
Computer-Driven Development of an in Silico Tool for Finding Selective Histone Deacetylase 1 Inhibitors

 ,
, 
 ,
, 
Abstract
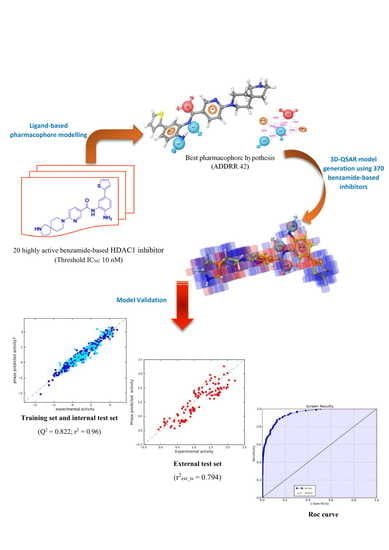
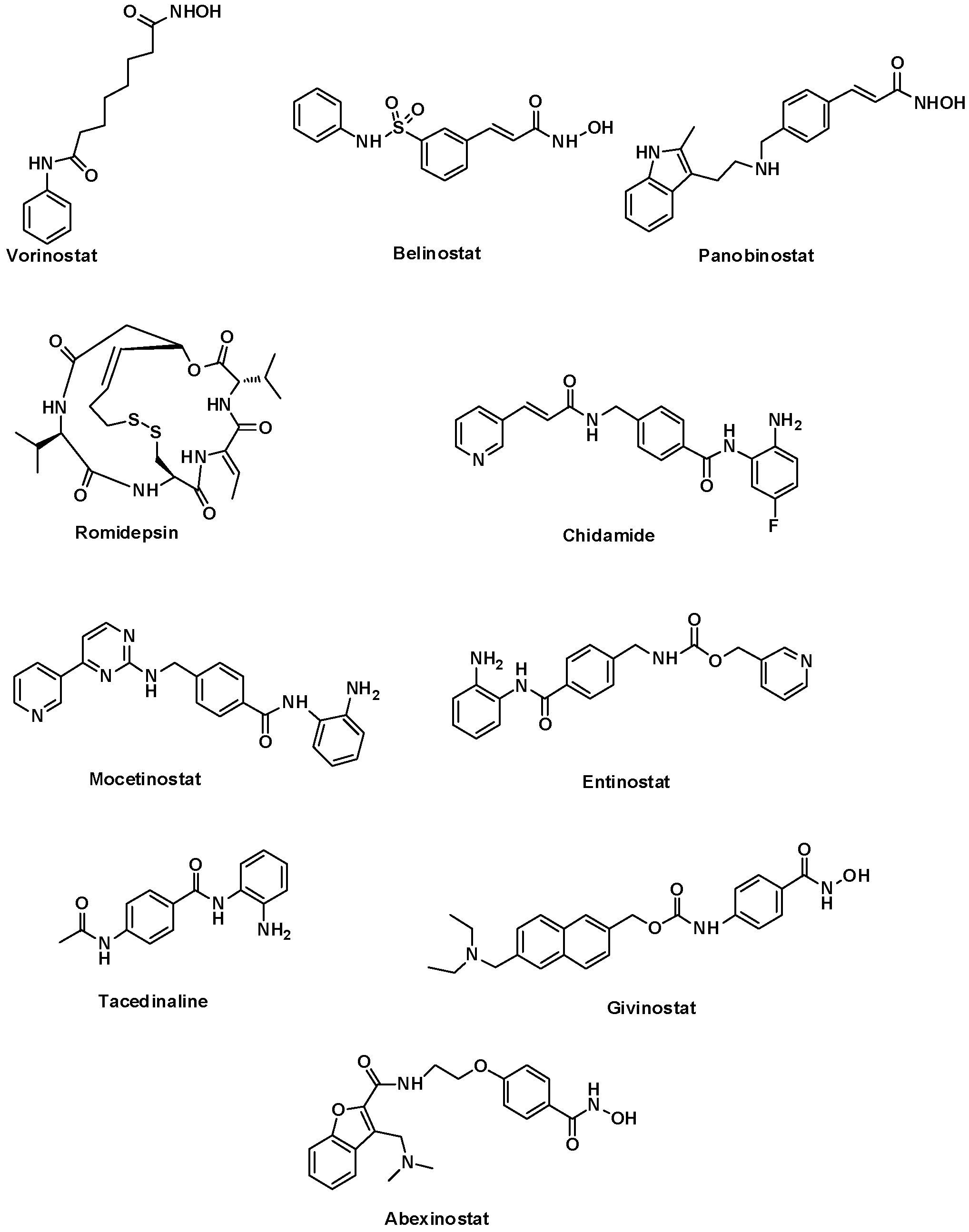
1. Introduction
2. Results and Discussion
2.1. Data Set Preparation
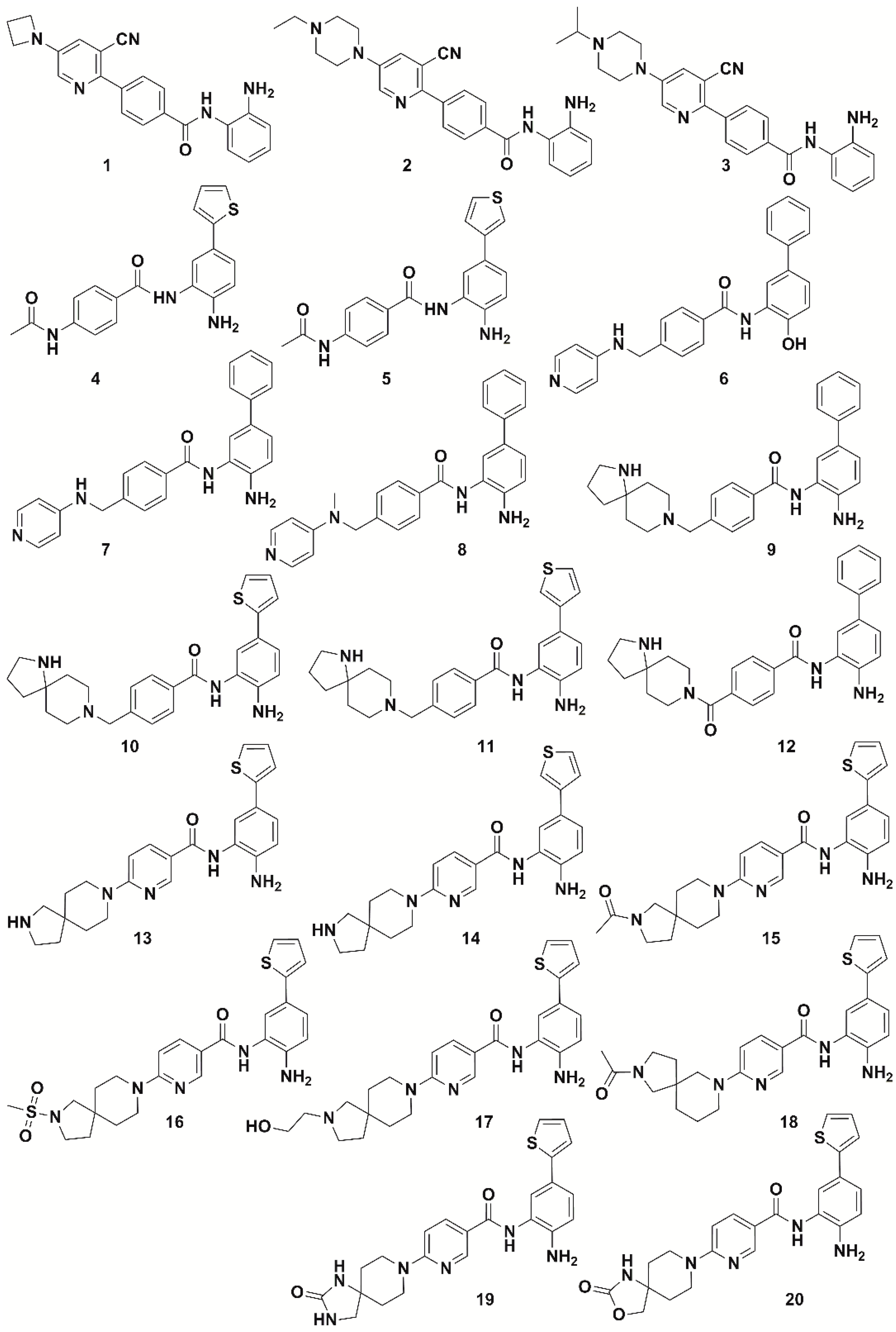
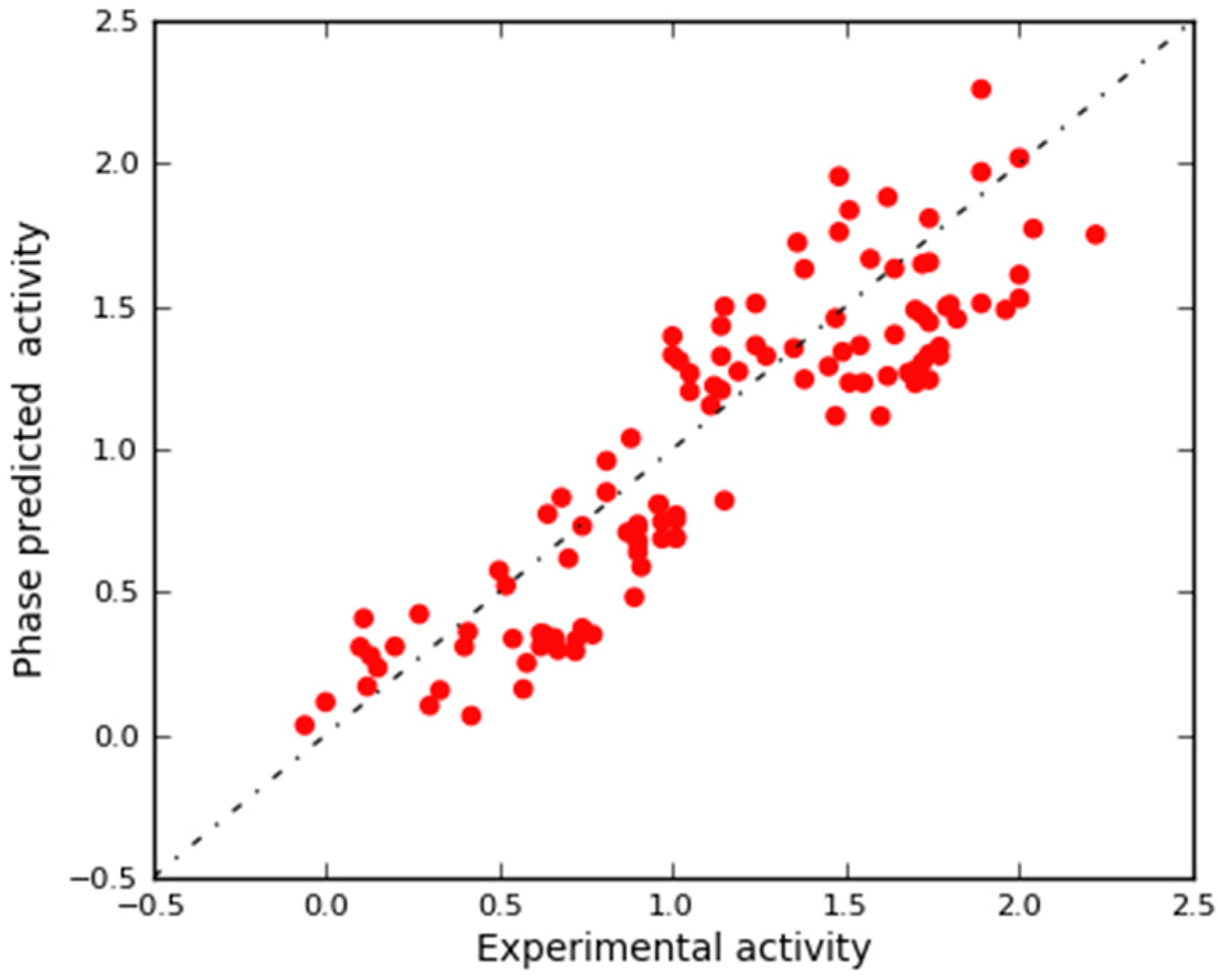
2.2. Pharmacophore Modeling and 3D-QSAR Model Generation
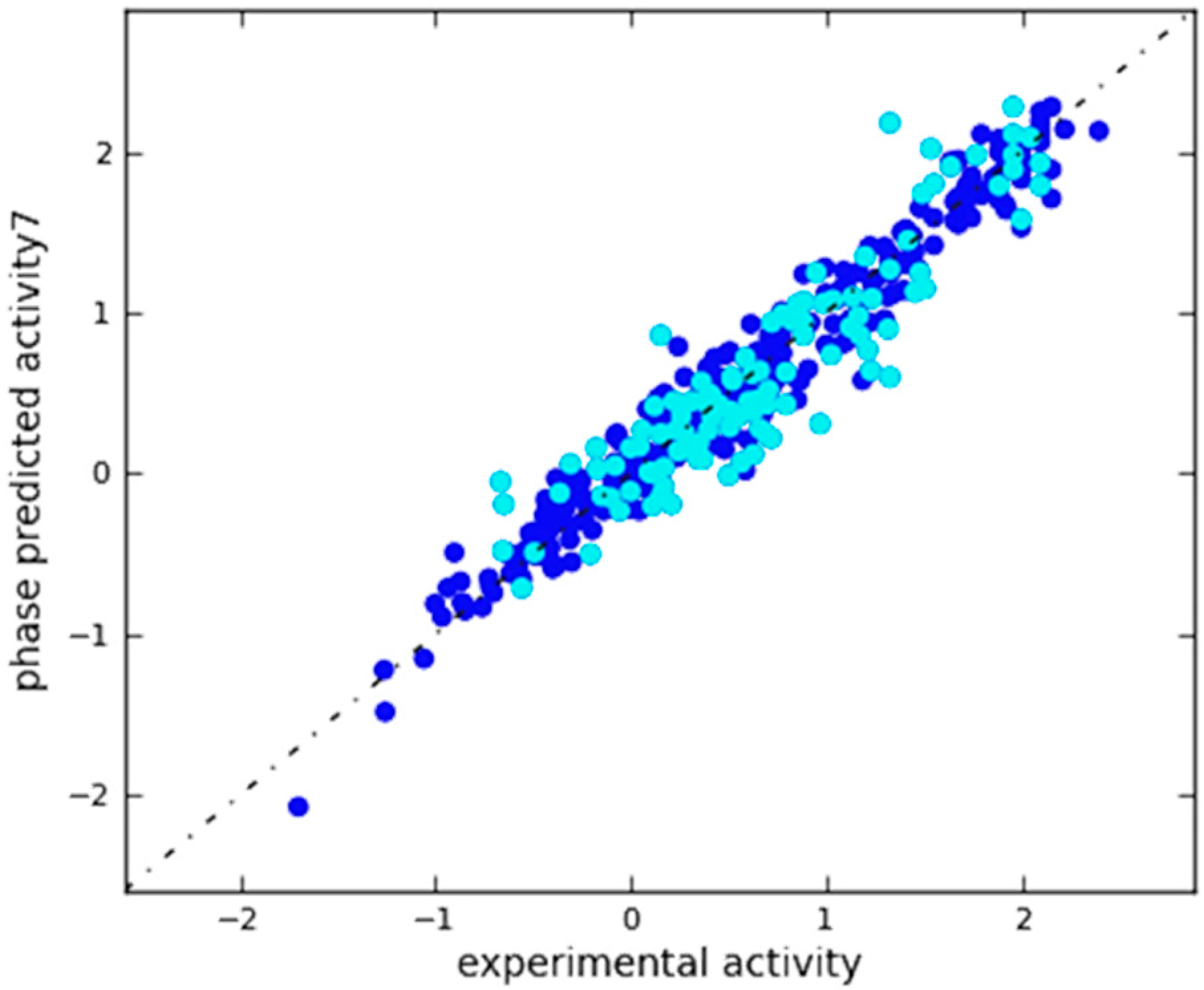
2.3. In Silico 3D-QSAR Model Validation
2.3.1. Validation Using External Test Set
2.3.2. Validation Using Decoy Set and Receiver-Operating Characteristic (ROC) Curve Approach
3. Materials and Methods
3.1. Hardware and Software Specifications
3.2. Ligands and Data Set Preparation
3.3. 3D-QSAR Model Generation
3.4. In Silico 3D-QSAR Model Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Conway, S.J.; Woster, P.M.; Greenlee, W.J.; Georg, G.; Wang, S. Epigenetics: Novel Therapeutics Targeting Epigenetics. J. Med. Chem. 2016, 59, 1247–1248. [Google Scholar] [CrossRef]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef]
- Bertrand, P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Paris, M.; Porcelloni, M.; Binaschi, M.; Fattori, D. Histone deacetylase inhibitors: From bench to clinic. J. Med. Chem 2008, 51, 1505–1529. [Google Scholar] [CrossRef]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Rodriquez, M.; Aquino, M.; Bruno, I.; De Martino, G.; Taddei, M.; Gomez-Paloma, L. Chemistry and biology of chromatin remodeling agents: State of art and future perspectives of HDAC inhibitors. Curr. Med. Chem. 2006, 13, 1119–1139. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Marks, P.A.; Dokmanovic, M. Histone deacetylase inhibitors: Discovery and development as anticancer agents. Expert Opin. Investig. Drugs 2005, 14, 1497–1511. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Brittain, D.; Weinmann, H.; Ottow, E. Recent Advances in the Medicinal Chemistry of Histone Deacetylase Inhibitors. Annu. Rep. Med. Chem. 2007, 42, 337–348. [Google Scholar] [CrossRef]
- Miller, T.A. Patent status of histone deacetylase inhibitors. Expert Opin. Ther. Pat. 2005, 14, 791–804. [Google Scholar] [CrossRef]
- Gregoretti, I.; Lee, Y.-M.; Goodson, H.V. Molecular Evolution of the Histone Deacetylase Family: Functional Implications of Phylogenetic Analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2006, 109, 31–39. [Google Scholar] [CrossRef]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Panobinostat: First Global Approval. Drugs 2015, 75, 695–704. [Google Scholar] [CrossRef]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef]
