
Rational Design, Synthesis, Characterization and Evaluation of Iodinated 4,4′-Bipyridines as New Transthyretin Fibrillogenesis Inhibitors

 ,
,  ,
,  ,
, 
 ,
,  ,
,  ,
, 
 and
and
Abstract
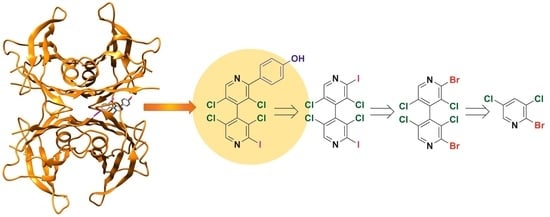
1. Introduction
2. Results and Discussion
2.1. Rational Design
2.1.1. Conceptual Basis
- (i)
- 4,4′-Bipyridines 1–6 consist of two linked heteroaromatic rings that accommodate six (1–5) and five (6) (Figure 4b,c) highly oriented halogens as substituents. 4,4′-Bipyridyl derivatives have been almost unexplored in the field of TTR stabilization to date, and only the 5-cyano-2-methyl-3,4’-bipyridin-6(1H)-one milrinone was found to be a strong competitive inhibitor of T4 binding to TTR [57];
- (ii)
- The two electron-poor bipyridyl rings are characterized by positive VS,max ranging from 72.06 to 96.58 kJ/mol (Supplementary Materials, Table S1), similar to the values calculated for tafamidis, which contains an electron-poor substructure centred on the oxazole ring (98.49; 98.88 kJ/mol) (Table S2);
- (iii)
- The presence of four 3,3′,5,5′-substituents induces chirality by restricted rotation around the 4,4′-bond (atropisomerism), introducing stereochemical features that could be fruitfully exploited to improve binding efficacy and selectivity toward TTR. So far, few studies considered chiral inhibitors and their stereochemical properties [35]. Moreover, the atropisomeric structure of the 3,3′,5,5′-tetrasubstituted-4,4′-bipyridyl ring mimics the skewed conformation of T4;
- (iv)
- The two pyridyl nitrogens can work as HB acceptors, exhibiting VS,min values ranging from −122.43 to −117.07 kJ/mol (Supplementary Materials, Table S1). Furthermore, nitrogen is known to be a high-impact design element because substitution of a CH group with a N atom, in aromatic and heteroaromatic ring frameworks, often improves the pharmacological profile of a molecular structure at different levels [58];
- (v)
- Iodine is considered a powerful XB donor due to its polarizability, in particular when it is bound to electron-withdrawing groups (EWGs). T4 presents multiple iodine atoms which are accommodated in the hBPs of the T4 binding site of TTR. For T4, we calculated VS,max ranging from 109.11 to 143.05 kJ/mol (Supplementary Materials, Table S2). In compounds 1 and 3–6, iodine substituents are activated as XB donors due to the EWG effect exerted by the tetrachlorinated heteroaromatic scaffold, with VS,max values ranging from 133.42 to 167.06 kJ/mol (Table S1). In this regard, it is worth mentioning that in our previous studies [48,49,50,51,52,59], the function of compounds 1–5 as XB donors was demonstrated by theoretical calculations, and HPLC, NMR, and X-ray diffraction (XRD) analysis involving oxygen and nitrogen as XB acceptors. On the other hand, iodine is able to fill the hBPs better than other halogens due to its volume, so optimizing the strength of van der Waals interactions [42];
- (vi)
- In compounds 2–6, the chlorine substituents could also function as potential XB donors, even if lower VS,max values ranging from 70.96 to 98.81 kJ/mol were calculated (Supplementary Materials, Table S1) due to the lower polarizability and the higher electronegativity of chlorine with respect to iodine. Moreover, COX-1 being characterized by smaller hydrophobic binding pockets with respect to TTR [34,35], the presence of bulkier chlorines, with respect to hydrogens, might reduce binding affinity of the small molecule towards COX-1, thus increasing selectivity in TTR binding;
- (vii)
- Since compounds 3–5 contain the same type and number of halogenated substituents but a different substitution pattern, it is possible to evaluate the scaffold effect on iodine properties and activity. Recently, Boeckler and co-workers demonstrated by theoretical calculations that the attachment position of a halogen within a heteroaromatic scaffold can influence size and shape of the σ-hole and, in turn, the strength of the XB [56];
- (viii)
- All haogens can also act as hydrophobic sites and HB acceptors due to a belt of higher electron density located around the σ-hole (Figure 4a).
