Does Antibody Stabilize the Ligand Binding in GP120 of HIV-1 Envelope Protein? Evidence from MD Simulation


Abstract
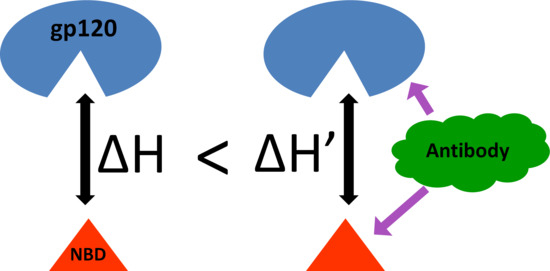
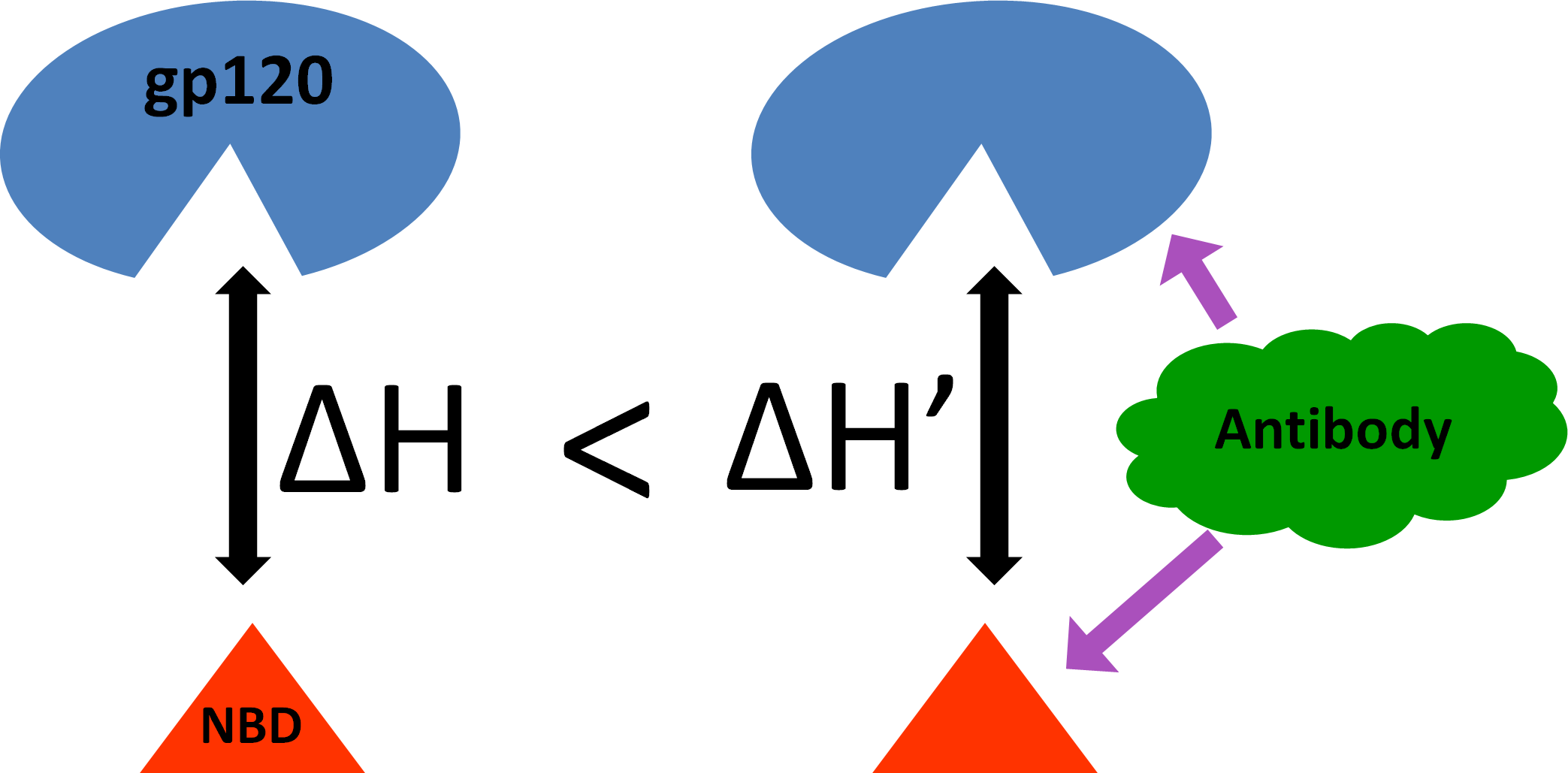
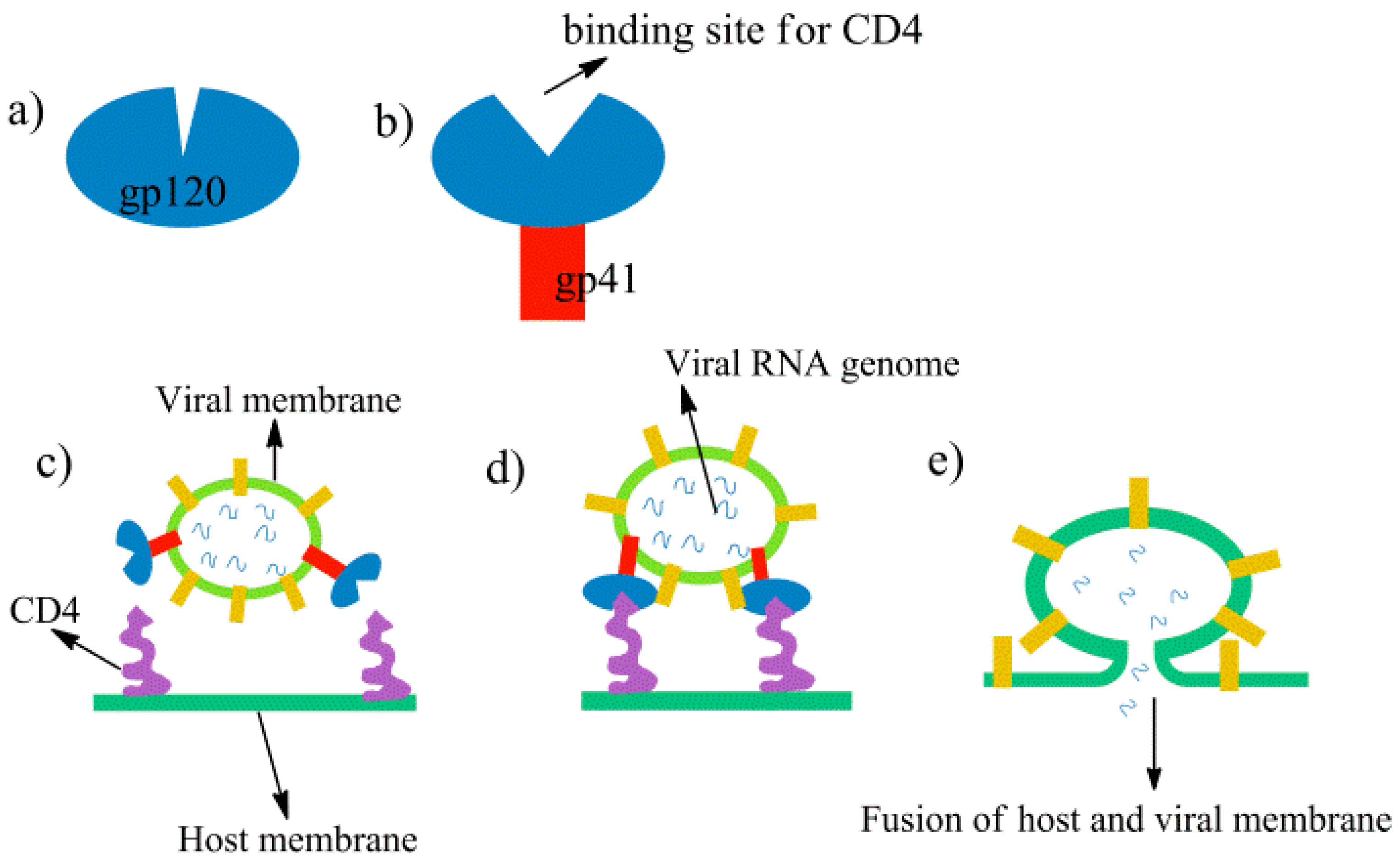
1. Introduction
2. Methods
2.1. System Setup
2.2. MD Simulations
2.3. Clustering of the Trajectories
2.4. Free Energy Calculations
3. Results and Discussion
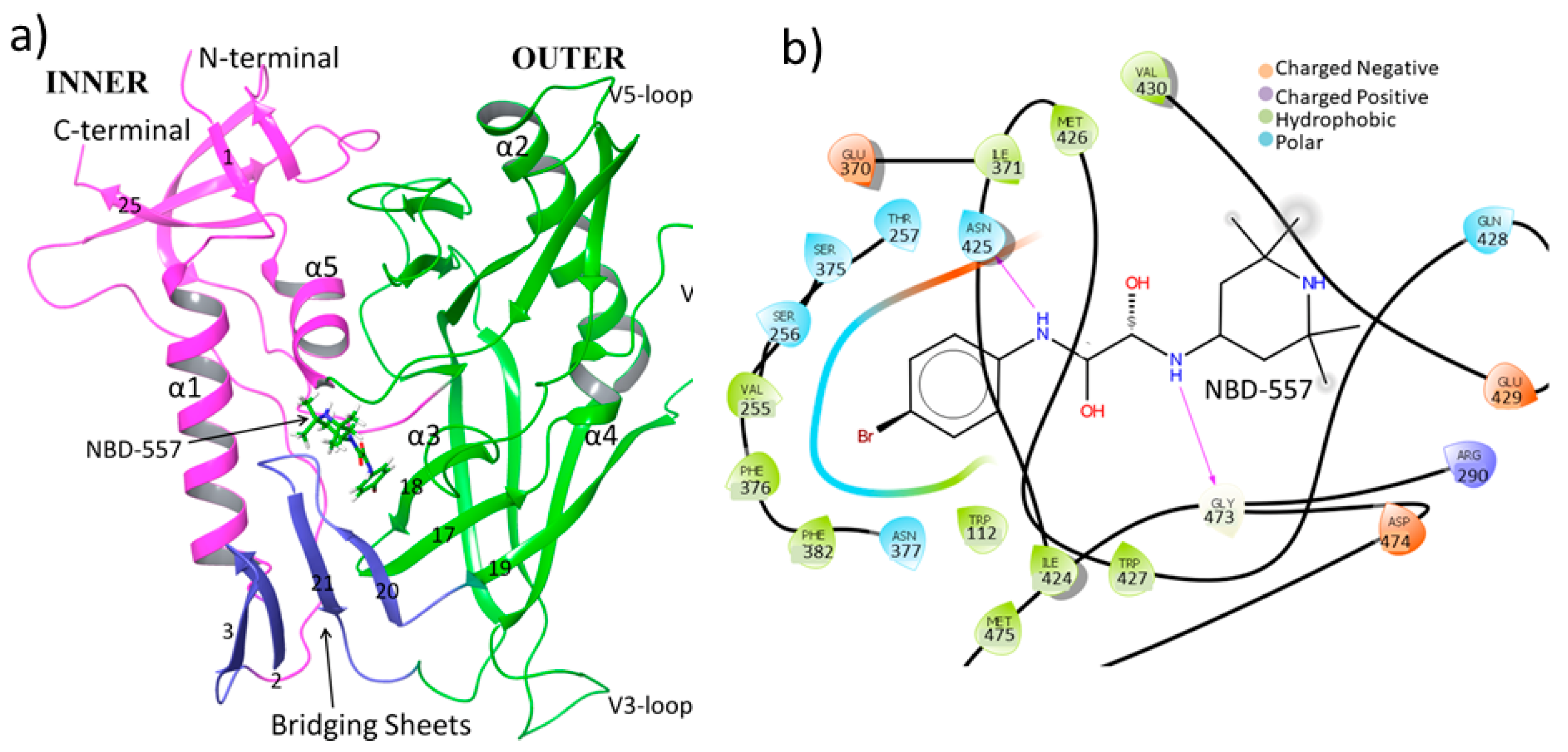
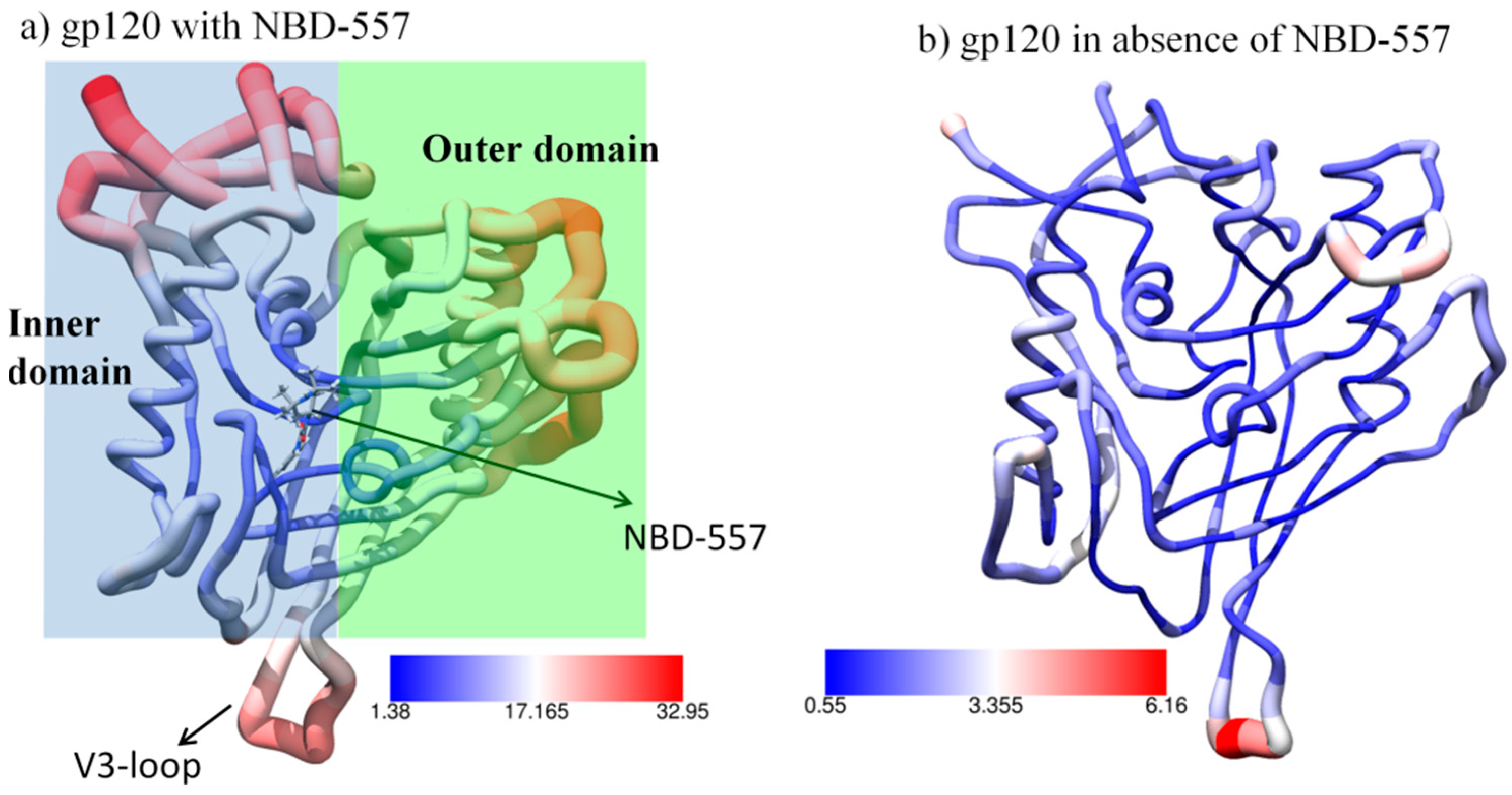
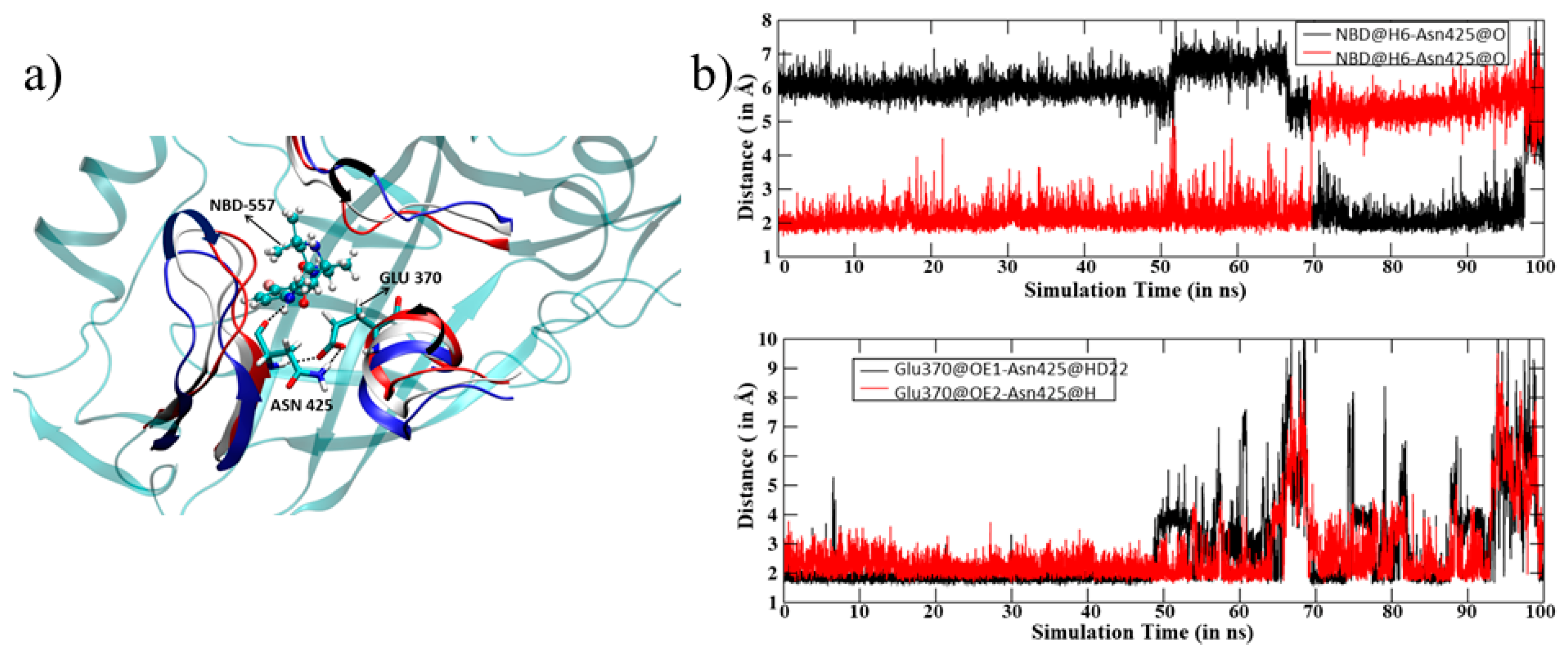
3.1. MD Simulation of gp120 with NBD-557
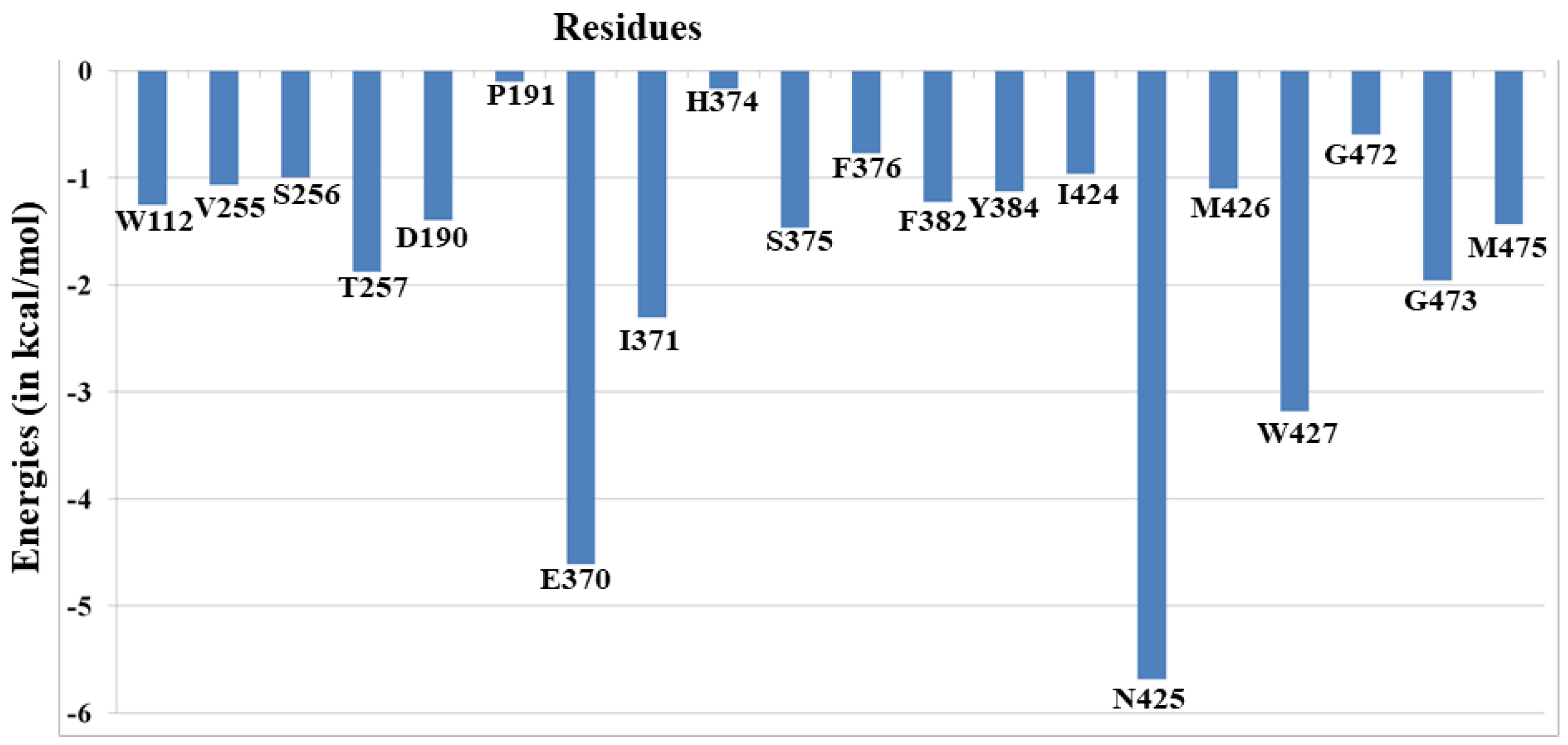
3.2. Thermo-Chemistry of gp120 Complex with NBD-557
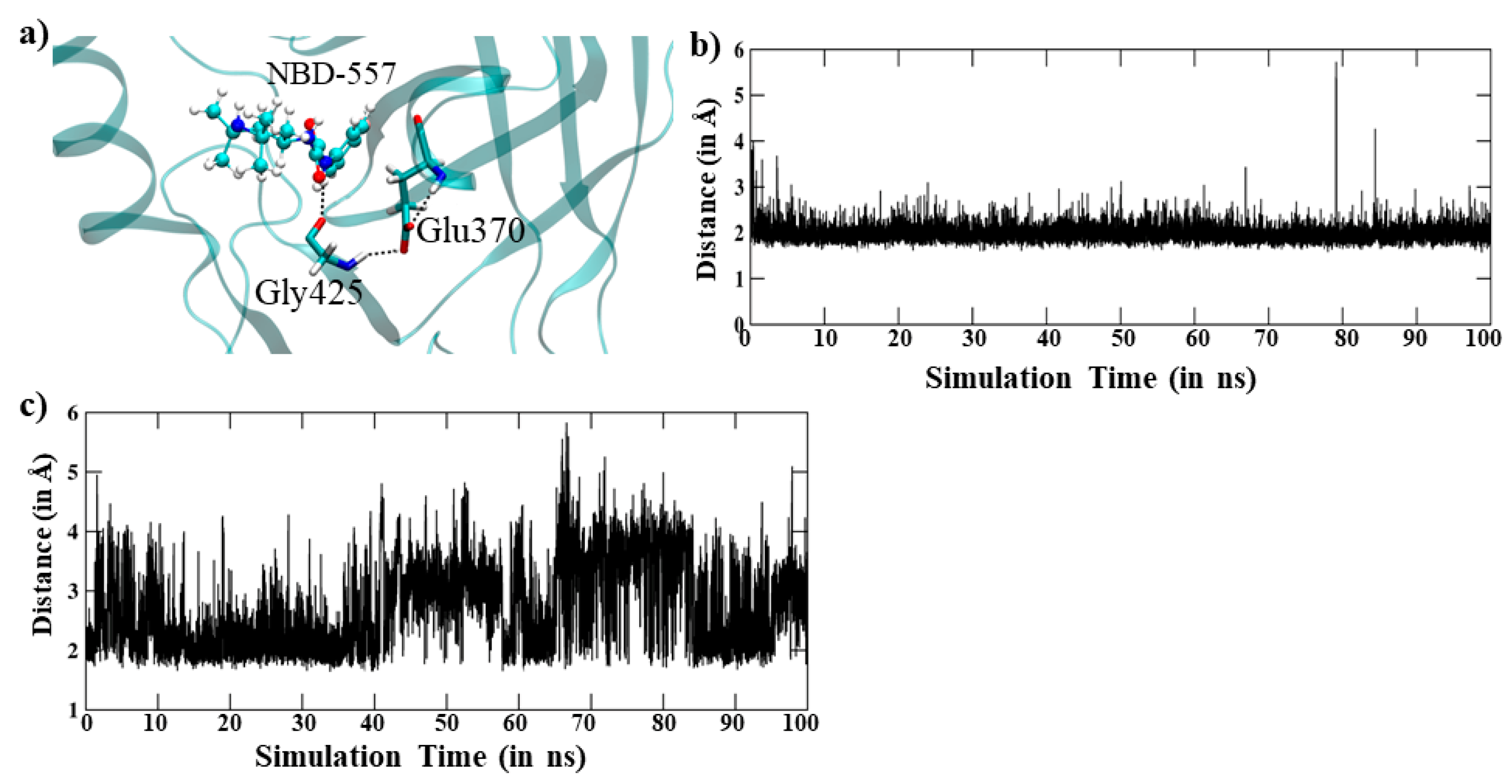
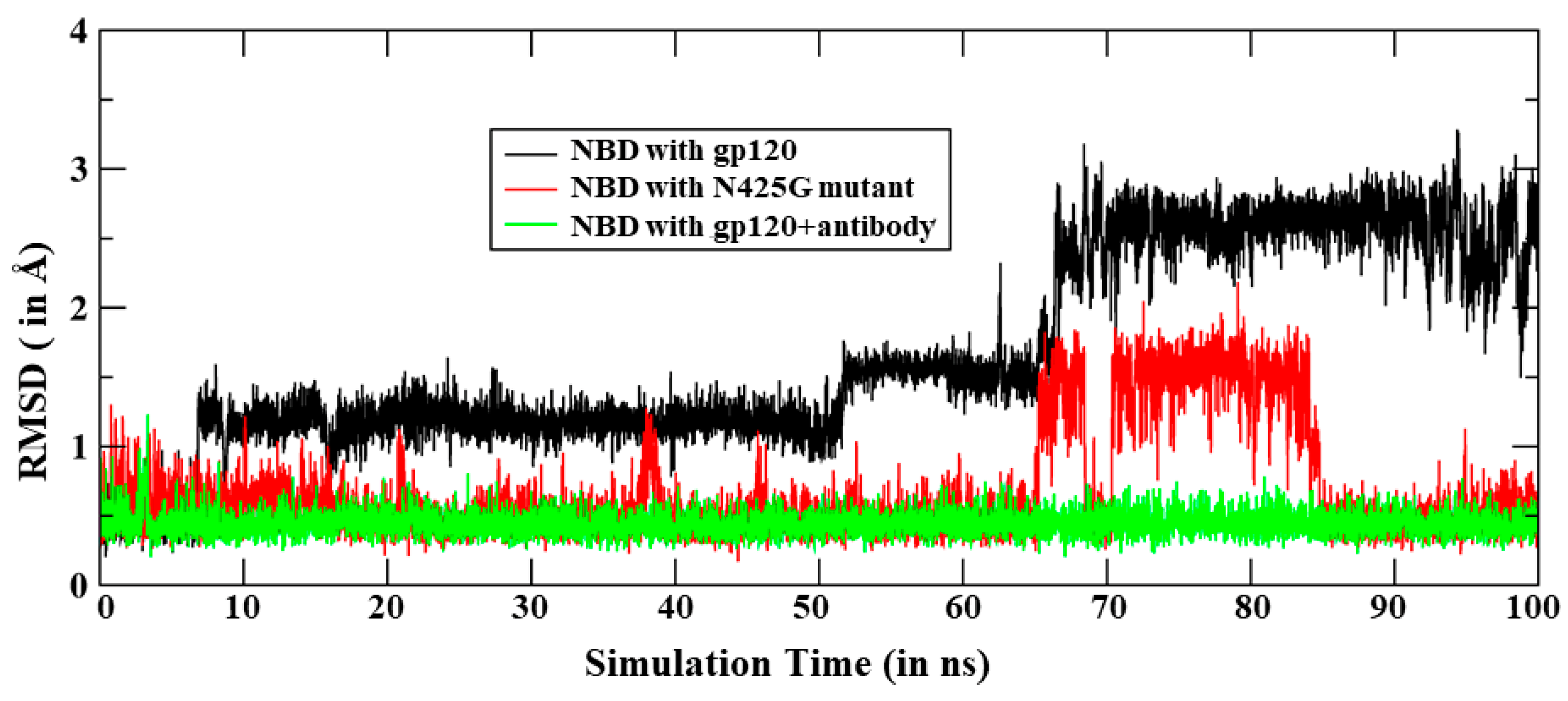
3.3. Dynamics of the In-Silico Mutant Asn425Gly
3.4. Effect of Antibody Binding on gp120 Interaction with NBD-557
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- Rife, B.; Salemi, M. On the early dynamics and spread of HIV-1. Trends. Microbiol. 2015, 23, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Marston, H.D. Ending the HIV–AIDS Pandemic—Follow the Science. N. Engl. J. Med. 2015, 373, 2197–2199. [Google Scholar] [CrossRef] [PubMed]