- Qiao, Z.; Ren, S.; Li, W.; Wang, X.; He, M.; Guo, Y.; Sun, L.; He, Y.; Ge, Y.; Yu, Q. Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 95–101. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Assouline, S.; Cortes, J.; Estrov, Z.; Kantarjian, H.; Yang, H.; Newsome, W.M.; Miller, W.H.; Rousseau, C.; Kalita, A.; et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood 2008, 112, 981–989. [Google Scholar] [CrossRef]
- Knipstein, J.; Gore, L. Entinostat for treatment of solid tumors and hematologic malignancies. Expert Opin. Investig. Drugs 2011, 20, 1455–1467. [Google Scholar] [CrossRef]
- Gediya, L.K.; Belosay, A.; Khandelwal, A.; Purushottamachar, P.; Njar, V.C.O. Improved synthesis of histone deacetylase inhibitors (HDIs) (MS-275 and CI-994) and inhibitory effects of HDIs alone or in combination with RAMBAs or retinoids on growth of human LNCaP prostate cancer cells and tumor xenografts. Bioorg. Med. Chem. 2008, 16, 3352–3360. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G.; Vannucchi, A.M.; Martinelli, V.; Ruggeri, M.; Nobile, F.; Specchia, G.; Pogliani, E.M.; Olimpieri, O.M.; Fioritoni, G.; Musolino, C.; et al. A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br. J. Haematol. 2013, 161, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Evens, A.M.; Balasubramanian, S.; Vose, J.M.; Harb, W.; Gordon, L.I.; Langdon, R.; Sprague, J.; Sirisawad, M.; Mani, C.; Yue, J.; et al. A Phase I/II Multicenter, Open-Label Study of the Oral Histone Deacetylase Inhibitor Abexinostat in Relapsed/Refractory Lymphoma. Clin. Cancer Res. 2015, 22, 1059–1066. [Google Scholar] [CrossRef]
- Bruserud, O.; Stapnes, C.; Ersvær, E.; Gjertsen, B.; Ryningen, A. Histone Deacetylase Inhibitors in Cancer Treatment: A Review of the Clinical Toxicity and the Modulation of Gene Expression in Cancer Cells. Curr. Pharm. Biotechnol. 2007, 8, 388–400. [Google Scholar] [CrossRef]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef]
- Karagiannis, T.C.; El-Osta, A. Will broad-spectrum histone deacetylase inhibitors be superseded by more specific compounds? Leukemia 2006, 21, 61–65. [Google Scholar] [CrossRef]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Brindisi, M.; Saraswati, A.P.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Old but Gold: Tracking the New Guise of Histone Deacetylase 6 (HDAC6) Enzyme as a Biomarker and Therapeutic Target in Rare Diseases. J. Med. Chem. 2020, 63, 23–39. [Google Scholar] [CrossRef]
- Bieliauskas, A.V.; Pflum, M.K.H. Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 2008, 37, 1402. [Google Scholar] [CrossRef]