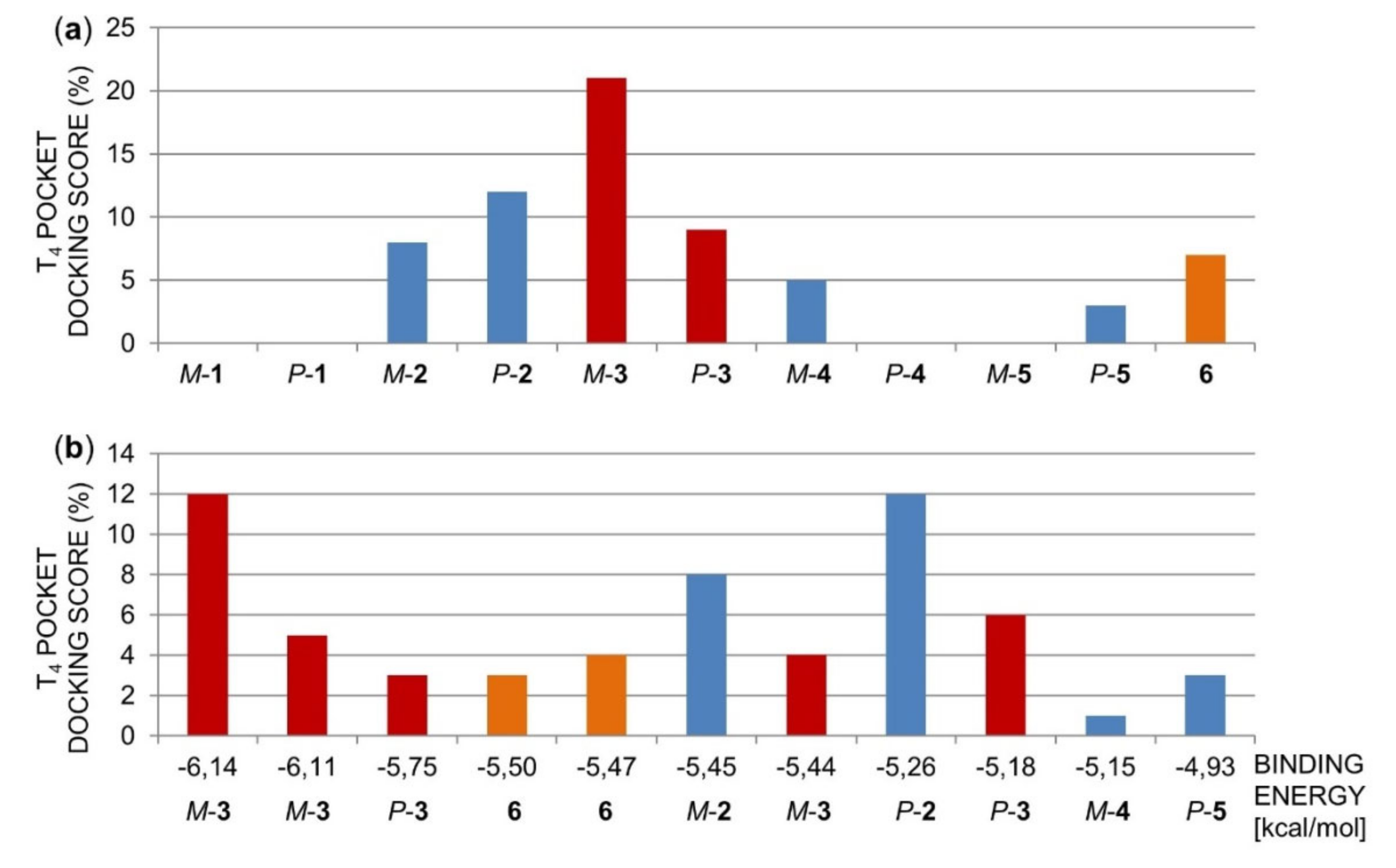
2.1.2. Molecular Docking of Polyhalogenated 4,4′-Bipyridines 1–6 in the TTR Tetramer
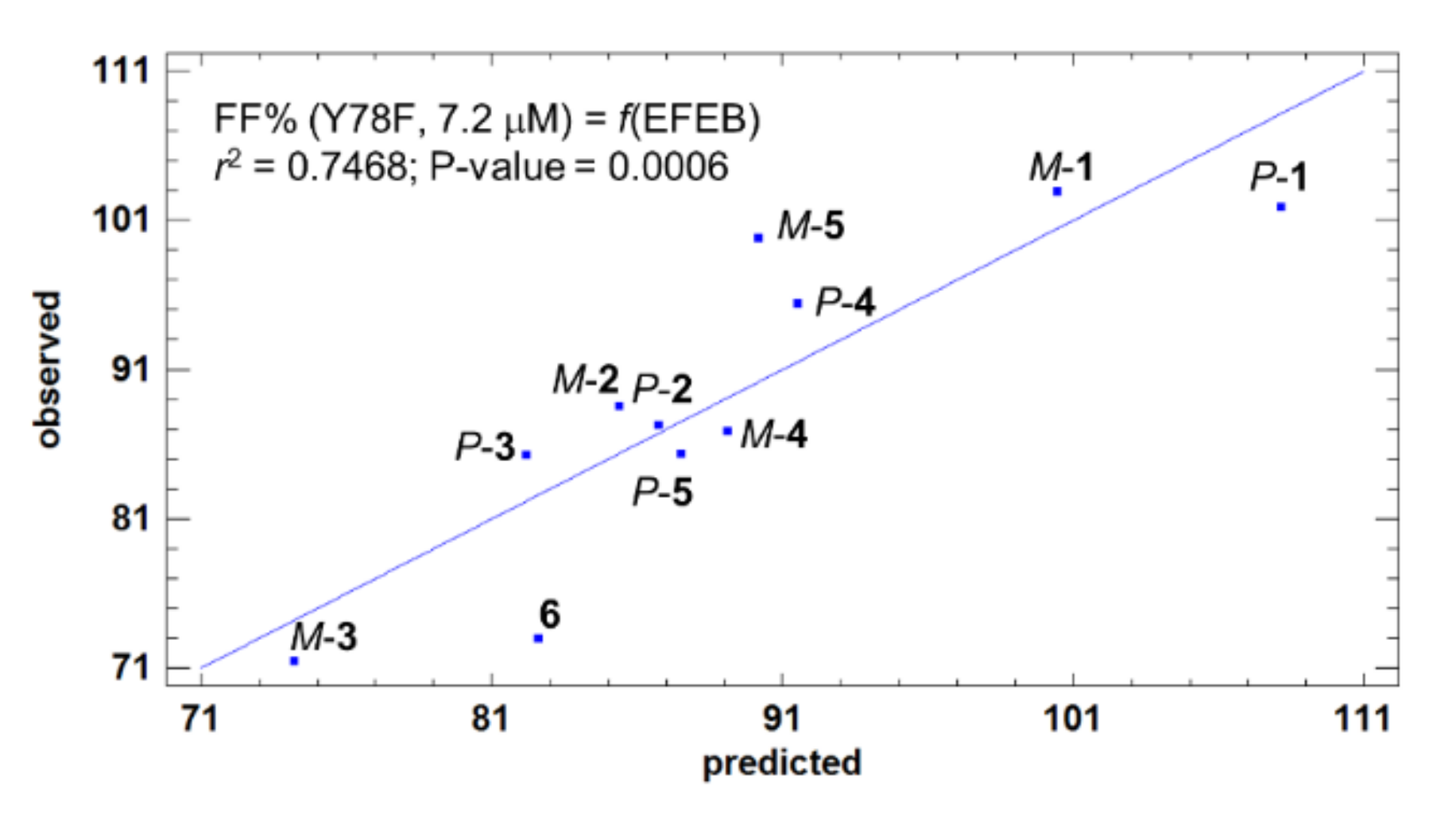
2.1.3. Acid-Mediated TTR FF Assay for Compounds 1–6 and Structure-Activity Relationships
2.2. Chemistry
2.2.1. Syntheses of 4,4′-Bipyridines 1–10
2.2.2. Enantioseparation of 4,4′-Bipyridines 7–10 on Chiral Stationary Phases
2.2.3. Absolute Configuration Assignment of 7–10
2.3. Biological Evaluation of Compounds 7–10 and Structure-Activity Relationships
- (i)
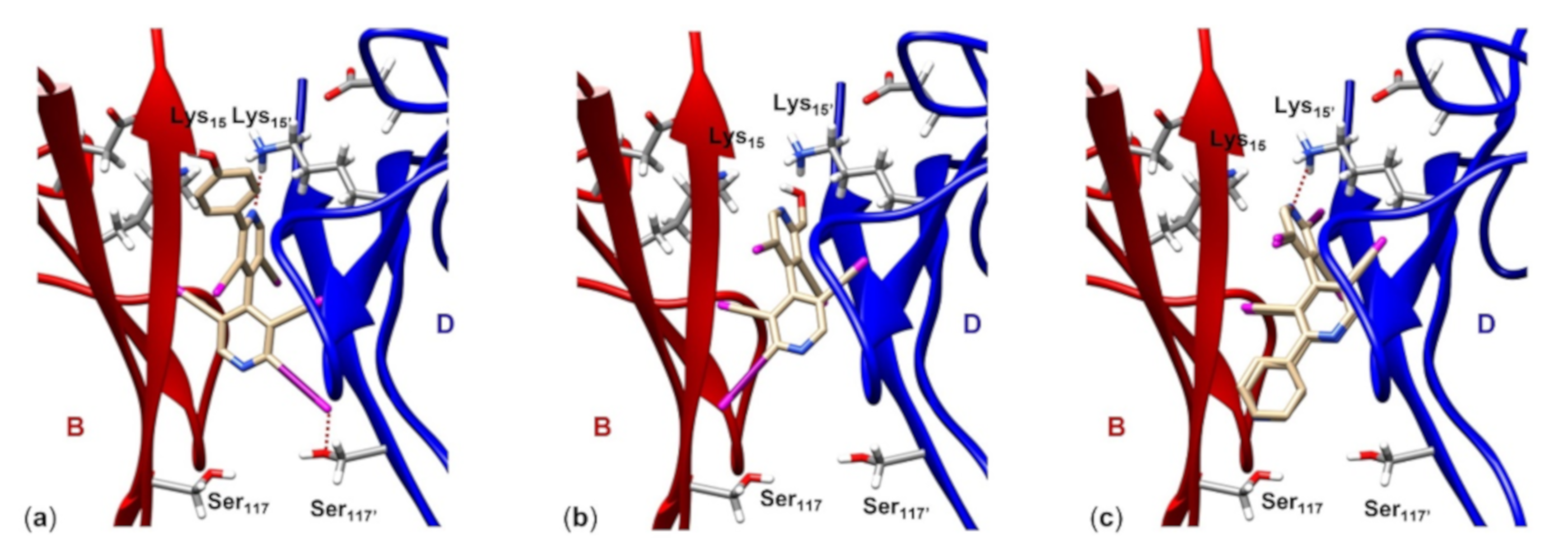
- Compound (M)-9 (Figure 7a) fully occupies the T4 pocket (molecular length 13.2 Å, VS isosurface enclosed volume 367.0 Å3), where the 4-hydroxyphenyl group is oriented toward B:Thr106 and B:Val121 (hBP1) and the four chlorines at the 3,3,5,5′-positions of the heteroaromatic ring anchor the residues B:Ala108 and D:Ala108’ (hBP2). Moreover, the N’pyr (VS,min = -128.3 kJ/mol) forms a HB with the ε-ammonium group of D:Lys15’ (2.6 Å), and 2-iodine (VS,max = 129.8 kJ/mol) showed a short contact with the side chain oxygen of D:Ser117’’ (3.2 Å) (hBP3). Interestingly, the comparison of the respective VS isosurface evidences for compound 9 a shape similarity to both T4 and tafamidis (Supplementary Information, Figure S1);
- (ii)
- Compound 10 (Figure 7b) is the smallest molecule of the series with a molecular length of about 9.80 Å and a volume of 303.1 Å3, therefore it is not able to fully occupy the T4 pocket, 2-iodine showing a longer contact with B:Ser117 (4.4 Å) despite a positive VS,max value (141.6 kJ/mol). Moreover, the best conformation of compound 10 is characterized by an intramolecular HB involving the hydroxyl group and N’pyr (VS,min = −29.12 kJ/mol). The latter is therefore less prone to interact with the side chain of D:Lys15’;
- (iii)
- For both compounds 7 and 8 (Figure 7c), reverse binding modes were found by molecular docking, with the Npyr interacting with D:Lys15’ in all cases. The basic difference between the two compounds, which could justify the different inhibition activity, lies in the presence of a Npyr, as a HB acceptor, at the 2′-position of compound 8 which makes possible an interaction with B:Ser117, thus anchoring the two monomers B and D.
