Structural Elucidation of Malonylcommunol and 6β-Hydroxy-trans-communic Acid, Two Undescribed Diterpenes from Salvia cinnabarina. First Examples of Labdane Diterpenoids from a Mexican Salvia Species

 , , , and
, , , and
Abstract
1. Introduction
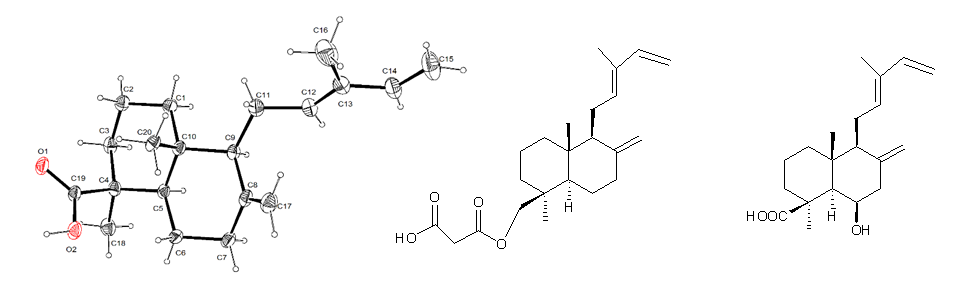
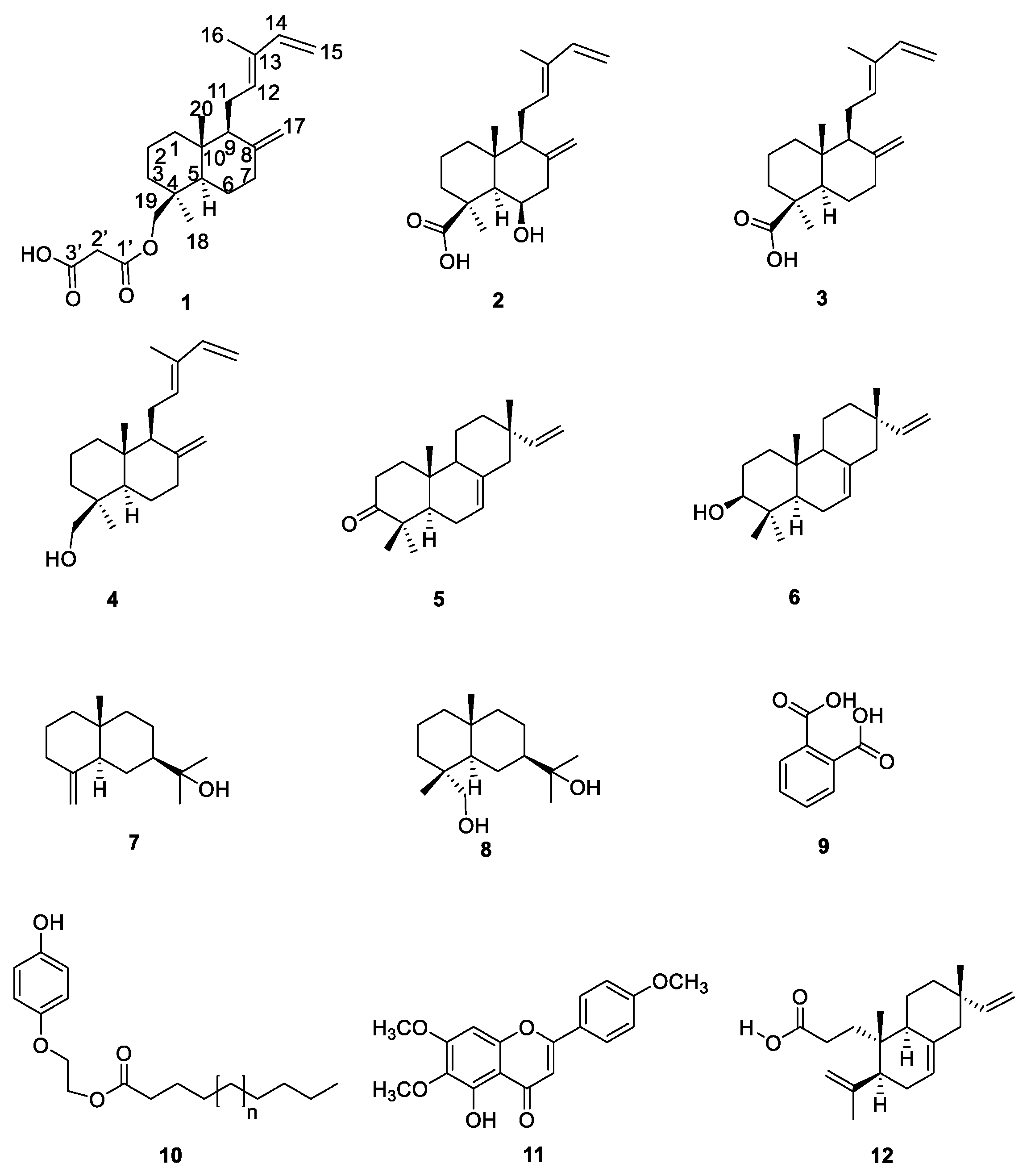
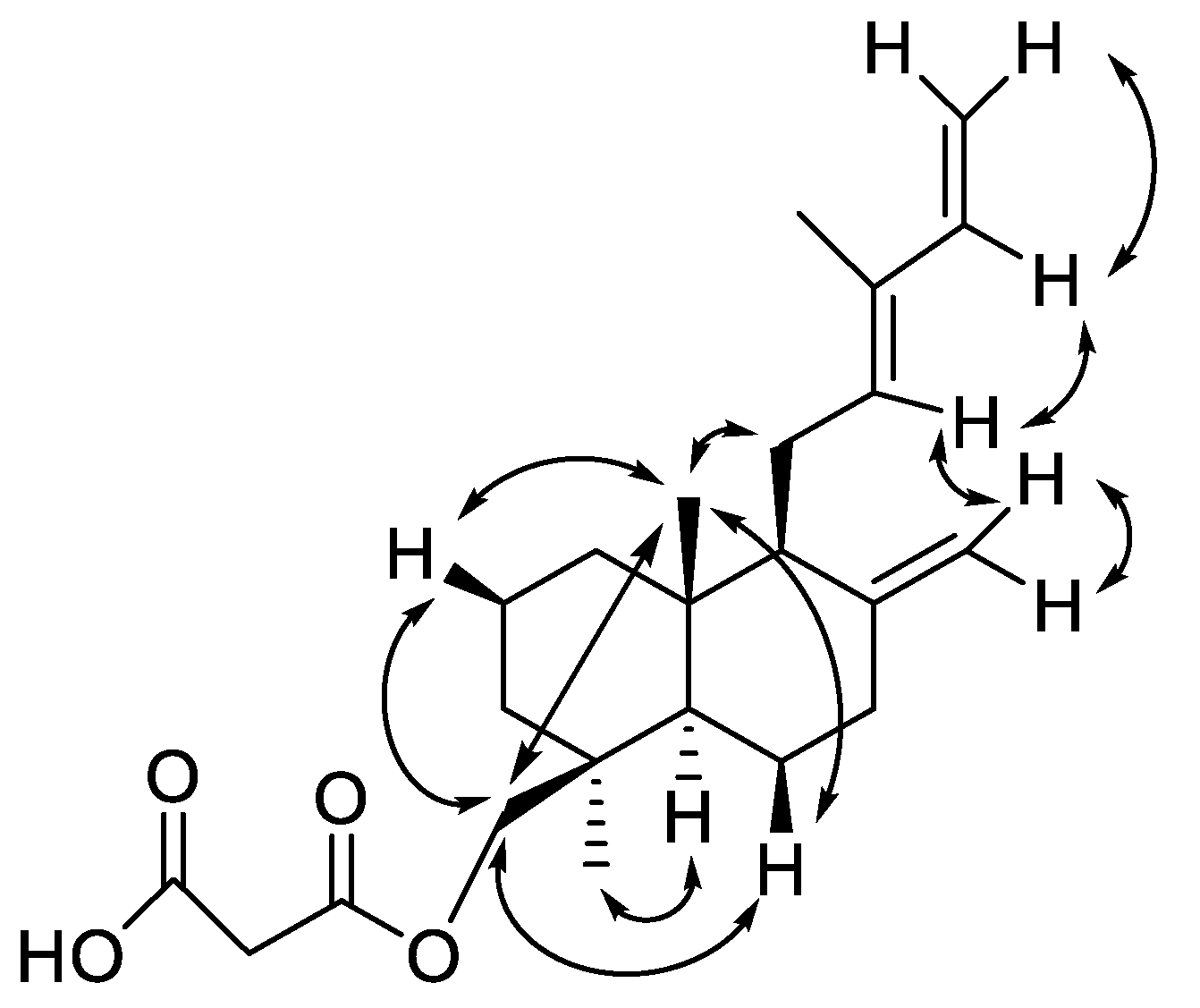
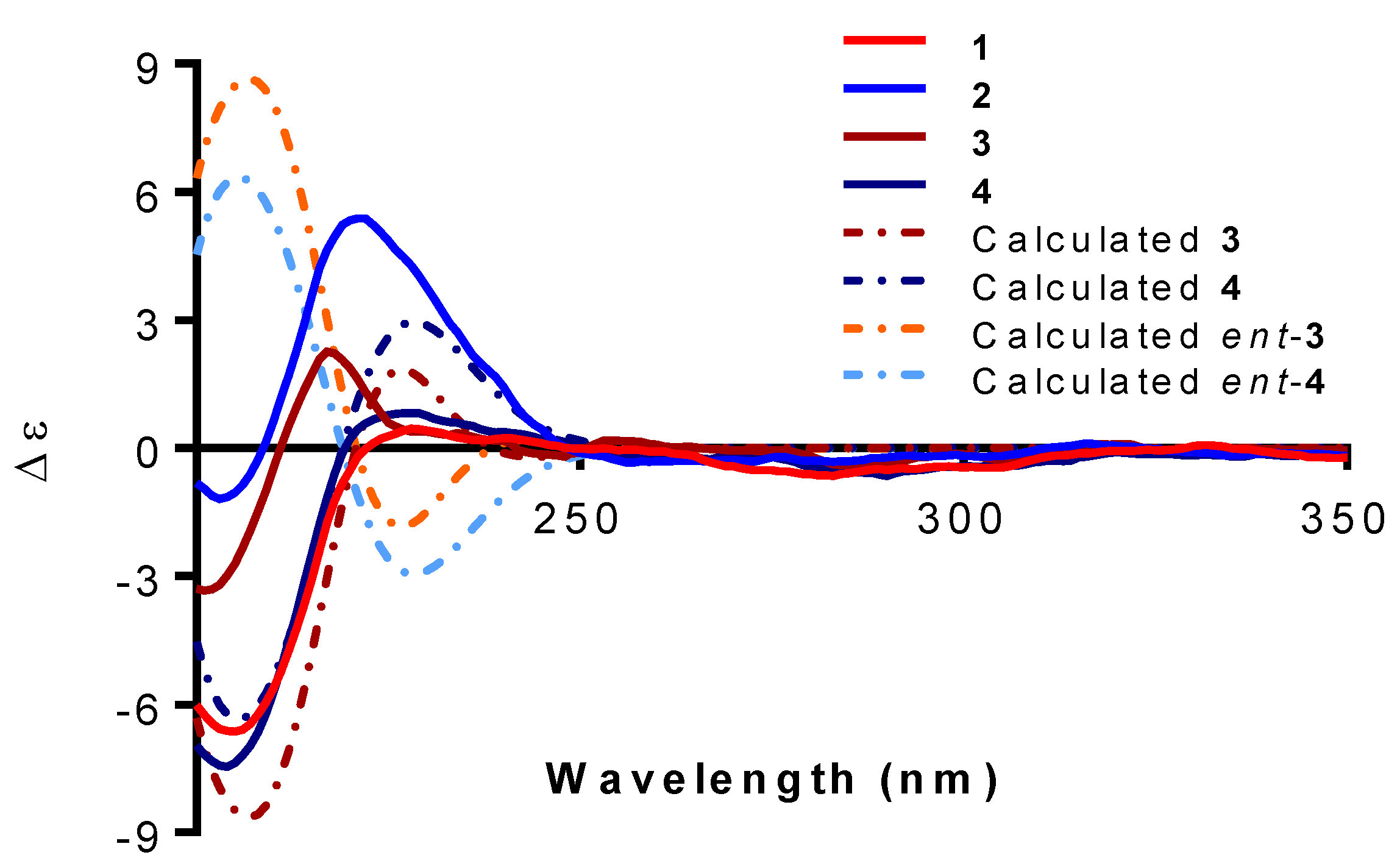
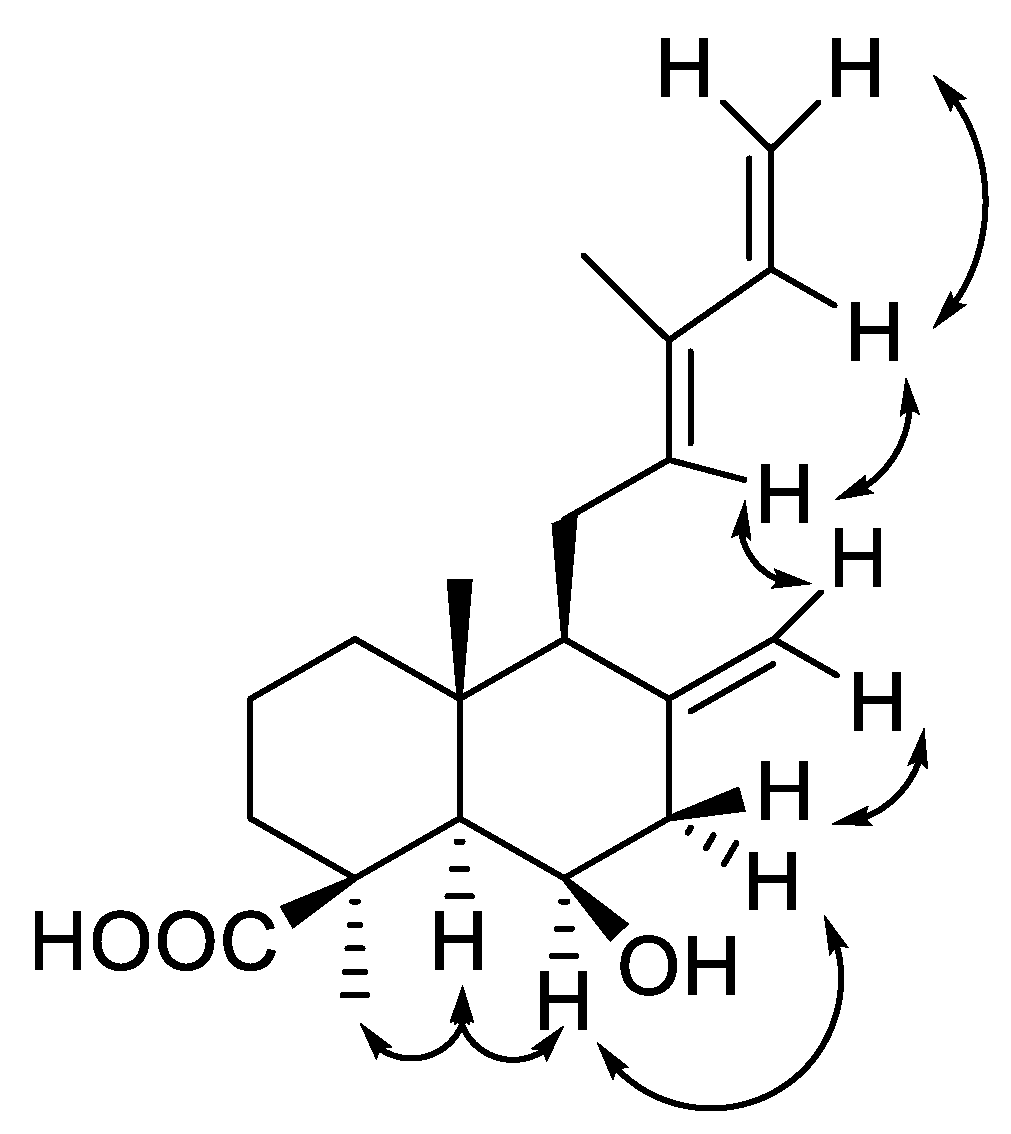
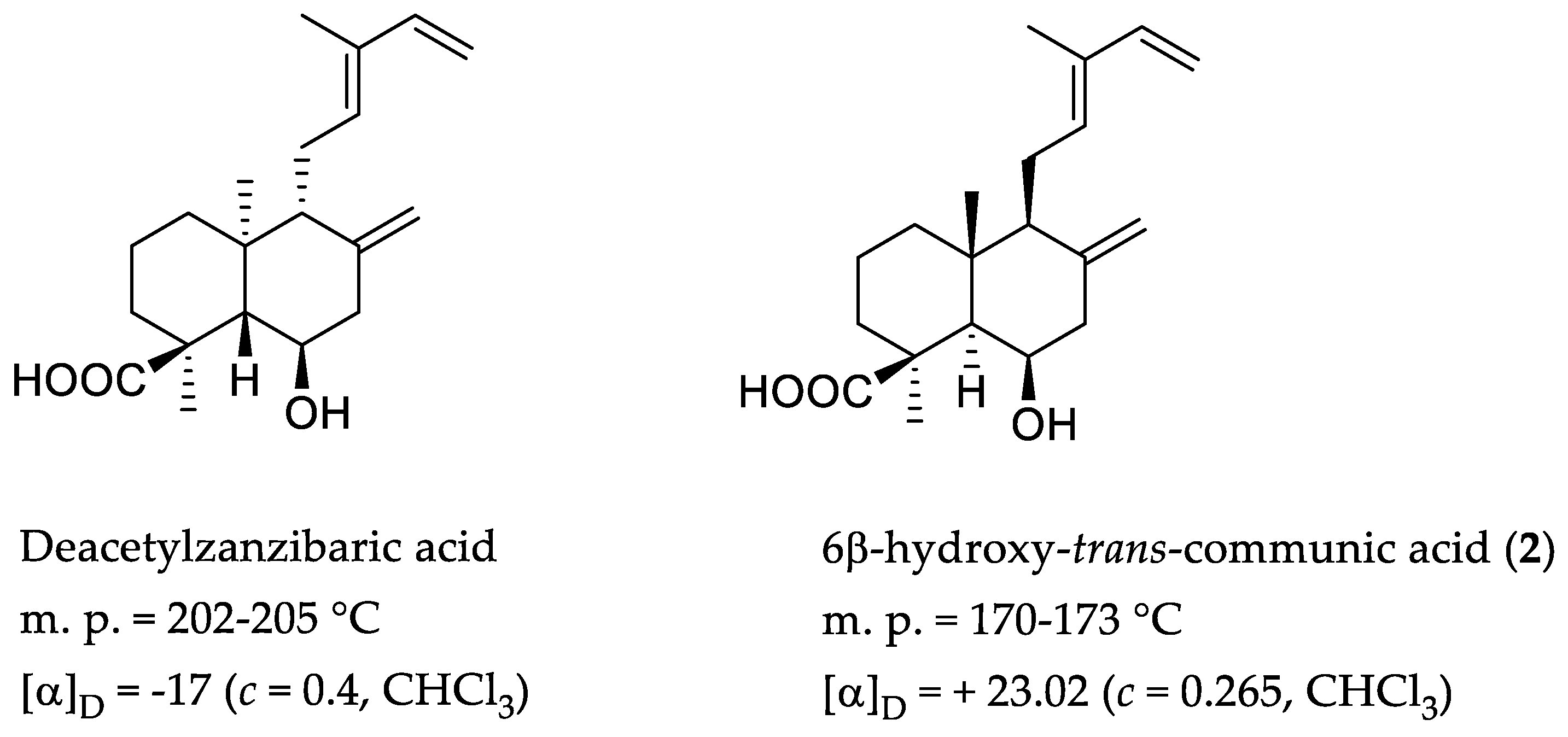
2. Results and Discussion
2.1. Biological Activity
2.1.1. Anti-Inflammatory Activity
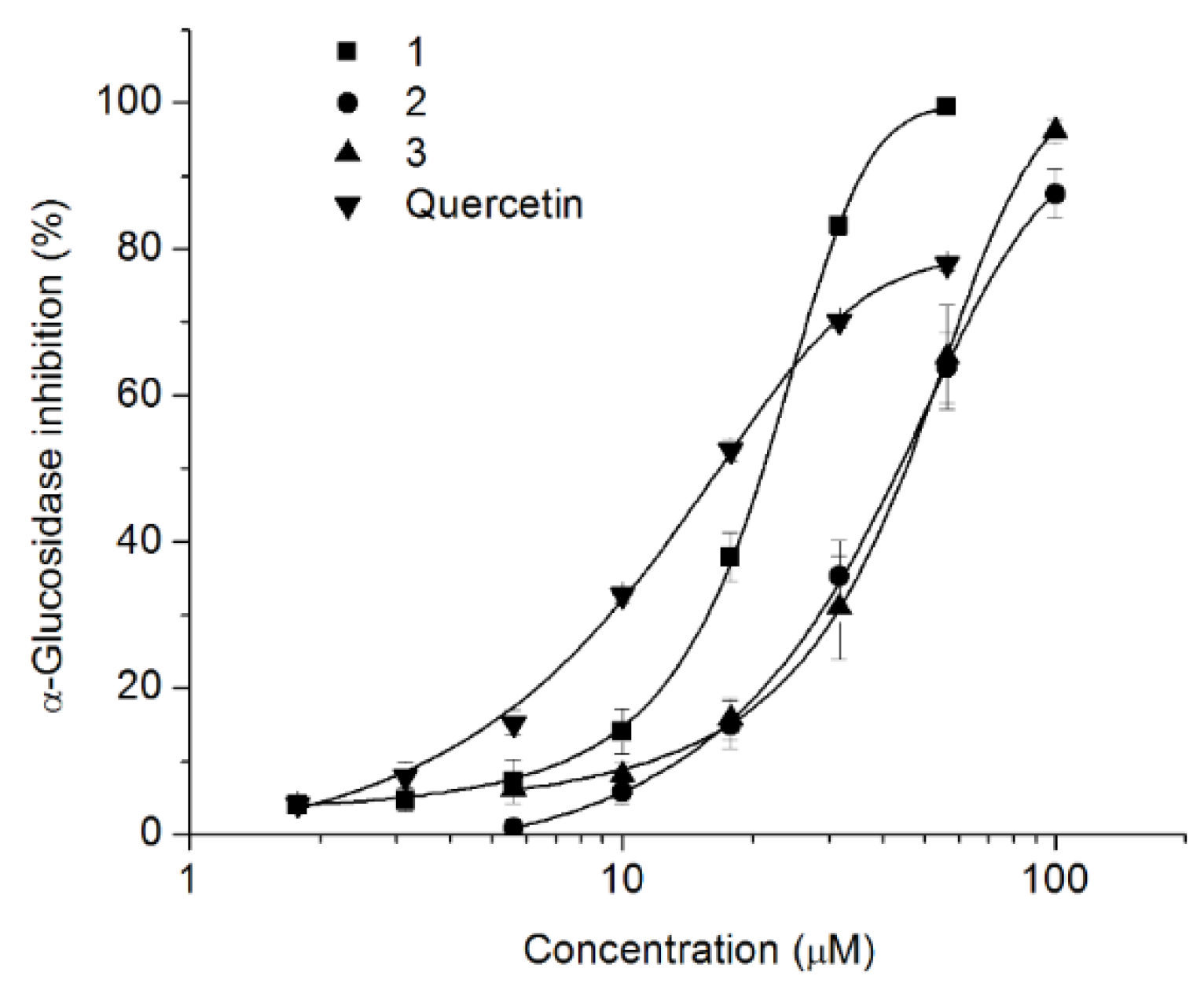
2.1.2. Inhibition of α-Glucosidase Activity
3. Materials and Methods
3.1. Experimental
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Computational Details
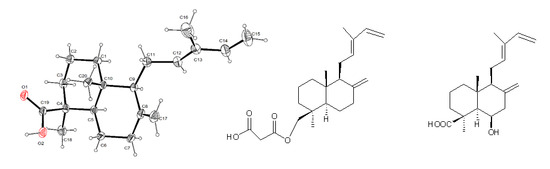
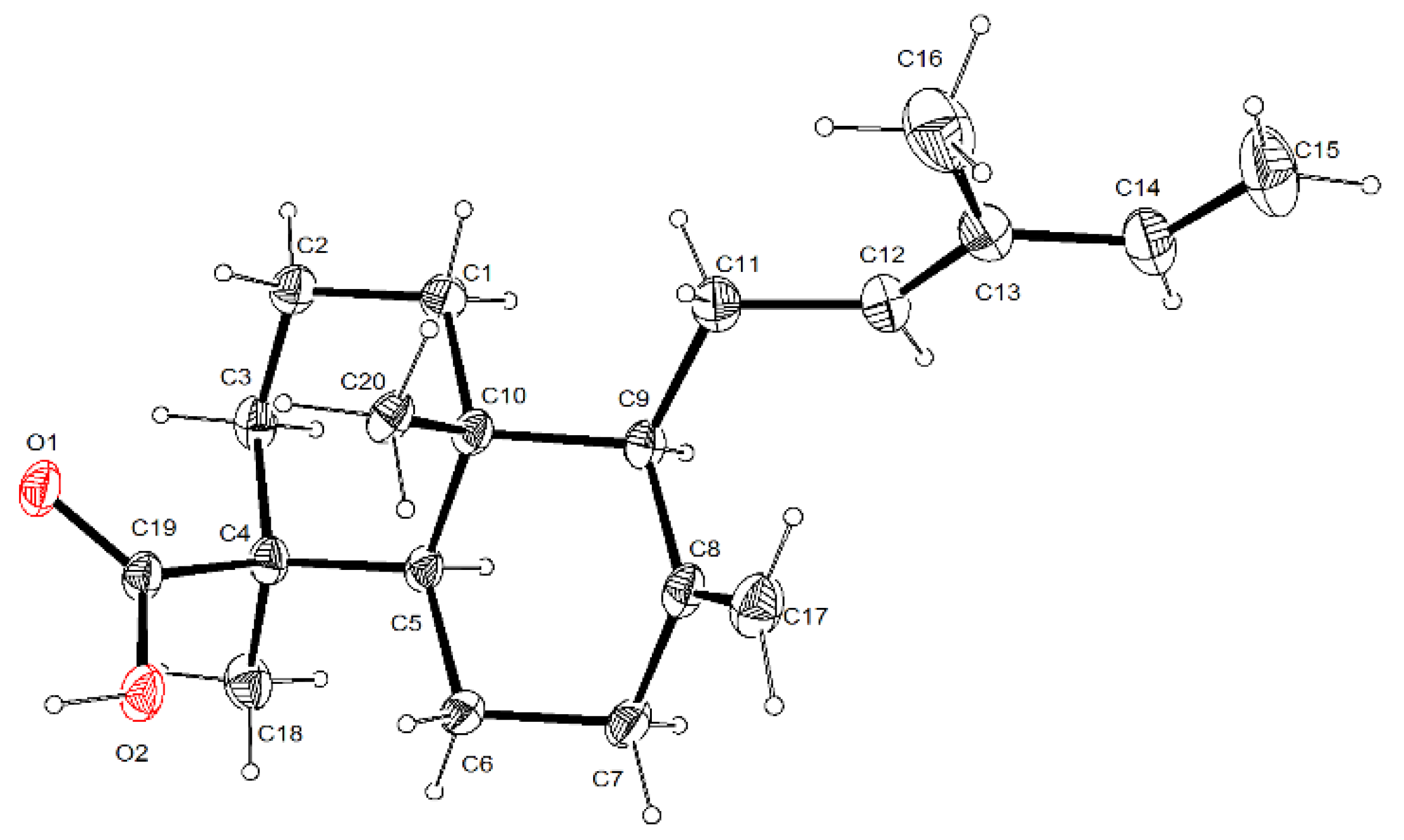
3.5. Single-Crystal X-ray Diffraction Analysis for Trans-Communic Acid (3)
3.6. TPA-Induced Edema Model
3.7. Inhibition of α-Glucosidase
3.7.1. Inhibition of Yeast α-Glucosidase
3.7.2. Mammalian α-Glucosidadse Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gonzalez-Gallegos, J.G.; Aguilar-Santelises, R. Salvia tilantongensis (Lamiaceae), una especie nueva de la Mixteca alta de Oaxaca, México. Acta Bot. 2014, 109, 1–22. [Google Scholar] [CrossRef][Green Version]
- Jenks, A.A.; Kim, S.C. Medicinal plant complexes of Salvia subgenus Calosphace: An ethnobotanical study of new world sages. J. Ethnopharmacol. 2013, 146, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Cornejo-Tenorio, G.; Ibarra-Manríquez, G. Diversidad y distribución del género Salvia (Lamiaceae) en Michoacán, México Diversity and distribution of the genus Salvia (Lamiaceae) in Michoacan, Mexico. Rev. Mex. Biodivers. 2011, 82, 1279–1296. [Google Scholar]
- De La Cruz-Jiménez, L.; Guzmán-Lucio, M.; Viveros-Valdez, E. Traditional medicinal plants used for the treatment of gastrointestinal diseases in Chiapas, México. World Appl. Sci. J. 2014, 31, 508–515. [Google Scholar]
- Ascrizzi, R.; Cioni, P.L.; Amadei, L.; Maccioni, S.; Flamini, G. Geographical patterns of in vivo spontaneously emitted volatile organic compounds in Salvia species. Microchem. J. 2017, 133, 13–21. [Google Scholar] [CrossRef]
- Bisio, A.; Ciarallo, G.; Romussi, G.; Fontana, N.; Mascolo, N.; Capasso, R.; Biscardi, D. Chemical composition of essential oils from some Salvia species. Phyther. Res. 1998, 12, 117–120. [Google Scholar] [CrossRef]