- Guha, M. HDAC inhibitors still need a home run, despite recent approval. Nat. Rev. Drug Discov. 2015, 14, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Ononye, S.N.; van Heyst, M.; Falcone, E.M.; Anderson, A.C.; Wright, D.L. Toward isozyme-selective inhibitors of histone deacetylase as therapeutic agents for the treatment of cancer. Pharm. Pat. Anal. 2012, 1, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Han, Y.; Jiang, Q.; Wang, C.; Chen, X.; Li, X.; Xu, F.; Jiang, Y.; Wang, Q.; Xu, W. Trend of Histone Deacetylase Inhibitors in Cancer Therapy: Isoform Selectivity or Multitargeted Strategy. Med. Res. Rev. 2015, 35, 63–84. [Google Scholar] [CrossRef] [PubMed]
- Glaser, K.B.; Li, J.; Staver, M.J.; Wei, R.-Q.; Albert, D.H.; Davidsen, S.K. Role of Class I and Class II histone deacetylases in carcinoma cells using siRNA. Biochem. Biophys. Res. Commun. 2003, 310, 529–536. [Google Scholar] [CrossRef]
- Hu, E.; Dul, E.; Sung, C.-M.; Chen, Z.; Kirkpatrick, R.; Zhang, G.-F.; Johanson, K.; Liu, R.; Lago, A.; Hofmann, G.; et al. Identification of Novel Isoform-Selective Inhibitors within Class I Histone Deacetylases. J. Pharmacol. Exp. Ther. 2003, 307, 720–728. [Google Scholar] [CrossRef]
- Kawai, H.; Li, H.; Avraham, S.; Jiang, S.; Avraham, H.K. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor? Int. J. Cancer 2003, 107, 353–358. [Google Scholar] [CrossRef]
- Rikimaru, T.; Taketomi, A.; Yamashita, Y.-I.; Shirabe, K.; Hamatsu, T.; Shimada, M.; Maehara, Y. Clinical Significance of Histone Deacetylase 1 Expression in Patients with Hepatocellular Carcinoma. Oncology 2007, 72, 69–74. [Google Scholar] [CrossRef]
- Senese, S.; Zaragoza, K.; Minardi, S.; Muradore, I.; Ronzoni, S.; Passafaro, A.; Bernard, L.; Draetta, G.F.; Alcalay, M.; Seiser, C.; et al. Role for Histone Deacetylase 1 in Human Tumor Cell Proliferation. Mol. Cell Biol. 2007, 27, 4784–4795. [Google Scholar] [CrossRef]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2007, 409, 581–589. [Google Scholar] [CrossRef]
- Shen, S.; Kozikowski, A.P. Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors-What Some May Have Forgotten or Would Rather Forget? ChemMedChem 2016, 11, 15–21. [Google Scholar] [CrossRef]
- Beckers, T.; Burkhardt, C.; Wieland, H.; Gimmnich, P.; Ciossek, T.; Maier, T.; Sanders, K. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int. J. Cancer 2007, 121, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Bonfils, C.; Kalita, A.; Dubay, M.; Siu, L.L.; Carducci, M.A.; Reid, G.; Martell, R.E.; Besterman, J.M.; Li, Z. Evaluation of the Pharmacodynamic Effects of MGCD0103 from Preclinical Models to Human Using a Novel HDAC Enzyme Assay. Clin. Cancer Res. 2008, 14, 3441–3449. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.J.; Herman, D.; Gottesfeld, J.M. Pimelic Diphenylamide 106 Is a Slow, Tight-binding Inhibitor of Class I Histone Deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.K.; Richon, V.M.; O’Connor, O.; Curley, T.; MacGregor-Curtelli, B.; Tong, W.; Klang, M.; Schwartz, L.; Richardson, S.; Rosa, E.; et al. Phase I clinical trial of histone deacetylase inhibitor: Suberoylanilide hydroxamic acid administered intravenously. Clin. Cancer Res. 2003, 9, 3578–3588. [Google Scholar]
- Nagaoka, Y.; Maeda, T.; Kawai, Y.; Nakashima, D.; Oikawa, T.; Shimoke, K.; Ikeuchi, T.; Kuwajima, H.; Uesato, S. Synthesis and cancer antiproliferative activity of new histone deacetylase inhibitors: Hydrophilic hydroxamates and 2-aminobenzamide-containing derivatives. Eur. J. Med. Chem. 2006, 41, 697–708. [Google Scholar] [CrossRef]
- Moradei, O.M.; Mallais, T.C.; Frechette, S.; Paquin, I.; Tessier, P.E.; Leit, S.M.; Fournel, M.; Bonfils, C.; Trachy-Bourget, M.-C.; Liu, J.; et al. Novel Aminophenyl Benzamide-Type Histone Deacetylase Inhibitors with Enhanced Potency and Selectivity. J. Med. Chem. 2007, 50, 5543–5546. [Google Scholar] [CrossRef]
- Suzuki, T.; Miyata, N. Non-hydroxamate Histone Deacetylase Inhibitors. Curr. Med. Chem. 2005, 12, 2867–2880. [Google Scholar] [CrossRef]
- Wang, D.-F.; Helquist, P.; Wiech, N.L.; Wiest, O. Toward Selective Histone Deacetylase Inhibitor Design: Homology Modeling, Docking Studies, and Molecular Dynamics Simulations of Human Class I Histone Deacetylases. J. Med. Chem. 2005, 48, 6936–6947. [Google Scholar] [CrossRef]
- Chemi, G.; Brogi, S. Breakthroughs in Computational Approaches for Drug Discovery. J. Drug Res. Dev. 2017, 3. [Google Scholar] [CrossRef]
- Verma, J.; Khedkar, V.; Coutinho, E. 3D-QSAR in Drug Design—A Review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef]
- Yang, S.-Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Corelli, F.; Di Marzo, V.; Ligresti, A.; Mugnaini, C.; Pasquini, S.; Tafi, A. Three-dimensional quantitative structure–selectivity relationships analysis guided rational design of a highly selective ligand for the cannabinoid receptor 2. Eur. J. Med. Chem. 2011, 46, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Papazafiri, P.; Roussis, V.; Tafi, A. 3D-QSAR using pharmacophore-based alignment and virtual screening for discovery of novel MCF-7 cell line inhibitors. Eur. J. Med. Chem. 2013, 67, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Zaccagnini, L.; Brogi, S.; Brindisi, M.; Gemma, S.; Chemi, G.; Legname, G.; Campiani, G.; Butini, S. Identification of novel fluorescent probes preventing