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Syntheses of 3,3′,5,5′-Tetrachloro-2,2′-diiodo-4,4′-bipyridine (3) and 3,3′,5,5′-tetrachloro-2-iodo- 4,4′-bipyridine (6)
3.1.3. General Procedure for the Synthesis of Bipyridines 7, 8 and 14
3,3′,5,5′-Tetrachloro-2-iodo-2′-phenyl-4,4′-bipyridine (7)
3,3′,5,5′-Tetrachloro-2-iodo-2′-(4-pyridyl)-4,4′-bipyridine (8)
3,3′,5,5′-Tetrachloro-2-iodo-2′-(4-((tert-butyldimethylsilyl)oxy)phenyl)-4,4′-bipyridine (14)
3.1.4. 3,3′,5,5′-Tetrachloro-2-iodo-2′-(4-hydroxyphenyl)-4,4′-bipyridine (9)
3.1.5. Synthesis of 3,3’,5,5’-Tetrachloro-2-iodo-2’-hydroxymethyl-4,4’-bipyridine (10)
3.1.6. Multimilligram Enantioseparation of 7–10
3.2. Biological Details
3.2.1. TTR Expression and Purification
3.2.2. Acid-mediated TTR Fibril Formation Assay
3.3. Computationals
3.3.1. Electrostatic Potential Isosurfaces
3.3.2. In Silico Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wojtczak, A.; Cody, V.; Luft, J.R.; Pangborn, W. Structures of human transthyretin complexed with thyroxine at 2.0 Å resolution and 3′,5′-dinitro-N-acetyl-L-thyronine at 2.2 Å resolution. Acta Cryst. 1996, 52, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Wojtczak, A.; Neumann, P.; Cody, V. Structure of a new polymorphic monoclinic form of human transthyretin at 3 Å resolution reveals a mixed complex between unliganded and T4-bound tetramers of TTR. Acta Cryst. 2001, 57, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Palaninathan, S.K. Nearly 200 X-Ray crystal structures of transthyretin: What do they tell us about this protein and the design of drugs for TTR amyloidosis? Curr. Med. Chem. 2012, 19, 2324–2342. [Google Scholar] [CrossRef] [PubMed]
- Blaney, J.M.; Jorgensen, E.C.; Connolly, M.L.; Ferrin, T.E.; Langridge, R.; Oatley, S.J.; Burridge, J.M.; Blake, C.C.F. Computer graphics in drug design: Molecular modelling of thyroid hormone-prealbumin interactions. J. Med. Chem. 1982, 25, 785–790. [Google Scholar] [CrossRef]
- Khanam, H.; Ali, A.; Asif, M. Shamsuzzaman Neurodegenerative diseases linked to misfolded proteins and their therapeutic approaches: A review. Eur. J. Med. Chem. 2016, 124, 1121–1141. [Google Scholar] [CrossRef]
- Pande, M.; Srivastava, R. Molecular and clinical insights into protein misfolding and associated amyloidosis. Eur. J. Med. Chem. 2019, 184, 111753. [Google Scholar] [CrossRef]
- McCutchen, S.L.; Lai, Z.; Miroy, G.J.; Kelly, J.W.; Colón, W. Comparison of lethal and nonlethal transthyretin variants and their relationship to amyloid disease. Biochemistry 1995, 34, 13527–13536. [Google Scholar] [CrossRef]
- Redondo, C.; Damas, A.M.; Olofsson, A.; Lundgren, E.; Saraiva, M.J.M. Search for intermediate structures in transthyretin fibrillogenesis: Soluble tetrameric TYR78Phe TTR expresses a specific epitope present only in amyloid fibrils. J. Mol. Biol. 2000, 304, 461–470. [Google Scholar] [CrossRef]
- Gimeno, A.; Santos, L.M.; Alemi, M.; Rivas, J.; Blasi, D.; Cotrina, E.Y.; Llop, J.; Valencia, G.; Cardoso, I.; Quintana, J.; et al. Insights on the interaction between transthyretin and Aβ in solution. A saturation transfer difference (STD) NMR analysis of the role of iododiflunisal. J. Med. Chem. 2017, 60, 5749–5758. [Google Scholar] [CrossRef]
- Rios, X.; Gómez-Vallejo, V.; Martín, A.; Cossío, U.; Morcillo, M.A.; Alemi, M.; Cardoso, I.; Quintana, J.; Jimeénez-Barbero, J.; Cotrina, E.Y.; et al. Radiochemical examination of transthyretin (TTR) brain penetration assisted by iododiflunisal, a TTR tetramer stabilizer and a new candidate drug for AD. Sci. Rep. 2019, 9, 13672. [Google Scholar] [CrossRef]
- Miroy, G.J.; Lai, Z.; Lashuel, H.A.; Peterson, S.A.; Strang, C.; Kelly, J.W. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc. Natl. Acad. Sci. USA 1996, 93, 15051–15056. [Google Scholar] [CrossRef]
- Hammarström, P.; Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science 2003, 299, 713–716. [Google Scholar] [CrossRef]
- Yee, A.W.; Aldeghi, M.; Blakeley, M.P.; Ostermann, A.; Mas, P.J.; Moulin, M.; de Sanctis, D.; Bowler, M.W.; Mueller-Dieckmann, C.; Mitchell, E.P.; et al. A molecular mechanism for transthyretin amyloidogenesis. Nat. Commun. 2019, 10, 925. [Google Scholar] [CrossRef]