- Romussi, G.; Ciarallo, G.; Bisio, A.; Fontana, N.; De Simone, F.; De Tommasi, N.; Mascolo, N.; Pinto, L. A new diterpenoid with antispasmodic activity from Salvia cinnabarina. Planta Med. 2001, 67, 153–155. [Google Scholar] [CrossRef]
- Bisio, A.; Pagano, B.; Romussi, A.; Bruno, O.; De Tommasi, N.; Romussi, G.; Mattia, C.A. Relative stereochemistry of a diterpene from Salvia cinnabarina. Molecules 2007, 12, 2279–2287. [Google Scholar] [CrossRef]
- Capasso, R.; Izzo, A.A.; Romussi, G.; Capasso, F.; De Tommasi, N.; Bisio, A.; Mascolo, N.A. Secoisopimarane diterpenoid from Salvia cinnabarina inhibits rat urinary bladder contractility in vitro. Planta Med. 2004, 70, 185–188. [Google Scholar]
- Capasso, R.; Izzo, A.A.; Capasso, F.; Romussi, G.; Bisio, A.; Mascolo, N.A. Diterpenoid from Salvia cinnabarina inhibits mouse intestinal motility in vivo. Planta Med. 2004, 70, 375–377. [Google Scholar]
- Alieri, A.; Maione, F.; Bisio, A.; Romussi, G.; Mascolo, N.; Cicala, C. Effect of a diterpenoid from Salvia cinnabarina on arterial blood pressure in rats. Phyther. Res. 2007, 21, 690–692. [Google Scholar] [CrossRef] [PubMed]
- Maione, F.; Bonito, M.C.; Colucci, M.; Cozzolino, V.; Bisio, A.; Romussi, G.; Cicala, C.; Pieretti, S.; Mascolo, N. First evidence for an anxiolytic effect of a diterpenoid from Salvia cinnabarina. Nat. Prod. Commun. 2009, 4, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Di Sotto, A.; Mastrangelo, S.; Romussi, G.; Bisio, A.; Mazzanti, G. Antimutagenic activity of a secoisopimarane diterpenoid from Salvia cinnabarina M. Martens et Galeotti in the bacterial reverse mutation assay. Food Chem. Toxicol. 2009, 47, 2092–2096. [Google Scholar] [CrossRef]
- Di Sotto, A.; Carbone, F.; Hrelia, P.; Maffei, F.; Castelli, F.; Sarpietro, M.G.; Mazzanti, G. Anticlastogenic effect in human lymphocytes by the sodium Salt of 3,4-secoisopimar-4(18),7,15-trien-3-oic acid. J. Nat. Prod. 2012, 75, 1294–1298. [Google Scholar] [CrossRef] [PubMed]
- Bustos-Brito, C.; Joseph-Nathan, P.; Burgueño-Tapia, E.; Martínez-Otero, D.; Nieto-Camacho, A.; Calzada, F.; Yépez-Mulia, L.; Esquivel, B.; Quijano, L. Structure and Absolute Configuration of Abietane Diterpenoids from Salvia clinopodioides: Antioxidant, Antiprotozoal, and Antipropulsive Activities. J. Nat. Prod. 2019, 82, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Arya, V.P.; Erdtman, H.; Kubota, T. Chemistry of the natural order Cupressales-41. The structure and stereochemistry of communic acid. Tetrahedron 1961, 16, 255–263. [Google Scholar] [CrossRef]