PrP Sc replication in prion diseases. Eur. J. Med. Chem. 2017, 127, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Brindisi, M.; Joshi, B.P.; Sanna Coccone, S.; Parapini, S.; Basilico, N.; Novellino, E.; Campiani, G.; Gemma, S.; Butini, S. Exploring clotrimazole-based pharmacophore: 3D-QSAR studies and synthesis of novel antiplasmodial agents. Bioorg. Med. Chem. Lett. 2015, 25, 5412–5418. [Google Scholar] [CrossRef] [PubMed]
- Chemi, G.; Gemma, S.; Campiani, G.; Brogi, S.; Butini, S.; Brindisi, M. Computational Tool for Fast in silico Evaluation of hERG K+ Channel Affinity. Front. Chem. 2017, 5. [Google Scholar] [CrossRef]
- Liu, B.; Lu, A.J.; Liao, C.Z.; Liu, H.B.; Zhou, J.J. 3D-QSAR of Sulfonamide Hydroxamic Acid HDAC Inhibitors. Acta Phys. Chim. Sin. 2005, 21, 333–337. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, J.; Guo, Z.; Chu, F.; Cheng, Y.; Wu, S. Exploration of a binding mode of indole amide analogues as potent histone deacetylase inhibitors and 3D-QSAR analyses. Bioorg. Med. Chem. 2005, 13, 5424–5434. [Google Scholar] [CrossRef]
- Juvale, D.C.; Kulkarni, V.V.; Deokar, H.S.; Wagh, N.K.; Padhye, S.B.; Kulkarni, V.M. 3D-QSAR of histone deacetylase inhibitors: Hydroxamate analogues. Org. Biomol. Chem. 2006, 4, 2858. [Google Scholar] [CrossRef]
- Ragno, R.; Simeoni, S.; Valente, S.; Massa, S.; Mai, A. 3-D QSAR Studies on Histone Deacetylase Inhibitors. A GOLPE/GRID Approach on Different Series of Compounds. J. Chem. Inf. Model. 2006, 46, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, H.; Tang, W.; Zhu, C.; Jiang, Y.; Zou, J.; Yu, Q.; You, Q. 3D-QSAR studies of HDACs inhibitors using pharmacophore-based alignment. Eur. J. Med. Chem. 2009, 44, 2868–2876. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-F.; Wiest, O.; Helquist, P.; Lan-Hargest, H.-Y.; Wiech, N.L. QSAR Studies of PC-3 cell line inhibition activity of TSA and SAHA-like hydroxamic acids. Bioorg. Med. Chem. Lett. 2004, 14, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Liao, C.; Li, Z.; Ning, Z.; Hu, W.; Lu, X.; Shi, L.; Zhou, J. Quantitative Structure-Activity Relationship Study of Histone Deacetylase Inhibitors. Anti-Cancer Agents Med. Chem. 2004, 4, 273–299. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Abdizadeh, T.; Kalani, M.R.; Abnous, K.; Tayarani-Najaran, Z.; Khashyarmanesh, B.Z.; Abdizadeh, R.; Ghodsi, R.; Hadizadeh, F. Design, synthesis and biological evaluation of novel coumarin-based benzamides as potent histone deacetylase inhibitors and anticancer agents. Eur. J. Med. Chem. 2017, 132, 42–62. [Google Scholar] [CrossRef]
