
Biodegradable Cell Microcarriers Based on Chitosan/Polyester Graft-Copolymers

 ,
,  , ,
, ,  ,
, 
Abstract
1. Introduction
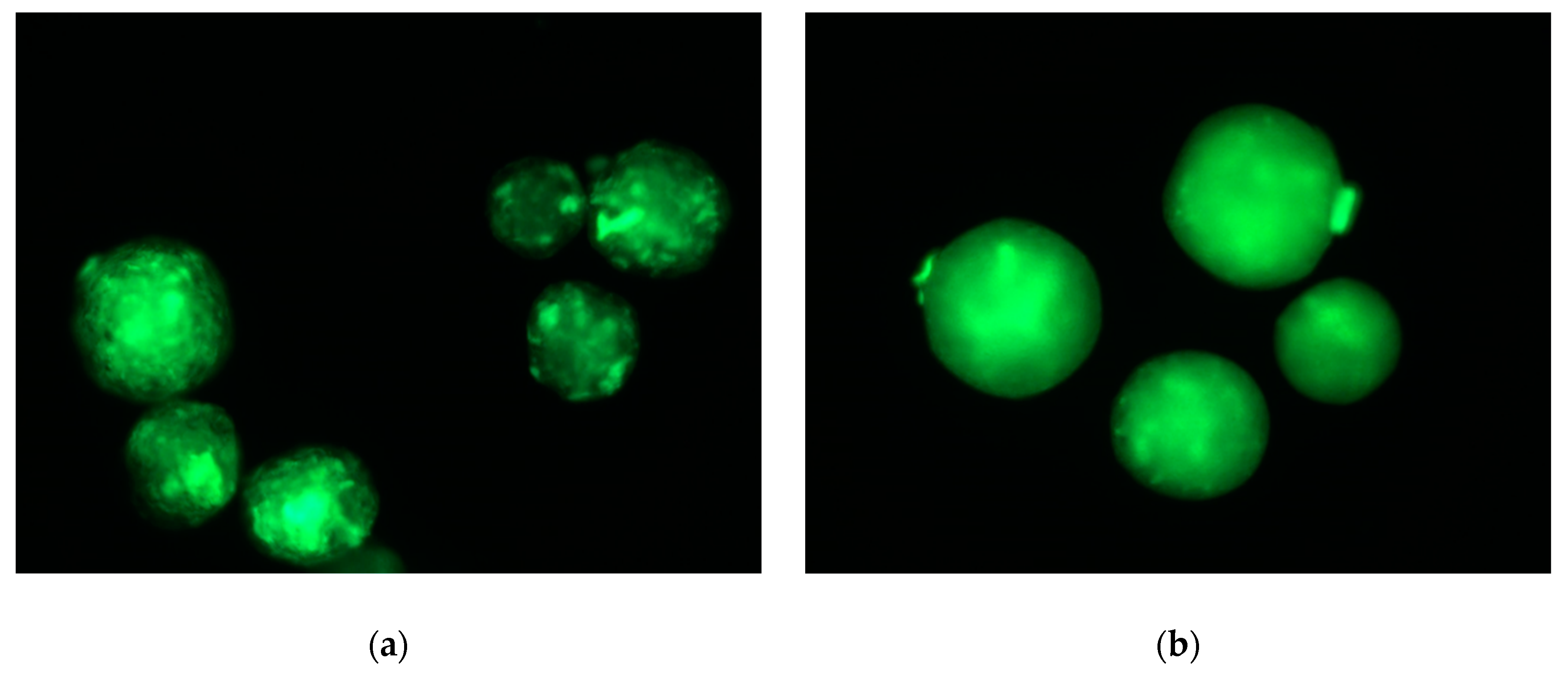
2. Results and Discussion
2.1. Microsphere Formation, Size Distribution and Recovery Yield
2.2. In Vitro Analysis of the Biocompatibility of the Microparticles
3. Materials and Methods
3.1. Copolymer’s Synthesis and Characterization
3.2. Preparation and Characterization of the Microspheres
3.3. Cell Cultivation on the Microparticles
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Van Wezel, A.L. Growth of Cell-Strains and Primary Cells on Micro-Carriers in Homogeneous Culture. Nature 1967, 216, 64–65. [Google Scholar] [CrossRef]
- Markvicheva, E.; Grandfils, C. Microcarriers for Animal Cell Culture. In Fundamentals of Cell Immobilisation Biotechnology; Nedovic, V., Willaert, R., Eds.; Kluwer Academic Publishers: Berlin, Germany, 2004; pp. 141–161. [Google Scholar]
- Chen, A.K.-L.; Chen, X.; Choo, A.B.H.; Reuveny, S.; Oh, S.K.W. Critical Microcarrier Properties Affecting the Expansion of Undifferentiated Human Embryonic Stem Cells. Stem Cell Res. 2011, 7, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Baakdhah, T.; van der Kooy, D. Expansion of Retinal Stem Cells and Their Progeny Using Cell Microcarriers in a Bioreactor. Biotechnol. Prog. 2019, 35, e2800. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Y.; Chen, J.-Y.; Tong, X.-M.; Mei, J.-G.; Chen, Y.-F.; Mou, X.-Z. Recent Advances in the Use of Microcarriers for Cell Cultures and Their Ex Vivo and in Vivo Applications. Biotechnol. Lett. 2020, 42, 1–10. [Google Scholar] [CrossRef]
- Neto, M.D.; Oliveira, M.B.; Mano, J.F. Microparticles in Contact with Cells: From Carriers to Multifunctional Tissue Modulators. Trends Biotechnol. 2019, 37, 1011–1028. [Google Scholar] [CrossRef] [PubMed]
- Campos, E.; Branquinho, J.; Carreira, A.S.; Carvalho, A.; Coimbra, P.; Ferreira, P.; Gil, M.H. Designing Polymeric Microparticles for Biomedical and Industrial Applications. Eur. Polym. J. 2013, 49, 2005–2021. [Google Scholar] [CrossRef]
- Zhang, Z.; Eyster, T.W.; Ma, P.X. Nanostructured Injectable Cell Microcarriers for Tissue Regeneration. Nanomedicine 2016, 11, 1611–1628. [Google Scholar] [CrossRef]
- Hernández, R.M.; Orive, G.; Murua, A.; Pedraz, J.L. Microcapsules and Microcarriers for in Situ Cell Delivery. Adv. Drug Deliv. Rev. 2010, 62, 711–730. [Google Scholar] [CrossRef]
- Tavassoli, H.; Alhosseini, S.N.; Tay, A.; Chan, P.P.Y.; Oh, S.K.W.; Warkiani, M.E. Large-Scale Production of Stem Cells Utilizing Microcarriers: A Biomaterials Engineering Perspective from Academic Research to Commercialized Products. Biomaterials 2018, 181, 333–346. [Google Scholar] [CrossRef]
- Li, B.; Wang, X.; Wang, Y.; Gou, W.; Yuan, X.; Peng, J.; Guo, Q.; Lu, S. Past, present, and future of microcarrier- based tissue engineering. J. Orthop. Trans. 2015, 3, 51–57. [Google Scholar] [CrossRef]