- Rosenblum, H.; Castano, A.; Alvarez, J.; Goldsmith, J.; Helmke, S.; Maurer, M.S. TTR (Transthyretin) stabilizers are associated with improved survival in patients with TTR cardiac amyloidosis. Circ. Heart Fail. 2018, 11, e004769. [Google Scholar] [CrossRef]
- Oza, V.B.; Smith, C.; Raman, P.; Koepf, E.K.; Lashuel, H.A.; Petrassi, H.M.; Chiang, K.P.; Powers, E.T.; Sacchettini, J.C.; Kelly, J.W. Synthesis, structure, and activity of diclofenac analogues as transthyretin amyloid fibril formation inhibitors. J. Med. Chem. 2002, 45, 321–332. [Google Scholar] [CrossRef]
- Adamski-Werner, S.L.; Palaninathan, S.K.; Sacchettini, J.C.; Kelly, J.W. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J. Med. Chem. 2004, 47, 355–374. [Google Scholar] [CrossRef]
- Gales, L.; Macedo-Ribeiro, S.; Arsequell, G.; Valencia, G.; Saraiva, M.J.; Damas, A.M. Human transthyretin in complex with iododiflunisal: Structural features associated with a potent amyloid inhibitor. Biochem. J. 2005, 388, 615–621. [Google Scholar] [CrossRef]
- Johnson, S.M.; Wiseman, R.L.; Sekijima, Y.; Green, N.S.; Adamski-Werner, S.L.; Kelly, J.W. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: A focus on the transthyretin amyloidosis. Acc. Chem. Res. 2005, 38, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Sato, T.; Kugimiya, M.; Sho, M.; Nakamura, T.; Ikemizu, S.; Chirifu, M.; Mizuguchi, M.; Nabeshima, Y.; Suwa, Y.; et al. The crystal structure of the green tea polyphenol (-)-epigallocatechin gallate-transthyretin complex reveals a novel binding site distinct from the thyroxine binding site. Biochemistry 2010, 49, 6104–6114. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Kosada, Y.; Mizuguchi, M. Inhibitory activities of propolis and its promising component, caffeic acid phenethyl ester, against amyloidogenesis of human transthyretin. J. Med. Chem. 2014, 57, 8928–8935. [Google Scholar] [CrossRef] [PubMed]
- Ortore, G.; Orlandini, E.; Braca, A.; Ciccone, L.; Rossello, A.; Martinelli, A.; Nencetti, S. Targeting different transthyretin binding sites with unusual natural compounds. ChemMedChem 2016, 11, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Park, H.; Lee, S.K.; Park, S.J.; Koo, T.-S.; Kang, N.S.; Hong, K.B.; Choi, S. Systemic optimization and structural evaluation of quinoline derivatives as transthyretin amyloidogenesis inhibitors. Eur. J. Med. Chem. 2016, 123, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.P.; Windsor, I.W.; Forest, K.T.; Raines, R.T. Stilbene boronic acids form a covalent bond with human transthyretin and inhibit its aggregation. J. Med. Chem. 2017, 60, 7820–7834. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Mizuguchi, M. Inhibition of the amyloidogenesis of transthyretin by natural products and synthetic compounds. Biol. Pharm. Bull. 2018, 41, 979–984. [Google Scholar] [CrossRef]
- Yokoyama, T.; Mizuguchi, M. Crown ethers as transthyretin amyloidogenesis inhibitors. J. Med. Chem. 2019, 62, 2076–2082. [Google Scholar] [CrossRef]
- Green, N.S.; Palaninathan, S.K.; Sacchettini, J.C.; Kelly, J.W. Synthesis and characterization of potent bivalent amyloidosis inhibitors that bind prior to transthyretin tetramerization. J. Am. Chem. Soc. 2003, 125, 13404–13414. [Google Scholar] [CrossRef]
- Corazza, A.; Verona, G.; Waudby, C.A.; Mangione, P.P.; Bingham, R.; Uings, I.; Canetti, D.; Nocerino, P.; Taylor, G.W.; Pepys, M.B.; et al. Binding of monovalent and bivalent ligands by transthyretin causes different short- and long-distance conformational changes. J. Med. Chem. 2019, 62, 8274–8283. [Google Scholar] [CrossRef]
- Inoue, M.; Ueda, M.; Higashi, T.; Anno, T.; Fujisawa, K.; Motoyama, K.; Mizuguchi, M.; Ando, Y.; Jono, H.; Arima, H. Therapeutic potential of polyamidoamine dendrimer for amyloidogenic transthyretin amyloidosis. ACS Chem. Neurosci. 2019, 10, 2584–2590. [Google Scholar] [CrossRef]
- Bulawa, C.E.; Connelly, S.; DeVit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar] [CrossRef]
- Zhao, Y.; Xin, Y.; Song, Z.; He, Z.; Hu, W. Tafamidis, a noninvasive therapy for delaying transthyretin familial amyloid polyneuropathy: Systematic review and meta-analysis. J. Clin. Neurol. 2019, 15, 108–115. [Google Scholar] [CrossRef]