- Hamblett, C.L.; Methot, J.L.; Mampreian, D.M.; Sloman, D.L.; Stanton, M.G.; Kral, A.M.; Fleming, J.C.; Cruz, J.C.; Chenard, M.; Ozerova, N.; et al. The discovery of 6-amino nicotinamides as potent and selective histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 5300–5309. [Google Scholar] [CrossRef]
- Sixto-López, Y.; Bello, M.; Correa-Basurto, J. Insights into structural features of HDAC1 and its selectivity inhibition elucidated by Molecular dynamic simulation and Molecular Docking. J. Biomol. Struct. Dyn. 2018, 37, 584–610. [Google Scholar] [CrossRef]
- Brindisi, M.; Cavella, C.; Brogi, S.; Nebbioso, A.; Senger, J.; Maramai, S.; Ciotta, A.; Iside, C.; Butini, S.; Lamponi, S.; et al. Phenylpyrrole-based HDAC inhibitors: Synthesis, molecular modeling and biological studies. Future Med. Chem. 2016, 8, 1573–1587. [Google Scholar] [CrossRef]
- Brindisi, M.; Senger, J.; Cavella, C.; Grillo, A.; Chemi, G.; Gemma, S.; Cucinella, D.M.; Lamponi, S.; Sarno, F.; Iside, C.; et al. Novel spiroindoline HDAC inhibitors: Synthesis, molecular modelling and biological studies. Eur. J. Med. Chem. 2018, 157, 127–138. [Google Scholar] [CrossRef]
- Braga, R.C.; Andrade, C.H. Assessing the Performance of 3D Pharmacophore Models in Virtual Screening: How Good are They? Curr. Top. Med. Chem. 2013, 13, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Dror, O.; Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Novel Approach for Efficient Pharmacophore-Based Virtual Screening: Method and Applications. J. Chem. Inf. Model. 2009, 49, 2333–2343. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Singh, D.K.; Meena, S.; Datta, D.; Siddiqi, M.I.; Banerjee, D. Pharmacophore-Based Screening and Identification of Novel Human Ligase I Inhibitors with Potential Anticancer Activity. J. Chem. Inf. Model. 2014, 54, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Sakkiah, S.; Thangapandian, S.; John, S.; Lee, K.W. Pharmacophore based virtual screening, molecular docking studies to design potent heat shock protein 90 inhibitors. Eur. J. Med. Chem. 2011, 46, 2937–2947. [Google Scholar] [CrossRef]
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.W. Pharmacophore-based virtual screening and Bayesian model for the identification of potential human leukotriene A4 hydrolase inhibitors. Eur. J. Med. Chem. 2011, 46, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.-P.; Bertrand, H.-O. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef]
- Zhao, W.; Hevener, K.E.; White, S.W.; Lee, R.E.; Boyett, J.M. A statistical framework to evaluate virtual screening. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef]
- Maestro; Version 10.1; Schrödinger, LLC: New York, NY, USA, 2015.