- Jiang, T.; Nukavarapu, S.P.; Deng, M.; Jabbarzadeh, E.; Kofron, M.D.; Doty, S.B.; Abdel-Fattah, W.I.; Laurencin, C.T. Chitosan–poly(lactide-co-glycolide) microsphere-based scaffolds for bone tissue engineering: In vitro degradation and in vivo bone regeneration studies. Acta Biomater. 2010, 6, 3457–3470. [Google Scholar] [CrossRef] [PubMed]
- Lao, L.; Tan, H.; Wang, Y.; Gao, C. Chitosan modified poly(l-lactide) microspheres as cell microcarriers for cartilage tissue engineering. Colloid Surf. B 2008, 66, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Derakhti, S.; Safiabadi-Tali, S.H.; Amoabediny, G.; Sheikhpour, M. Attachment and Detachment Strategies in Microcarrier-Based Cell Culture Technology: A Comprehensive Review. Mater. Sci. Eng. C 2019, 103, 109782. [Google Scholar]
- Shekaran, A.; Lam, A.; Sim, E.; Jialing, L.; Jian, L.; Wen, J.T.P.; Chan, J.K.Y.; Choolani, M.; Reuveny, S.; Birch, W.; et al. Biodegradable ECM-Coated PCL Microcarriers Support Scalable Human Early MSC Expansion and in Vivo Bone Formation. Cytotherapy 2016, 18, 1332–1344. [Google Scholar] [CrossRef]
- Li, L.; Song, K.; Chen, Y.; Wang, Y.; Shi, F.; Nie, Y.; Liu, T. Design and Biophysical Characterization of Poly (L-Lactic) Acid Microcarriers with and without Modification of Chitosan and Nanohydroxyapatite. Polymers 2018, 10, 1061. [Google Scholar] [CrossRef]
- Privalova, A.; Markvicheva, E.; Sevrin, C.; Drozdova, M.; Kottgen, C.; Gilbert, B.; Ortiz, M.; Grandfils, C. Biodegradable Polyester-Based Microcarriers with Modified Surface Tailored for Tissue Engineering. J. Biomed. Mater. Res. A 2015, 103, 939–948. [Google Scholar] [CrossRef]
- Bee, S.-L.; Abdul Hamid, Z.A.; Mariatti, M.; Yahaya, B.H.; Lim, K.; Bee, S.-T.; Sin, L.T. Approaches to Improve Therapeutic Efficacy of Biodegradable PLA/PLGA Microspheres: A Review. Polym. Rev. 2018, 58, 495–536. [Google Scholar] [CrossRef]
- Jiang, T.; Abdel-Fattah, W.I.; Laurencin, C.T. In vitro evaluation of chitosan/poly(lactic acid-glycolic acid) sintered microsphere scaffolds for bone tissue engineering. Biomaterials 2006, 27, 4894–4903. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, J.; He, J.; Zhang, J.; Gan, Z. Fabrication, hydrolysis and cell cultivation of microspheres from cellulose-graft-poly(l-lactide) copolymers. RSC Adv. 2016, 6, 17617–17623. [Google Scholar] [CrossRef]
- Demina, T.S.; Akopova, T.A.; Vladimirov, L.V.; Zelenetskii, A.N.; Markvicheva, E.A.; Grandfils, C. Polylactide-Based Microspheres Prepared Using Solid-State Copolymerized Chitosan and d,l-Lactide. Mater. Sci. Eng. C 2016, 59, 333–338. [Google Scholar]
- Demina, T.S.; Sevrin, C.; Kapchiekue, C.; Akopova, T.A.; Grandfils, C. Chitosan-g-Polyester Microspheres: Effect of Length and Composition of Grafted Chains. Macromol. Mater. Eng. 2019, 304. [Google Scholar] [CrossRef]
- Rosca, I.D.; Watari, F.; Uo, M. Microparticle Formation and Its Mechanism in Single and Double Emulsion Solvent Evaporation. J. Control. Release 2004, 99, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Helou, M.A.; Anjum, N.; Guedeau-Boudeville, M.-A.; Rosticher, M.; Mourchid, A. Structure and Mechanical Properties of Polylactide Copolymer Microspheres and Capsules. Polymer 2010, 51, 5440–5447. [Google Scholar] [CrossRef]