- Falk, R.H. Tafamidis for transthyretin amyloid cardiomyopathy: The solution or just the beginning of the end? Eur. Heart J. 2019, 40, 1009–1012. [Google Scholar] [CrossRef] [PubMed]
- Sant’Anna, R.; Gallego, P.; Robinson, L.Z.; Pereira-Henriques, A.; Ferreira, N.; Pinheiro, F.; Esperante, S.; Pallares, I.; Huertas, O.; Rosário Almeida, M.; et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nature Commun. 2016, 7, 10787. [Google Scholar] [CrossRef] [PubMed]
- Rezania, K.; Saadat, L. Neurological manifestations of transthyretin-related amyloidosis. In Amyloid Diseases; Kurouski, D., Ed.; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Klabunde, T.; Petrassi, H.M.; Oza, V.B.; Raman, P.; Kelly, J.W.; Sacchettini, J.C. Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol. 2000, 7, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Palaninathan, S.K.; Mohamedmohaideed, N.N.; Orlandini, E.; Ortore, G.; Nencetti, S.; Lapucci, A.; Rossello, A.; Freundlich, J.S.; Sacchettini, J.C. Novel transthyretin amyloid fibril formation inhibitors: Synthesis, biological evaluation, and X-ray structural analysis. PLoS ONE 2009, 4, e6290. [Google Scholar] [CrossRef]
- Choi, S.; Reixach, N.; Connelly, S.; Johnson, S.M.; Wilson, I.A.; Kelly, J.W. A substructure combination strategy to create potent and selective transthyretin kinetic stabilizers that prevent amyloidogenesis and cytotoxicity. J. Am. Chem. Soc. 2010, 132, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Nencetti, S.; Rossello, A.; Stura, E.A.; Orlandini, E. Synthesis and structural analysis of halogen substituted fibril formation inhibitors of human transthyretin (TTR). J. Enzyme Inhib. Med. Chem. 2016, 31, 40–51. [Google Scholar] [CrossRef][Green Version]
- Ortore, G.; Martinelli, A. Identification of transthyretin fibril formation inhibitors using structure-based virtual screening. ChemMedChem 2017, 12, 1327–1334. [Google Scholar] [CrossRef]
- Loconte, V.; Menozzi, I.; Ferrari, A.; Folli, C.; Imbimbo, B.P.; Zanotti, G.; Berni, R. Structure-activity relationships of flurbiprofen analogues as stabilizers of the amyloidogenic protein transthyretin. J. Struct. Biol. 2019, 208, 165–173. [Google Scholar] [CrossRef]
- Gales, L.; Almeida, M.R.; Arsequell, G.; Valencia, G.; Saraiva, M.J.; Damas, A.M. Iodination of salicylic acid improves its binding to transthyretin. Biochim. Biophys. Acta 2008, 1784, 512–517. [Google Scholar] [CrossRef]
- Mairal, T.; Nieto, J.; Pinto, M.; Almeida, M.R.; Gales, L.; Ballesteros, A.; Barluenga, J.; Pérez, J.J.; Vázquez, J.T.; Centeno, N.B.; et al. Iodine atoms: A new molecular feature for the design of potent transthyretin fibrillogenesis inhibitors. PLoS ONE 2009, 4, e4124. [Google Scholar] [CrossRef]
- Cotrina, E.Y.; Pinto, M.; Bosch, L.; Vilaà, M.; Blasi, D.; Quintana, J.; Centeno, N.B.; Arsequell, G.; Planas, A.; Valencia, G. Modulation of the fibrillogenesis inhibition properties of two transthyretin ligands by halogenation. J. Med. Chem. 2013, 56, 9110–9121. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.A.; Oliveira, S.M.; Guido, L.F.; Magalhães, A.; Valencia, G.; Arsequell, G.; Saraiva, M.J.; Cardoso, I. Transthyretin stabilization by iododiflunisal promotes amyloid-β peptide clearance, decreases its deposition, and ameliorates cognitive deficits in an Alzheimer’s disease mouse model. J. Alzheimers Dis. 2014, 39, 357–370. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Quirante, J.; Nieto, J.; Almeida, M.R.; Saraiva, M.J.; Planas, A.; Arsequell, G.; Valencia, G. Isatin derivatives, a novel class of transthyretin fibrillogenesis inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 5270–5273. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Cody, V.; Murray-Rust, P. Iodine···X(O, N, S) intermolecular contacts: Models of thyroid hormone-protein binding interactions using information from the Cambridge Crystallographic Data files. J. Mol. Struct. 1984, 112, 189–199. [Google Scholar] [CrossRef]
- Mamane, V.; Aubert, E.; Peluso, P.; Cossu, S. Lithiation of prochiral 2,2′-dichloro-5,5′-dibromo-4,4′-bipyridine as a tool for the synthesis of chiral polyhalogenated 4,4′-bipyridines. J. Org. Chem. 2013, 78, 7683–7689. [Google Scholar] [CrossRef]
- Mamane, V.; Peluso, P.; Aubert, E.; Cossu, S.; Pale, P. Chiral hexalogenated 4,4′-bipyridines. J. Org. Chem. 2016, 81, 4576–4587. [Google Scholar] [CrossRef]