- Barré-Sinoussi, F.; Ross, A.L.; Delfraissy, J.-F. Past, present and future: 30 years of HIV research. Nat. Rev. Microbiol. 2013, 11, 877–883. [Google Scholar]
- Blumenthal, R.; Durell, S.; Viard, M. HIV Entry and Envelope Glycoprotein-mediated Fusion. J. Biol. Chem. 2012, 287, 40841–40849. [Google Scholar] [CrossRef]
- Jones, P.L.; Korte, T.; Blumenthal, R. Conformational changes in cell surface HIV-1 envelope glycoproteins are triggered by cooperation between cell surface CD4 and co-receptors. J. Biol. Chem. 1998, 273, 404–409. [Google Scholar] [CrossRef]
- Chien, M.P.; Jiang, S.; Chang, D.K. The function of coreceptor as a basis for the kinetic dissection of HIV type 1 envelope protein-mediated cell fusion. FASEB J. 2008, 22, 1179–1192. [Google Scholar] [CrossRef]
- Sougrat, R.; Bartesaghi, A.; Lifson, J.D.; Bennett, A.E.; Bess, J.W.; Zabransky, D.J.; Subramaniam, S. Electron tomography of the contact between T cells and SIV/HIV-1: Implications for viral entry. PLoS Pathog. 2007, 3, e63. [Google Scholar] [CrossRef]
- Dobrowsky, T.M.; Daniels, B.R.; Siliciano, R.F.; Sun, S.X.; Wirtz, D. Organization of cellular receptors into a nanoscale junction during HIV-1 adhesion. PLoS Comput. Biol. 2010, 6, e1000855. [Google Scholar] [CrossRef]