- Andrews, D.M.; Gibson, K.M.; Graham, M.A.; Matusiak, Z.S.; Roberts, C.A.; Stokes, E.S.; Brady, M.C.; Chresta, C.M. Design and campaign synthesis of pyridine-based histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2525–2529. [Google Scholar] [CrossRef]
- Methot, J.L.; Chakravarty, P.K.; Chenard, M.; Close, J.; Cruz, J.C.; Dahlberg, W.K.; Fleming, J.; Hamblett, C.L.; Hamill, J.E.; Harrington, P.; et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 973–978. [Google Scholar] [CrossRef]
- Kattar, S.D.; Surdi, L.M.; Zabierek, A.; Methot, J.L.; Middleton, R.E.; Hughes, B.; Szewczak, A.A.; Dahlberg, W.K.; Kral, A.M.; Ozerova, N.; et al. Parallel medicinal chemistry approaches to selective HDAC1/HDAC2 inhibitor (SHI-1:2) optimization. Bioorg. Med. Chem. Lett. 2009, 19, 1168–1172. [Google Scholar] [CrossRef]
- Methot, J.L.; Hamblett, C.L.; Mampreian, D.M.; Jung, J.; Harsch, A.; Szewczak, A.A.; Dahlberg, W.K.; Middleton, R.E.; Hughes, B.; Fleming, J.C.; et al. SAR profiles of spirocyclic nicotinamide derived selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 6104–6109. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Xie, N.; Xu, M.; Qian, P.; Zhao, Y.; Li, S. Design, synthesis and antiproliferative activities of novel benzamides derivatives as HDAC inhibitors. Eur. J. Med. Chem. 2015, 100, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Y.; Qian, P.; Wang, Y.; Jiang, F.; Yao, Z.; Hu, W.; Zhao, Y.; Li, S. Design, synthesis and bioevalution of novel benzamides derivatives as HDAC inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Siliphaivanh, P.; Harrington, P.; Witter, D.J.; Otte, K.; Tempest, P.; Kattar, S.; Kral, A.M.; Fleming, J.C.; Deshmukh, S.V.; Harsch, A.; et al. Design of novel histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 4619–4624. [Google Scholar] [CrossRef] [PubMed]
- Andrews, D.M.; Stokes, E.S.; Carr, G.R.; Matusiak, Z.S.; Roberts, C.A.; Waring, M.J.; Brady, M.C.; Chresta, C.M.; East, S.J. Design and campaign synthesis of piperidine- and thiazole-based histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2580–2584. [Google Scholar] [CrossRef]
- Hirata, Y.; Hirata, M.; Kawaratani, Y.; Shibano, M.; Taniguchi, M.; Yasuda, M.; Ohmomo, Y.; Nagaoka, Y.; Baba, K.; Uesato, S. Anti-tumor activity of new orally bioavailable 2-amino-5-(thiophen-2-yl)benzamide-series histone deacetylase inhibitors, possessing an aqueous soluble functional group as a surface recognition domain. Bioorg. Med. Chem. Lett. 2012, 22, 1926–1930. [Google Scholar] [CrossRef]
- Kiyokawa, S.; Hirata, Y.; Nagaoka, Y.; Shibano, M.; Taniguchi, M.; Yasuda, M.; Baba, K.; Uesato, S. New orally bioavailable 2-aminobenzamide-type histone deacetylase inhibitor possessing a (2-hydroxyethyl)(4-(thiophen-2-yl)benzyl)amino group. Bioorg. Med. Chem. 2010, 18, 3925–3933. [Google Scholar] [CrossRef]
- Mahboobi, S.; Dove, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T. Design of chimeric histone deacetylase- and tyrosine kinase-inhibitors: A series of imatinib hybrides as potent inhibitors of wild-type and mutant BCR-ABL, PDGF-Rbeta, and histone deacetylases. J. Med. Chem. 2009, 52, 2265–2279. [Google Scholar] [CrossRef]
- Rajak, H.; Kumar, P.; Parmar, P.; Thakur, B.S.; Veerasamy, R.; Sharma, P.C.; Sharma, A.K.; Gupta, A.K.; Dangi, J.S. Appraisal of GABA and PABA as linker: Design and synthesis of novel benzamide based histone deacetylase inhibitors. Eur. J. Med. Chem. 2012, 53, 390–397. [Google Scholar] [CrossRef]
- Valente, S.; Trisciuoglio, D.; De Luca, T.; Nebbioso, A.; Labella, D.; Lenoci, A.; Bigogno, C.; Dondio, G.; Miceli, M.; Brosch, G.; et al. 1,3,4-Oxadiazole-containing histone deacetylase inhibitors: Anticancer activities in cancer cells. J. Med. Chem. 2014, 57, 6259–6265. [Google Scholar] [CrossRef]
- Rusche, J.R.; Peet, N.P.; Hopper, A.T. Compositions Including 6-Aminohexanoic Acid Derivatives as HDAC Inhibitors. U.S. Patent 9,265,734, 23 February 2016. [Google Scholar]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- LigPrep; Version 3.3; Schrödinger, LLC: New York, NY, USA, 2015.