- Abboud, M.; Mamane, V.; Aubert, E.; Lecomte, C.; Fort, Y. Synthesis of polyhalogenated 4,4′-bipyridines via a simple dimerization procedure. J. Org. Chem. 2010, 75, 3224–3231. [Google Scholar] [CrossRef]
- Mamane, V.; Aubert, E.; Peluso, P.; Cossu, S. Synthesis, resolution, and absolute configuration of chiral 4,4′-bipyridines. J. Org. Chem. 2012, 77, 2579–2583. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Aubert, E.; Dessì, A.; Dallocchio, R.; Dore, A.; Pale, P.; Cossu, S. Insights into halogen bond-driven enantioseparations. J. Chromatogr. A 2016, 146, 228–238. [Google Scholar] [CrossRef]
- Dallocchio, R.; Dessì, A.; Solinas, M.; Arras, A.; Cossu, S.; Aubert, E.; Mamane, V.; Peluso, P. Halogen bond in high-performance liquid chromatography enantioseparations: Description, features and modelling. J. Chromatogr. A 2018, 1563, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Connelly, S.; Choi, S.; Johnson, S.M.; Kelly, J.W.; Wilson, I.A. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidosis. Curr. Opin. Struct. Biol. 2010, 20, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Tran, K.-A.; Lane, P.; Murray, J.S.; Politzer, P. Comparative analysis of electrostatic potential maxima and minima on molecular surfaces, as determined by three methods and a variety of basis sets. J. Comput. Sci. 2016, 17, 273–284. [Google Scholar] [CrossRef]
- Lange, A.; Heidrich, J.; Zimmermann, M.O.; Exner, T.E.; Boeckler, F.M. Scaffold effect on halogen bonding strength. J. Chem. Inf. Model. 2019, 59, 885–894. [Google Scholar] [CrossRef]
- Wojtczak, A.; Luft, J.R.; Cody, V. Structural aspects of inotropic bipyridine binding. J. Biol. Chem. 1993, 268, 6202–6206. [Google Scholar] [CrossRef]
- Pennington, L.D.; Moustakas, D.T. The necessary nitrogen atom: A versatile high-impact design element for multiparameter optimization. J. Med. Chem. 2017, 60, 3552–3579. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Dallocchio, R.; Dessì, A.; Villano, R.; Sanna, D.; Aubert, E.; Pale, P.; Cossu, S. Polysaccharide-based chiral stationary phases as halogen bond acceptors: A novel strategy for detection of stereoselective σ-hole bonds in solution. J. Sep. Sci. 2018, 41, 1247–1256. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. Docking screens for novel ligands conferring new biology. J. Med. Chem. 2016, 59, 4103–4120. [Google Scholar] [CrossRef]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef] [PubMed]
- De Lima, W.E.A.; Pereira, A.F.; de Castro, A.A.; de Cunha, E.F.F.; Ramalho, T.C. Flexibility in the molecular design of acetylcholinesterase reactivators: Probing representative conformations by chemometric techniques and docking/QM calculations. Lett. Drug Des. Discov. 2016, 13, 360–371. [Google Scholar] [CrossRef]
- Fine, J.; Konc, J.; Samudrala, R.; Chopra, G. CANDOCK: Chemical atomic network-based hierarchical flexible docking algorithm using generalized statistical potentials. J. Chem. Inf. Model. 2020, 60, 1509–1527. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wu, Y.; Xu, L.; Jin, J. Theoretical studies on the selectivity mechanisms of glycogen synthase kinase 3β (GSK3β) with pyrazine ATP-competitive inhibitors by 3DQSAR, molecular docking, molecular dynamics simulation and free energy calculations. Curr. Comput.-Aided Drug Des. 2020, 16, 17–30. [Google Scholar] [CrossRef]
- Shen, C.; Wang, Z.; Yao, X.; Li, Y.; Lei, T.; Wang, E.; Xu, L.; Zhu, F.; Li, D.; Hou, T. Comprehensive assessment of nine docking programs on type II kinase inhibitors: Prediction accuracy of sampling power, scoring power and screening power. Brief. Bioinform. 2020, 21, 282–297. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera - a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A. Molecular mechanical study of halogen bonding in drug discovery. J. Comput. Chem. 2011, 32, 2564–2574. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A. Molecular mechanical perspective on halogen bonding. J. Mol. Model. 2012, 18, 4625–4638. [Google Scholar] [CrossRef] [PubMed]