- Faia-Torres, A.B.; Charnley, M.; Goren, T.; Guimond-Lischer, S.; Rottmar, M.; Maniura-Weber, K.; Spencer, N.D.; Reis, R.L.; Textor, M.; Neves, N.M. Osteogenic differentiation of human mesenchymal stem cells in the absence of osteogenic supplements: A surface-roughness gradient study. Acta Biomater. 2015, 28, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.; Rosales-Ib’anez, R.; Pozos-Guill’en, A.; de Bien, C.; Toye, D.; Flores, H.; Grandfls, C. DPSC colonization of functionalized 3D textiles. J. Biomed. Mater. Res. Part B 2017, 105, 785–794. [Google Scholar] [CrossRef]
- Akopova, T.A.; Demina, T.S.; Shchegolikhin, A.N.; Kurkin, T.S.; Grandfils, C.; Perov, N.S.; Kechekyan, A.S.; Zelenetskii, A.N. A Novel Approach to Design Chitosan-Polyester Materials for Biomedical Applications. Int. J. Polym. Sci. 2012. [Google Scholar] [CrossRef]
- Demina, T.S.; Kuryanova, A.S.; Aksenova, N.A.; Shubhyy, A.G.; Popyrina, T.N.; Sokovikov, Y.V.; Istranova, E.V.; Ivanov, P.L.; Timashev, P.S.; Akopova, T.A. Chitosan-g-Oligo/Polylactide Copolymer Non-Woven Fibrous Mats Containing Protein: From Solid-State Synthesis to Electrospinning. RSC Adv. 2019, 9, 37652–37659. [Google Scholar] [CrossRef]
- Akopova, T.A.; Zelenetskii, A.N.; Ozerin, A.N. Solid State Synthesis and Modification of Chitosan. In Focus on Chitosan Research; Ferguson, A.N., O’Neill, A.G., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2011; Chapter 8; pp. 223–254. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Component Content, wt.% | Temperature, °C | |||
|---|---|---|---|---|---|
| chitosan | poly(L-lactide) | PLGA | gelatin | ||
| CPLG | 60 | − | 40 | − | 60 |
| CPG | 35 | 52 | − | 13 | 100 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demina, T.S.; Drozdova, M.G.; Sevrin, C.; Compère, P.; Akopova, T.A.; Markvicheva, E.; Grandfils, C. Biodegradable Cell Microcarriers Based on Chitosan/Polyester Graft-Copolymers. Molecules 2020, 25, 1949. https://doi.org/10.3390/molecules25081949
Demina TS, Drozdova MG, Sevrin C, Compère P, Akopova TA, Markvicheva E, Grandfils C. Biodegradable Cell Microcarriers Based on Chitosan/Polyester Graft-Copolymers. Molecules. 2020; 25(8):1949. https://doi.org/10.3390/molecules25081949
Chicago/Turabian StyleDemina, Tatiana S., Maria G. Drozdova, Chantal Sevrin, Philippe Compère, Tatiana A. Akopova, Elena Markvicheva, and Christian Grandfils. 2020. "Biodegradable Cell Microcarriers Based on Chitosan/Polyester Graft-Copolymers" Molecules 25, no. 8: 1949. https://doi.org/10.3390/molecules25081949
APA StyleDemina, T. S., Drozdova, M. G., Sevrin, C., Compère, P., Akopova, T. A., Markvicheva, E., & Grandfils, C. (2020). Biodegradable Cell Microcarriers Based on Chitosan/Polyester Graft-Copolymers. Molecules, 25(8), 1949. https://doi.org/10.3390/molecules25081949