- Garbarino, J.A.; Molinari, A. A labdane diterpene from Calcolaria corymbosa. J. Nat. Prod. 1993, 56, 624–626. [Google Scholar] [CrossRef]
- Garbarino, J.A.; Molinari, A. Labdane diterpenes from Calceolaria densifolia. J. Nat. Prod. 1992, 55, 744–747. [Google Scholar] [CrossRef]
- Hansen, E.W.; Ruoff, P. Estimation of malonic acid and methylmalonic acid enolization rate constants by an isotopic-exchange reaction using 1H NMR spectroscopy. J. Phys. Chem. 1988, 92, 2641–2645. [Google Scholar] [CrossRef]
- Bohlmann, F.; Zdero, C.; Robinson, H.; King, R.M. Ein neues germacren-derivat sowie ein diterpenmalonat aus Baccharis-arten. Phytochemistry 1979, 18, 1993–1996. [Google Scholar] [CrossRef]
- Toyota, M.; Asakawa, Y. Diterpenoid constituents of the liverwort Nardia subclavata. Phytochemistry 1993, 34, 751–753. [Google Scholar] [CrossRef]
- Langenbahn, U.; Burkhardt, G.; Becker, H. Diterpene malonates and other terpenes from Nardia succulenta and N. scalaris. Phytochemistry 1993, 33, 1173–1179. [Google Scholar] [CrossRef]
- Urones, J.G.; Marcos, I.S.; Cubillo, I.; Garrido, N.M.; Basabe, P. Terpenoid compounds from Parentucellia latifolia. Phytochemistry 1990, 29, 2223–2228. [Google Scholar] [CrossRef]
- Urones, J.G.; Marcos, S.; Ferreras, J.F.; Barcala, P.B. Terpenoids from Nepeta tuberosa subsp. reticulata. Phytochemistry 1988, 27, 523–526. [Google Scholar] [CrossRef]
- King, R.M.; Zdero, C.; Bohlmann, F.; Paz, L.; Botanical, M.; Index-baccharis, W. Ent-clerodanes and other constituents from bolivian Baccharis species. Phytochemistry 1989, 28, 531–542. [Google Scholar]
- Bohlmann, F.; Wegner, P. Ent-beyer-15-ene derivatives from Nidorella anomala. Phytochemistry 1982, 21, 1175–1177. [Google Scholar] [CrossRef]
- Labbe, C.; Castillo, M.; Hernandez, M. Diterpenoids from Baccharis lejia. Phytochemistry 1991, 30, 1607–1611. [Google Scholar] [CrossRef]
- Hugel, G.O.G. Diterpenes de Trachylobium. IV.-Structure et stereochemie de l’acide zanzibarique. Bull. Soc. Chim. Fr. 1965, 10, 2903–2908. [Google Scholar]
- Fukushima, J.I.; Yatagai, M.; Ohira, T. Abietane-type and labdane-type diterpenoids from the cones of Chamaecyparis obtusa. J. Wood Sci. 2002, 48, 326–330. [Google Scholar] [CrossRef]
- Barrero, A.F.; Herrador, M.M.; Arteaga, P.; Arteaga, J.F.; Arteaga, A.F. Communic acids: Occurrence, properties and use as chirons for the synthesis of bioactive compounds. Molecules 2012, 17, 1448–1467. [Google Scholar] [CrossRef]