- Muñoz-Barroso, I.; Durell, S.; Sakaguchi, K.; Appella, E.; Blumenthal, R. Dilation of the human immune deficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J. Cell Biol. 1998, 140, 315–323. [Google Scholar] [CrossRef]
- Huang, C.C.; Tang, M.; Zhang, M.Y.; Majeed, S.; Montabana, E.; Stanfield, R.L.; Dimitrov, D.S.; Korber, B.; Sodroski, J.; Wilson, I.A.; et al. Structure of a V3-containing HIV-1 gp120 core. Science 2005, 310, 1025–1028. [Google Scholar] [CrossRef]
- Kwong, P.D.; Wyatt, R.; James Robinson, J.; Sweet, R.W.; Joseph Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 395, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Ma, L.; Jiang, S.; Liu, S.; He, Y.; Strick, N.; Neamati, N.; Debnath, A.K. Identification of N-phenyl-N′-(2,2,6,6tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 2005, 339, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-benzoyl-1-[(4methoxy-1 H-pyrrolo [2,3-b] pyridin-3-yl) oxoacetyl]-2-(R)-methylpiperazine (BMS-378806): A novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J. Med. Chem. 2003, 46, 4236–4239. [Google Scholar] [CrossRef]
- Herschhorn, A.; Gu, C.; Espy, N.; Richard, J.; Finzi, A.; Sodroski, J.G. A broad HIV-1 inhibitor blocks envelope glycoprotein transitions critical for entry. Nat. Chem. Biol. 2014, 10, 845–852. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Courter, J.R.; Madani, N.; Sodroski, J.; Schön, A.; Freire, E.; Kwong, P.D.; Hendrickson, W.A.; Chaiken, I.M.; LaLonde, J.M.; Smith, A.B., 3rd. Structure-based design, synthesis and validation of CD4-mimetic small molecule inhibitors of HIV-1 entry: Conversion of a viral entry agonist to an antagonist. Acc. Chem. Res. 2014, 47, 1228–1237. [Google Scholar] [CrossRef]
- Li, W.; Lu, L.; Li, W.; Jiang, S. Small-molecule HIV-1 entry inhibitors targeting gp120 and gp41: A patent review (2010–2015). Exp. Opin. Therap. Patent 2017. [Google Scholar] [CrossRef] [PubMed]
- Kuritzkes, D.R. HIV-1 entry inhibitors: An overview. Curr. Opin. 2009, 4, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Huang, B.; Zhan, P.; De Clercq, E.; Liu, X. Discovery of small molecular inhibitors targeting HIV-1 gp120-CD4 interaction drived from BMS-378806. Eur. J. Med. Chem. 2014, 86, 481–490. [Google Scholar] [CrossRef]
- Schön, A.; Madani, N.; Klein, J.C.; Hubicki, A.; Ng, D.; Yang, X.; Smith, A.B.; Sodroski, S.; Freire, E. Thermodynamics of Binding of a Low-Molecular-Weight CD4 Mimetic to HIV-1 gp120. Biochemistry 2006, 45, 10973–10980. [Google Scholar] [CrossRef]
