleCastellani, Maria Elena, and Jan R. R. Verlet. 2021. "Intramolecular Photo-Oxidation as a Potential Source to Probe Biological Electron Damage: A Carboxylated Adenosine Analogue as Case Study" Molecules 26, no. 10: 2877. https://doi.org/10.3390/molecules26102877
APA StyleCastellani, M. E., & Verlet, J. R. R. (2021). Intramolecular Photo-Oxidation as a Potential Source to Probe Biological Electron Damage: A Carboxylated Adenosine Analogue as Case Study. Molecules, 26(10), 2877. https://doi.org/10.3390/molecules26102877