- Arya, V.P.; Enzell, C.; Erdtaman, H.; Kubota, T. Communic acid, a new diterpene acid from Juniperus communis L. Acta Chem. Scand. 1961, 15, 225–226. [Google Scholar] [CrossRef]
- Lee, G.H.; Lin, C.C.; Cheng, Y.S.; Peng, S.M. Structure of methyl trans-communate. Acta Crystallogr. 1987, C43, 1382–1384. [Google Scholar]
- Wu, Y.B.; Ni, Z.Y.; Shi, Q.W.; Dong, M.; Kiyota, H.; Gu, Y.C.; Cong, B. Constituents from Salvia species and their biological activities. Chem. Rev. 2012, 112, 5967–6026. [Google Scholar] [CrossRef] [PubMed]
- Habibi, Z.; Eftekhar, F.; Samiee, K.; Rustaiyan, A. Structure and antibacterial activity of a new labdane diterpenoid from Salvia leriaefolia. J. Nat. Prod. 2000, 63, 270–271. [Google Scholar] [CrossRef] [PubMed]
- Jassbi, A.R.; Eghtesadi, F.; Hazeri, N.; Ma’sumi, H.; Valizadeh, J.; Chandran, J.N.; Schneider, B.; Baldwin, I.T. The roots of Salvia rhytidea: A rich source of biologically active diterpenoids. Nat. Prod. Res. 2017, 31, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Moridi Farimani, M.; Miran, M. Labdane diterpenoids from Salvia reuterana. Phytochemistry 2014, 108, 264–269. [Google Scholar] [CrossRef]
- Shpatov, A.V.; Popov, S.A.; Salnikova, O.I.; Khokhrina, E.A.; Shmidt, E.N.; Um, B.H. Low-volatile lipophilic compounds in needles, defoliated twigs, and outer bark of Pinus thunbergii. Nat. Prod. Commun. 2013, 8, 1759–1762. [Google Scholar] [CrossRef]
- Lin, T.C.; Fang, J.M.; Cheng, Y.S. Terpenes and lignans from leaves of Chamaecyparis formosensis. Phytochemistry 1999, 51, 793–801. [Google Scholar] [CrossRef]
- Kitajima, J.; Noda, N.; Ida, Y.; Komori, T.; Kawasaki, T. Studies on the constituents of the crude drug “Fritillariae bulbus.” IV. On the diterpenoid constituents of the crude drug “Fritillariae bulbus.”. Chem. Pharm. Bull. 1982, 30, 3922–3931. [Google Scholar] [CrossRef]
- Poulin, J.; Helwig, K. Inside amber: New insights into the macromolecular structure of Class Ib resinite. Org. Geochem. 2015, 86, 94–106. [Google Scholar] [CrossRef]
- Lago, J.H.G.; Brochini, C.B.; Roque, N.F. Terpenes from leaves of Guarea macrophylla (Meliaceae). Phytochemistry 2000, 55, 727–731. [Google Scholar] [CrossRef]
- Rather, M.A.; Hassan, T. Analysis of the diterpene rich essential oil of Nepeta clarkei hooke. from Kashmir himalayas by capillary GC-MS. Int. J. ChemTech Res. 2011, 3, 959–962. [Google Scholar]
- Zhao, J.; Zhu, H.J.; Zhou, X.J.; Yang, T.H.; Wang, Y.Y.; Su, J.; Li, Y.; Cheng, Y.X. Diterpenoids from the feces of Trogopterus xanthipes. J. Nat. Prod. 2010, 73, 865–869. [Google Scholar] [CrossRef]