- Kwon, Y.D.; Lalonde, J.M.; Yang, Y.; Elban, M.A.; Sugawara, A.; Courter, J.R.; Jones, D.M.; Smith, A.B.; Debnath, A.K.; Kwong, P.D. Crystal Structures of HIV-1 gp120 Envelope Glycoprotein in Complex with NBD Analogues That Target the CD4-Binding Site. PLoS ONE 2014, 9, e85940. [Google Scholar] [CrossRef]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber 14; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollmann, P.A. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J. Am. Chem. Soc. 1993, 115, 9620–9631. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the Cartesian equation of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond molecular dynamics simulations with AMBER on GPU. 2. Explicit solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Dubey, K.D.; Tiwari, R.K.; Ojha, R.P. Recent Advances in Protein–Ligand Interactions: MD Simulations and Binding Free Energy. Curr. Comput. Aided Drug Des. 2013, 9, 518–531. [Google Scholar] [CrossRef]
- Gohlke, H.; Case, D.A. Converging free energy estimates: MM-PB (GB) SA studies on the protein-protein complex RasRaf. J. Comput. Chem. 2003, 25, 238–250. [Google Scholar] [CrossRef]
- Fogolari, F.; Brigo, A.; Molinari, H. Protocols for MM/PBSA molecular dynamics simulations of proteins. Biophys. J. 2003, 85, 159–166. [Google Scholar] [CrossRef]
- Grochowaski, P.; Trylska, J. Continuum molecular electrostatics, salt effects, and counterion binding—A review of the Poisson Boltzmann theory and its modifications. Biopolymers 2007, 89, 93–113. [Google Scholar] [CrossRef]
- Tsui, V.; Case, D.A. Theory and application of generalized born solvation model in macromolecular simulations. Biopolymers 2001, 56, 271–291. [Google Scholar] [CrossRef]
- Dubey, K.D.; Chaubey, A.K.; Ojha, R.P. Stability of trimeric DENV envelope protein at low and neutral pH: An insight from MD study. Biochim. Biophys. Acta 2013, 1834, 53–64. [Google Scholar] [CrossRef]
- Dubey, K.D.; Chaubey, A.K.; Ojha, R.P. Role of pH on dimeric interactions for DENVenvelope protein: An insight from molecular dynamics study. Biochim. Biophys. Acta 2011, 1814, 1796–1801. [Google Scholar] [CrossRef]
- Dubey, K.D.; Tiwari, G.; Ojha, R.P. Targeting domain-III hinging of dengue envelope (DENV-2) protein by MD simulations, docking and free energy calculations. J. Mol. Model. 2017, 23, 102. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Virtual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Haung, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy Contribution | Values (in kcal Mol) | Standard Error |
|---|---|---|
| ∆EVDW | −36.94 | 2.19 |
| ∆EEEL | −5.22 | 1.93 |
| ∆ENPOLAR | 21.33 | 1.71 |
| ∆EDISPER | −4.32 | 0.17 |
| ∆Ggas | −42.16 | 3.04 |
| ∆G solv | 17 | 1.67 |
| ∆HTOTAL | −25.16 | 3.24 |
| ∆HEXPref | −24.50 |
| Energy Contribution | Values (In kcal Mol) | Standard Error |
|---|---|---|
| ∆EVDW | −46.19 | 3.23 |
| ∆EEEL | −14.88 | 3.3 |
| ∆ENPOLAR | 29.65 | 2.52 |
| ∆EDISPER | −5.28 | 0.32 |
| ∆Ggas | −61.07 | 5.25 |
| ∆Gsolv | 24.36 | 2.32 |
| ∆HTOTAL | −36.71 | 3.66 |
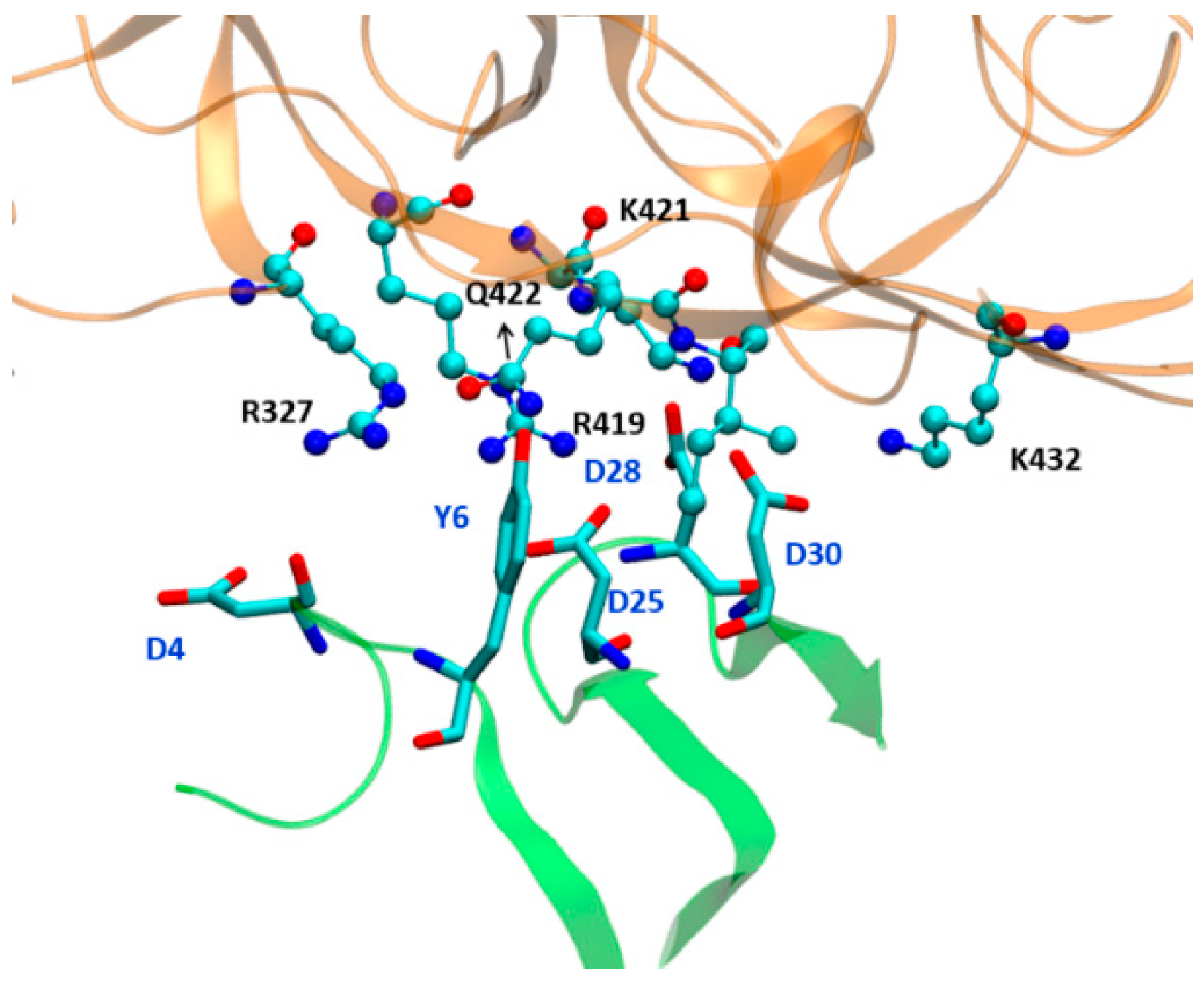
| Residues (Antibody) | Residues (gp120) | Energy (kcal/mol) |
|---|---|---|
| TYR6 | ARG327 | −3.928 |
| TYR6 | GLN422 | −2.059 |
| ASP25 | ARG419 | −18.346 |
| ASP25 | LYS421 | −2.437 |
| ASP28 | PRO369 | −1.679 |
| ASP28 | ARG419 | −7.616 |
| ASP28 | LYS421 | −10.152 |
| ASP30 | LYS421 | −11.001 |
| ASP30 | ILE 423 | −2.028 |
| ASP30 | LYS432 | −7.517 |
| MET32 | MET434 | −1.656 |
| Energy Contribution | Values (in kcal Mol) | Standard Error |
|---|---|---|
| ∆EVDW | −43.94 | 2.16 |
| ∆EEEL | −14.12 | 2.19 |