- Kolář, M.H.; Hobza, P.; Bronowska, K. Plugging the explicit σ-holes in molecular docking. Chem. Commun. 2013, 49, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Kolář, M.H.; Hobza, P. On extension of the current biomolecular empirical force field for the description of halogen bonds. J. Chem. Theory Comput. 2012, 8, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Ruiz, F.X.; Kadlčíková, A.; Řezáč, J.; Cousido-Siah, A.; Mitschler, A.; Haldar, S.; Lepšík, M.; Kolář, M.H.; Majer, P.; et al. The effect of halogen-to-hydrogen bond substitution on human aldose reductase inhibition. ACS Chem. Biol. 2015, 10, 1637–1642. [Google Scholar] [CrossRef]
- Arsequell, G.; Planas, A. Methods to evaluate the inhibition of TTR fibrillogenesis induced by small ligands. Curr. Med. Chem. 2012, 19, 2343–2455. [Google Scholar] [CrossRef]
- Dolado, I.; Nieto, J.; Saraiva, M.J.; Arsequell, G.; Valencia, G.; Planas, A. Kinetic assay for high-throughput screening of in vitro transthyretin amyloid fibrillogenesis inhibitors. J. Comb. Chem. 2005, 7, 246–252. [Google Scholar] [CrossRef]
- Weiss, R.; Aubert, E.; Peluso, P.; Cossu, S.; Pale, P.; Mamane, V. Chiral chalcogen bond donors based on the 4,4′-bipyridine scaffold. Molecules 2019, 24, 4484. [Google Scholar] [CrossRef]
- Jiang, X.; Buxbaum, J.N.; Kelly, J.W. The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc. Natl. Acad. Sci. USA 2001, 98, 14943–14948. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neil, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B. 01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyser. J. Comp.Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved marching tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity -a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. GaussView Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
Sample Availability: Samples of the compounds 1–10 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | FF% 1 (WT-TTR) | FF% 1 (Y78F-TTR) | ||
|---|---|---|---|---|
| 7.2 μM | 3.6 μM | 7.2 μM | 3.6 μM | |
| Diflunisal | 6 | 18 | 18 | 43 |
| (M)-1 | 99 | 100 | 100 | 100 |
| (P)-1 | 100 | 100 | 100 | 100 |
| (M)-2 | 93 | 98 | 89 | 94 |
| (P)-2 | 95 | 97 | 87 | 93 |
| (M)-3 | 94 | 94 | 72 | 62 |
| (P)-3 | 96 | 100 | 85 | 87 |
| (M)-4 | 96 | 97 | 87 | 91 |
| (P)-4 | 96 | 99 | 95 | 95 |
| (M)-5 | 90 | 97 | 85 | 91 |
| (P)-5 | 98 | 98 | 100 | 94 |
| 6 | 91 | 95 | 73 | 82 |
| Compound | FF% 1 (WT-TTR) | FF% 1 (Y78F-TTR) | FF%1 (V30M-TTR) | |||
|---|---|---|---|---|---|---|
| 7.2 μM | 3.6 μM | 7.2 μM | 3.6 μM | 7.2 μM | 3.6 μM | |
| Diflunisal | 6 | 18 | 18 | 43 | 36 | 33 |
| (M)-7 | 96 | 100 | 82 | 84 | nd | nd |
| (P)-7 | 88 | 95 | 71 | 88 | nd | nd |
| (M)-8 | 58 | 72 | 79 | 82 | 50 | 58 |
| (P)-8 | 42 | 67 | 51 | 67 | 38 | 43 |
| (M)-9 | 16 | 49 | 44 | 71 | 38 | 40 |
| (P)-9 | 17 | 49 | 44 | 72 | 39 | 41 |
| (M)-10 | 62 | 79 | 80 | 88 | 59 | 75 |
| (P)-10 | 80 | 89 | 85 | 91 | 67 | 72 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dessì, A.; Peluso, P.; Dallocchio, R.; Weiss, R.; Andreotti, G.; Allocca, M.; Aubert, E.; Pale, P.; Mamane, V.; Cossu, S. Rational Design, Synthesis, Characterization and Evaluation of Iodinated 4,4′-Bipyridines as New Transthyretin Fibrillogenesis Inhibitors. Molecules 2020, 25, 2213. https://doi.org/10.3390/molecules25092213
Dessì A, Peluso P, Dallocchio R, Weiss R, Andreotti G, Allocca M, Aubert E, Pale P, Mamane V, Cossu S. Rational Design, Synthesis, Characterization and Evaluation of Iodinated 4,4′-Bipyridines as New Transthyretin Fibrillogenesis Inhibitors. Molecules. 2020; 25(9):2213. https://doi.org/10.3390/molecules25092213
Chicago/Turabian StyleDessì, Alessandro, Paola Peluso, Roberto Dallocchio, Robin Weiss, Giuseppina Andreotti, Mariateresa Allocca, Emmanuel Aubert, Patrick Pale, Victor Mamane, and Sergio Cossu. 2020. "Rational Design, Synthesis, Characterization and Evaluation of Iodinated 4,4′-Bipyridines as New Transthyretin Fibrillogenesis Inhibitors" Molecules 25, no. 9: 2213. https://doi.org/10.3390/molecules25092213
APA StyleDessì, A., Peluso, P., Dallocchio, R., Weiss, R., Andreotti, G., Allocca, M., Aubert, E., Pale, P., Mamane, V., & Cossu, S. (2020). Rational Design, Synthesis, Characterization and Evaluation of Iodinated 4,4′-Bipyridines as New Transthyretin Fibrillogenesis Inhibitors. Molecules, 25(9), 2213. https://doi.org/10.3390/molecules25092213