- Bao, Y.; Wang, W.; Wu, H.; Qi, M.; Li, J.; Yang, Y. A new sesquiterpene from the barks of Manglietia hookeri. Nat. Prod. Res. 2016, 30, 2396–2401. [Google Scholar] [CrossRef]
- Koroch, A.R.; Simon, J.E.; Juliani, H.R. Essential oil composition of purple basils, their reverted green varieties (Ocimum basilicum) and their associated biological activity. Ind. Crops Prod. 2017, 107, 526–530. [Google Scholar] [CrossRef]
- Aydoǧmuş, Z.; Yeşilyurt, V.; Topcu, G. Constituents of Salvia microphylla. Nat. Prod. Res. 2006, 20, 775–781. [Google Scholar] [CrossRef]
- Archile, B.O.K.; Mathieu, T.; Alembert, T.T.; Pierre, T.; Michel, F. eacute d eacute rich Terpenoids from Phaulopsis imbricata (Acanthaceae). J. Med. Plants Res. 2016, 10, 122–129. [Google Scholar] [CrossRef][Green Version]
- Ulubelen, A.; Öztürk, S.; Iśildatici, S. A new flavone from Salvia triloba L.f (Labiatae). J. Pharm. Sci. 1968, 57, 1037–1038. [Google Scholar] [CrossRef]
- Lehbili, M.; Alabdul Magid, A.; Kabouche, A.; Voutquenne-Nazabadioko, L.; Abedini, A.; Morjani, H.; Gangloff, S.C.; Kabouche, Z. Antibacterial, antioxidant and cytotoxic activities of triterpenes and flavonoids from the aerial parts of Salvia barrelieri Etl. Nat. Prod. Res. 2018, 32, 2683–2691. [Google Scholar] [CrossRef]
- Hasan, M.R.; Al-Jaber, H.I.; Al-Qudah, M.A.; Abu Zarga, M.H. New sesterterpenoids and other constituents from Salvia Dominica growing wild in Jordan. Phytochem. Lett. 2016, 16, 12–17. [Google Scholar] [CrossRef]
- Srivedavyasasri, R.; Hayes, T.; Ross, S.A. Phytochemical and biological evaluation of Salvia apiana. Nat. Prod. Res. 2017, 31, 2058–2061. [Google Scholar] [CrossRef]
- Mofidi Tabatabaei, S.; Salehi, P.; Moridi Farimani, M.; Neuburger, M.; De Mieri, M.; Hamburger, M.; Nejad-Ebrahimi, S. A nor-diterpene from Salvia sahendica leaves. Nat. Prod. Res. 2017, 31, 1758–1765. [Google Scholar] [CrossRef]
- Mansourabadi, A.H.; Sadeghi, H.M.; Razavi, N.; Rezvani, E. Anti-inflammatory and Analgesic Properties of Salvigenin, Salvia officinalis Flavonoid Extracted. Adv. Herb. Med. 2015, 1, 31–41. [Google Scholar]
- Singh, N.; Mahmood, U.; Kaul, V.K.; Jirovetz, L. A new phthalic acid ester from Ajuga bracteosa. Nat. Prod. Res. 2006, 20, 593–597. [Google Scholar] [CrossRef]
- Pan, Y.P.; Ye, J.; Zhang, Y.; Jin, H.Z. Chemical Constituents of Hedyotis uncinella. Chem. Nat. Compd. 2017, 53, 738–739. [Google Scholar] [CrossRef]