| ∆ENPOLAR | 29.40 | 1.46 |
| ∆EDISPER | −4.90 | 0.15 |
| ∆Ggas | −58.07 | 2.75 |
| ∆G solv | 24.49 | 1.40 |
| ∆HTOTAL | −33.57 | 2.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, S.; Pandey, V.; Kumar Tiwari, R.; Ojha, R.P.; Dubey, K.D. Does Antibody Stabilize the Ligand Binding in GP120 of HIV-1 Envelope Protein? Evidence from MD Simulation. Molecules 2021, 26, 239. https://doi.org/10.3390/molecules26010239
Yadav S, Pandey V, Kumar Tiwari R, Ojha RP, Dubey KD. Does Antibody Stabilize the Ligand Binding in GP120 of HIV-1 Envelope Protein? Evidence from MD Simulation. Molecules. 2021; 26(1):239. https://doi.org/10.3390/molecules26010239
Chicago/Turabian StyleYadav, Shalini, Vishnudatt Pandey, Rakesh Kumar Tiwari, Rajendra Prasad Ojha, and Kshatresh Dutta Dubey. 2021. "Does Antibody Stabilize the Ligand Binding in GP120 of HIV-1 Envelope Protein? Evidence from MD Simulation" Molecules 26, no. 1: 239. https://doi.org/10.3390/molecules26010239
APA StyleYadav, S., Pandey, V., Kumar Tiwari, R., Ojha, R. P., & Dubey, K. D. (2021). Does Antibody Stabilize the Ligand Binding in GP120 of HIV-1 Envelope Protein? Evidence from MD Simulation. Molecules, 26(1), 239. https://doi.org/10.3390/molecules26010239