- Sirous, H.; Chemi, G.; Campiani, G.; Brogi, S. An integrated in silico screening strategy for identifying promising disruptors of p53-MDM2 interaction. Comput. Biol. Chem. 2019, 83, 107105. [Google Scholar] [CrossRef] [PubMed]
- Sirous, H.; Chemi, G.; Gemma, S.; Butini, S.; Debyser, Z.; Christ, F.; Saghaie, L.; Brogi, S.; Fassihi, A.; Campiani, G.; et al. Identification of Novel 3-Hydroxy-pyran-4-One Derivatives as Potent HIV-1 Integrase Inhibitors Using in silico Structure-Based Combinatorial Library Design Approach. Front. Chem. 2019, 7, 574. [Google Scholar] [CrossRef] [PubMed]
- Sirous, H.; Fassihi, A.; Brogi, S.; Campiani, G.; Christ, F.; Debyser, Z.; Gemma, S.; Butini, S.; Chemi, G.; Grillo, A.; et al. Synthesis, Molecular Modelling and Biological Studies of 3-hydroxypyrane- 4-one and 3-hydroxy-pyridine-4-one Derivatives as HIV-1 Integrase Inhibitors. Med. Chem. 2019, 15, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- MacroModel; Version 10.7; Schrödinger, LLC: New York, NY, USA, 2015.
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Phase; Version 4.2; Schrödinger, LLC: New York, NY, USA, 2015.
- Todeschini, R.; Ballabio, D.; Grisoni, F. Beware of Unreliable Q2! A Comparative Study of Regression Metrics for Predictivity Assessment of QSAR Models. J. Chem. Inf. Model. 2016, 56, 1905–1913. [Google Scholar] [CrossRef]
- Frechette, S.; Leit, S.; Woo, S.H.; Lapointe, G.; Jeannotte, G.; Moradei, O.; Paquin, I.; Bouchain, G.; Raeppel, S.; Gaudette, F.; et al. 4-(Heteroarylaminomethyl)-N-(2-aminophenyl)-benzamides and their analogs as a novel class of histone deacetylase inhibitors. Bioorg. Med. Chem. Lett 2008, 18, 1502–1506. [Google Scholar] [CrossRef]
- Marson, C.M.; Matthews, C.J.; Atkinson, S.J.; Lamadema, N.; Thomas, N.S. Potent and Selective Inhibitors of Histone Deacetylase-3 Containing Chiral Oxazoline Capping Groups and a N-(2-Aminophenyl)-benzamide Binding Unit. J. Med. Chem. 2015, 58, 6803–6818. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, X.; Ran, T.; Niu, J.; Zhao, S.; Lu, T.; Tang, W. Design, synthesis and biological evaluation of urea-based benzamides derivatives as HDAC inhibitors. Med. Chem. Res. 2017, 26, 2879–2888. [Google Scholar] [CrossRef]
- Harrington, P.; Kattar, S.; Miller, T.A.; Stanton, M.G.; Tempest, P.; Witter, D.J. 4-Carboxybenzylamino Derivatives as Histone Deacetylase Inhibitors. U.S. Patent 8,389,553, 5 March 2013. [Google Scholar]
- Kirchmair, J.; Markt, P.; Distinto, S.; Wolber, G.; Langer, T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection—What can we learn from earlier mistakes? J. Comput. Aided Mol. Des. 2008, 22, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HYPO ID | Survival | Survival—Inactive | Site | Vector | Volume | Selectivity | Matches | Energy | Activity | Inactive |
|---|---|---|---|---|---|---|---|---|---|---|
| ADDRR | 3.769 | 1.841 | 0.97 | 0.999 | 0.798 | 1.578 | 20 | 0.006 | 2.097 | 1.928 |
| LVs | R 2a | SD b | F c | P d | RMSE e | Q 2f | Q2F3 g | R-Pearson h |
|---|---|---|---|---|---|---|---|---|
| 1 | 0.3408 | 0.6978 | 132.9 | 4.699 × 10−22 | 0.5199 | 0.3886 | 0.635 | 0.6747 |
| 2 | 0.6273 | 0.5257 | 215.5 | 1.344 × 10−55 | 0.3979 | 0.6420 | 0.791 | 0.8282 |
| 3 | 0.7620 | 0.4209 | 272.2 | 3.629 × 10−79 | 0.3615 | 0.7045 | 0.823 | 0.8579 |
| 4 | 0.8775 | 0.3025 | 455.0 | 1.704 × 10−144 | 0.3514 | 0.7207 | 0.833 | 0.8690 |
| 5 | 0.9159 | 0.2512 | 551.0 | 9.327 × 10−134 | 0.2971 | 0.8004 | 0.881 | 0.9003 |
| 6 | 0.9433 | 0.2067 | 698.6 | 6.590 × 10−134 | 0.2865 | 0.8143 | 0.890 | 0.9100 |
| 7 | 0.9582 | 0.1778 | 822.1 | 4.377 × 10−169 | 0.2808 | 0.8217 | 0.894 | 0.9152 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirous, H.; Campiani, G.; Brogi, S.; Calderone, V.; Chemi, G. Computer-Driven Development of an in Silico Tool for Finding Selective Histone Deacetylase 1 Inhibitors. Molecules 2020, 25, 1952. https://doi.org/10.3390/molecules25081952
Sirous H, Campiani G, Brogi S, Calderone V, Chemi G. Computer-Driven Development of an in Silico Tool for Finding Selective Histone Deacetylase 1 Inhibitors. Molecules. 2020; 25(8):1952. https://doi.org/10.3390/molecules25081952
Chicago/Turabian StyleSirous, Hajar, Giuseppe Campiani, Simone Brogi, Vincenzo Calderone, and Giulia Chemi. 2020. "Computer-Driven Development of an in Silico Tool for Finding Selective Histone Deacetylase 1 Inhibitors" Molecules 25, no. 8: 1952. https://doi.org/10.3390/molecules25081952
APA StyleSirous, H., Campiani, G., Brogi, S., Calderone, V., & Chemi, G. (2020). Computer-Driven Development of an in Silico Tool for Finding Selective Histone Deacetylase 1 Inhibitors. Molecules, 25(8), 1952. https://doi.org/10.3390/molecules25081952