- Sivajothi, V.; Shruthi, S.D. In vitro and in silico anti-diabetic activity of phthalic acid isolated from phyllanthus rheedii. Int. J. Res. Ayurveda Pharm. 2013, 4, 889–892. [Google Scholar] [CrossRef]
- Wahidulla, S.; D’Souza, L.; Govenker, M. Lipid constituents of the red alga Acantophora spicifera. Phytochemistry 1998, 48, 1203–1206. [Google Scholar] [CrossRef]
- Bai, R.; Ma, F.; Liang, D.; Zhao, X. Phthalic acid induces oxidative stress and alters the activity of some antioxidant enzymes in roots of Malus prunifolia. J. Chem. Ecol. 2009, 35, 488–494. [Google Scholar] [CrossRef]
- Dorney, J.R.; Weber, J.B.; Overcash, M.R.; Strek, H.J. Plant Uptake and Soil Retention of Phthalic Acid Applied to Norfolk Sandy Loam. J. Agric. Food Chem. 1985, 33, 398–403. [Google Scholar] [CrossRef]
- Chen, J.J.; Wu, H.M.; Peng, C.F.; Chen, I.S.; Chu, S. Der seco-Abietane diterpenoids, a phenylethanoid derivative, and antitubercular constituents from Callicarpa pilosissima. J. Nat. Prod. 2009, 72, 223–228. [Google Scholar] [CrossRef]
- Ding, L.J.; Yuan, W.; Li, Y.X.; Liao, X.J.; Sun, H.; Peng, Q.; Han, B.N.; Lin, H.W.; Li, Z.Y.; Yang, F.; et al. Hypocrol A, a new tyrosol derivative from a sponge-derived strain of the fungus Hypocrea koningii. Nat. Prod. Res. 2016, 30, 1633–1638. [Google Scholar] [CrossRef]
- Enzell, C.R.; Thomas, B.R. The chemistry of the Order Araucariales 3. Structure and configuration of araucarolone and some related compounds from Agathis australis. Acta Chem. Scand. 1965, 19, 1875–1896. [Google Scholar] [CrossRef]
- Rivera-Chávez, J.; Zacatenco-Abarca, J.; Morales-Jiménez, J.; Martínez-Aviña, B.; Hernández-Ortega, S.; Aguilar-Ramírez, E. Cuautepestalorin, a 7,8-Dihydrochromene-Oxoisochromane Adduct Bearing a Hexacyclic Scaffold from Pestalotiopsis sp. IQ-011. Org. Lett. 2019, 21, 3558–3562. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic Circular Dichroism spectra. Quirality 2013, 4325, 243–249. [Google Scholar] [CrossRef]
- Rivera-Chávez, J.; Figueroa, M.; González, M.D.C.; Glenn, A.E.; Mata, R. α-Glucosidase Inhibitors from a Xylaria feejeensis Associated with Hintonia latiflora. J. Nat. Prod. 2015, 78, 730–735. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. A General Definition of Ring Puckering Coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Carlson, R.P.; Lynn, O.D.; Chang, J.; Lewis, A.J. Modulation of mouse ear edema by cyclooxygenase and lipoxygenase inhibitors and other pharmacologic agents. Agents Actions 1985, 17, 197–204. [Google Scholar] [CrossRef]
- Zhou, T.; Zhang, S.W.; Liu, S.S.; Cong, H.J.; Xuan, L.J. Daphnodorin dimers from Edgeworthia chrysantha with α-glucosidase inhibitory activity. Phytochem. Lett. 2010, 3, 242–247. [Google Scholar] [CrossRef]
- Ye, X.P.; Song, C.Q.; Yuan, P.; Mao, R.G. α-Glucosidase and α-Amylase Inhibitory Activity of Common Constituents from Traditional Chinese Medicine Used for Diabetes Mellitus. Chin. J. Nat. Med. 2010, 8, 349–352. [Google Scholar] [CrossRef]
- Jo, S.H.; Ka, E.H.; Lee, H.S.; Apostolidis, E.; Jang, H.D.; Kwon, Y.I. Comparison of antioxidant potential and rat intestinal α-glucosidases inhibitory activities of quercetin, rutin, and isoquercetin. Int. J. Appl. Res. Nat. Prod. 2009, 2, 52–60. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC | Type a | δH (J In Hz) | HMBC |
|---|---|---|---|---|
| 1a | 39.0 | CH2 | 1.83, brd (12.5) | 2, 3, 5, 20 |
| 1b | 1.13, td (12.5, 5.4) | 2, 3, 5, 10, 20 | ||
| 2 | 19.0 | CH2 | 1.52, m | 1, 3, 4 |
| 3a | 36.2 | CH2 | 1.73, brd (12.8) | 1, 2, 4, 5, 18, 19 |
| 3b | 1.04, td (12.8, 5.3) | 1, 2, 4, 18, 19 | ||
| 4 | 37.6 | C | ||
| 5 | 56.2 | CH | 1.30, brd (12.5) | 1, 6, 7, 18, 19, 20 |
| 6a | 24.3 | CH2 | 1.83, m | 5, 8 |
| 6b | 1.34, dq (12.5, 3.9) | 5, 7 | ||
| 7a | 38.3 | CH2 | 2.39, m | 5, 6, 9, 17, |
| 7b | 1.97, td (12.5, 4.6) | 5, 6, 8, 17 | ||
| 8 | 147.7 | C | ||
| 9 | 57.2 | CH | 1.75, brd (13.0) | 8, 11, 12, 17, 20 |
| 10 | 39.5 | C | ||
| 11a | 23.3 | CH2 | 2.36, m | 8, 9, 12, 13, 14,* 17 * |
| 11b | 2.14, m | 8, 9, 12, 13, 14 * | ||
| 12 | 133.8 | CH | 5.39, brt (6.2) | 9, 11, 14, 16 |
| 13 | 133.7 | C | ||
| 14 | 141.7 | CH | 6.32, dd (17.4, 10.7) | 12, 13, 16 |
| 15a | 110.1 | CH2 | 5.04, d (17.4) | 12,* 13, 14 |
| 15b | 4.88, d (10.7) | 12,* 13, 14 | ||
| 16 | 12.0 | CH3 | 1.74, s | 12, 13, 14 |
| 17a | 108.2 | CH2 | 4.82, brs | 6,* 7, 8 |
| 17b | 4.47, brs | 6,* 7, 8, 9 | ||
| 18 | 27.6 | CH3 | 0.97, s | 3, 5, 19, |
| 19a | 68.5 | CH2 | 4.38, d (10.9) | 3, 5, 3′, 18 |
| 19b | 3.95, d (10.9) | 3, 5, 3′, 18 | ||
| 20 | 15.4 | CH3 | 0.72, s | 1, 5, 9, 10, |
| 1′ | 169.6 | C | ||
| 2′ | 40.7 | CH2 | 3.41, brs | 1′, 3′, 19 |
| 3′ | 168.3 | C |
| Position | δC | Type a | δH (J In Hz) | HMBC |
|---|---|---|---|---|
| 1a | 20.2 | CH2 | 1.76, qd (14.3, 3.5) | 3 |
| 1b | 1.56, dt (14.3, 2.8) | 10 | ||
| 2a | 41.6 | CH2 | 1.88, brd (13.0) | 1, 3, 10, 20 * |
| 2b | 1.19, td (13.0, 3.7) | 1, 3, 9, 10, 20 * | ||
| 3a | 40.4 | CH2 | 2.38, brd (13.0) | 1, 2, 4 |
| 3b | 1.01, td (13.0, 3.7) | 1, 4, 5, 18, 19 | ||
| 4 | 46.6 | C | ||
| 5 | 57.5 | CH | 1.46, brs | 4, 6, 9, 10, 18, 19, 20 |
| 6 | 67.9 | CH | 4.52, brq (2.6) | 4, 5, 7, 8, 10 |
| 7a | 45.4 | CH2 | 2.50, dd (13.6, 2.6) | 5, 6, 8, 9, 17 |
| 7b | 2.34, brd (13.6) | 6, 8, 9, 17 | ||
| 8 | 133.9 | C | ||
| 9 | 56.7 | CH | 1.82, brd (11.1) | 5, 7, 8, 10, 11, 17, 20 |
| 10 | 41.3 | C | ||
| 11a | 23.4 | CH2 | 2.40, m | 8, 9, 12, 13, 15 |
| 11b | 2.23, ddd (16.6, 11.3, 6.4) | 9, 12, 13 | ||
| 12 | 132.9 | CH | 5.41, brt (6.4) | 9, 11, 14, 16 |
| 13 | 142.3 | C | ||
| 14 | 141.4 | CH | 6.32, dd (17.4, 10.8) | 12, 16 |
| 15a | 110.3 | CH2 | 5.06, d (17.4) | 12, 14 |
| 15b | 4.90, d (10.8) | 12, 14 | ||
| 16 | 11.9 | CH3 | 1.76, s | 12, 14 |
| 17a | 111.9 | CH2 | 5.03, brs | 7, 8, 9 |
| 17b | 4.76, brs | 7, 8, 9 | ||
| 18 | 28.4 | CH3 | 1.33, s | 3, 4, 5 |
| 19 | 180.2 | C | ||
| 20 | 15.8 | CH3 | 0.87, s | 9, 10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustos-Brito, C.; Nieto-Camacho, A.; Hernandez-Ortega, S.; Rivera-Chávez, J.; Quijano, L.; Esquivel, B. Structural Elucidation of Malonylcommunol and 6β-Hydroxy-trans-communic Acid, Two Undescribed Diterpenes from Salvia cinnabarina. First Examples of Labdane Diterpenoids from a Mexican Salvia Species. Molecules 2020, 25, 1808. https://doi.org/10.3390/molecules25081808
Bustos-Brito C, Nieto-Camacho A, Hernandez-Ortega S, Rivera-Chávez J, Quijano L, Esquivel B. Structural Elucidation of Malonylcommunol and 6β-Hydroxy-trans-communic Acid, Two Undescribed Diterpenes from Salvia cinnabarina. First Examples of Labdane Diterpenoids from a Mexican Salvia Species. Molecules. 2020; 25(8):1808. https://doi.org/10.3390/molecules25081808
Chicago/Turabian StyleBustos-Brito, Celia, Antonio Nieto-Camacho, Simón Hernandez-Ortega, José Rivera-Chávez, Leovigildo Quijano, and Baldomero Esquivel. 2020. "Structural Elucidation of Malonylcommunol and 6β-Hydroxy-trans-communic Acid, Two Undescribed Diterpenes from Salvia cinnabarina. First Examples of Labdane Diterpenoids from a Mexican Salvia Species" Molecules 25, no. 8: 1808. https://doi.org/10.3390/molecules25081808
APA StyleBustos-Brito, C., Nieto-Camacho, A., Hernandez-Ortega, S., Rivera-Chávez, J., Quijano, L., & Esquivel, B. (2020). Structural Elucidation of Malonylcommunol and 6β-Hydroxy-trans-communic Acid, Two Undescribed Diterpenes from Salvia cinnabarina. First Examples of Labdane Diterpenoids from a Mexican Salvia Species. Molecules, 25(8), 1808. https://doi.org/10.3390/molecules25081808