How Computational Chemistry and Drug Delivery Techniques Can Support the Development of New Anticancer Drugs



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Design of Anticancer Small Organic Molecules
2.1. Selected Examples of Anticancer Small Molecules Design
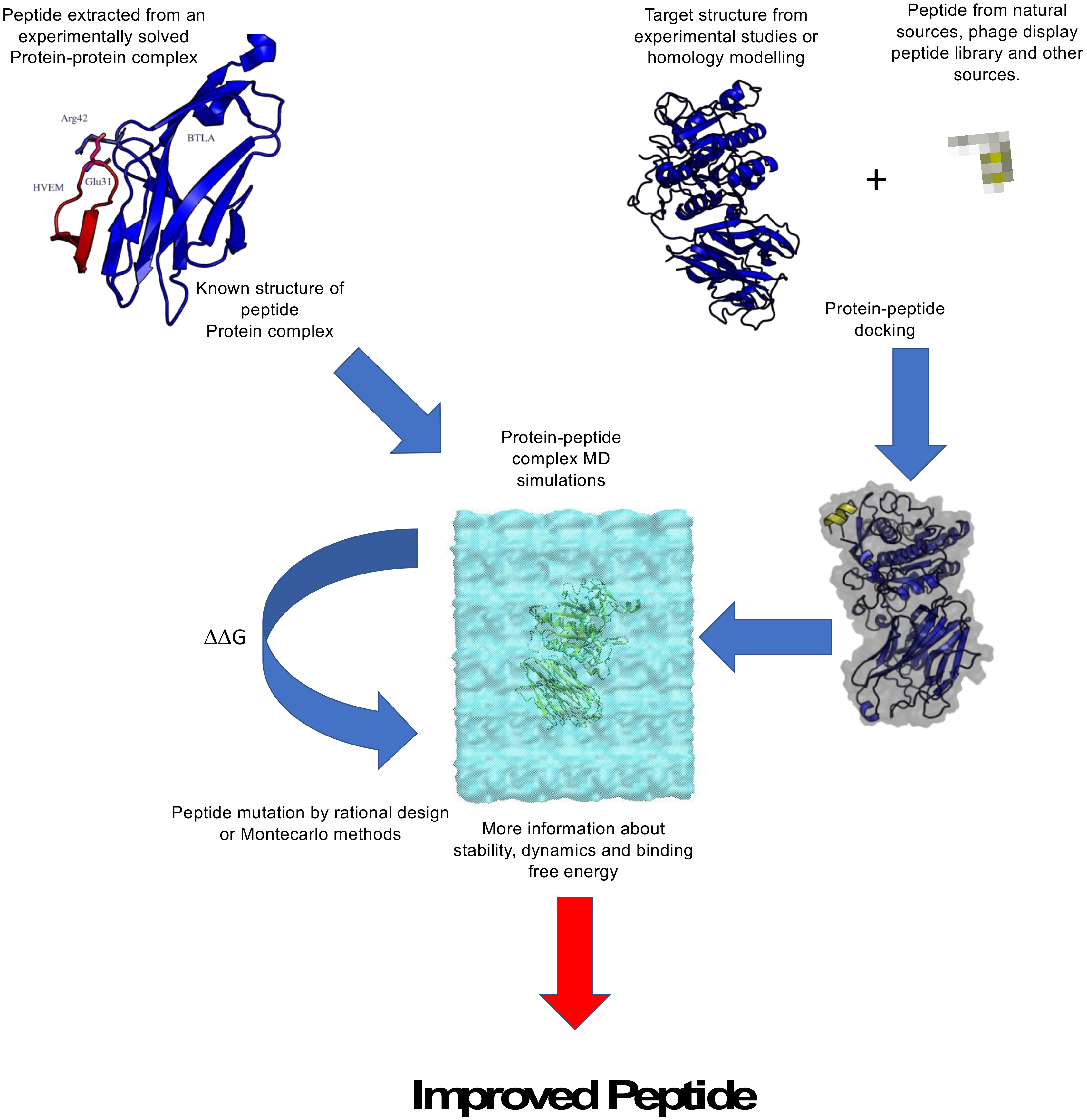
2.2. Computational Design of Anticancer Peptides
2.3. Selected Examples of Anticancer Peptide Design
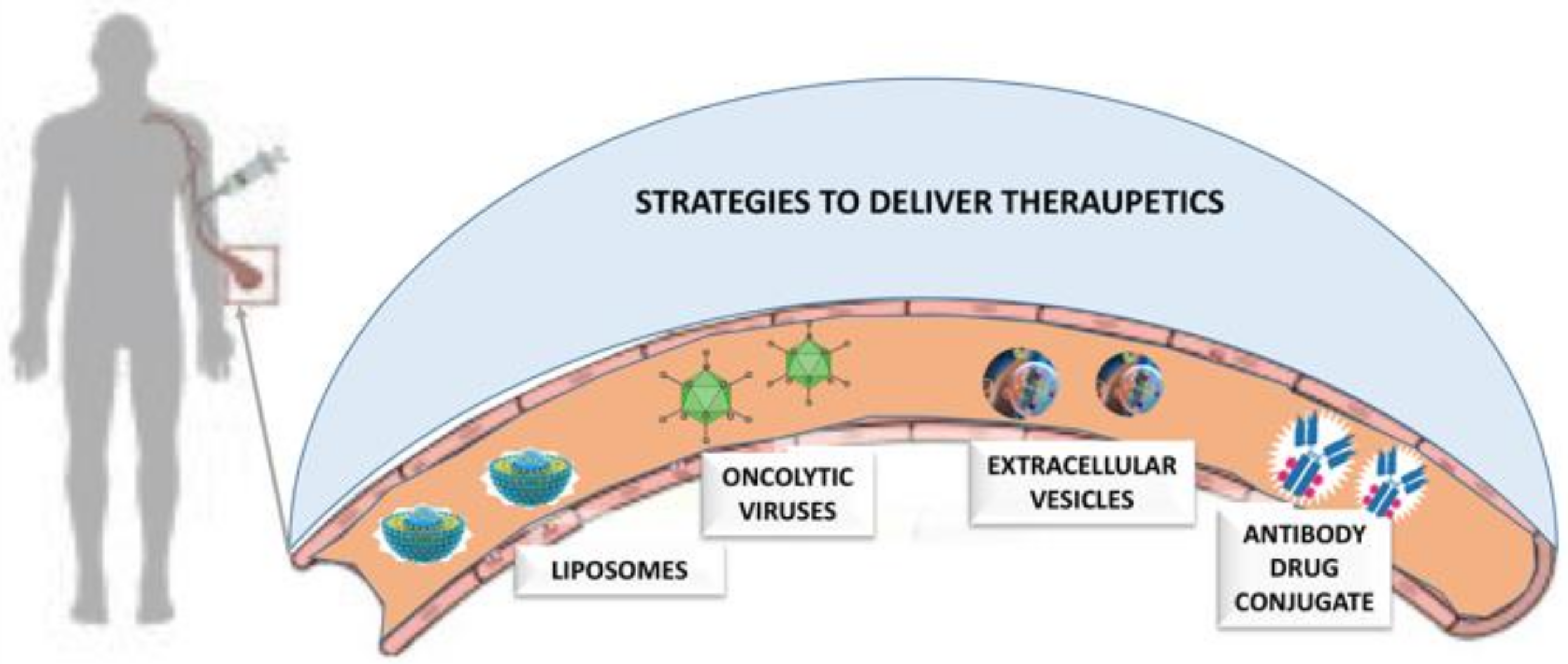
3. Advanced Drug Delivery Approaches
3.1. Oncolytic Viruses in Drug Delivery
3.2. Liposomes
3.3. Extracellular Vesicles and Cancer Drug Delivery
3.4. Antibody-Drug Conjugates
4. Computational Chemistry in the Design of DDS
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Xiaomei, M.; Herbert, Y. Global Burden of Cancer. Yale J. Biol. Med. 2006, 79, 85–94. [Google Scholar]
- Mullard, A. 2019 FDA drug approvals. Nat. Rev. Drug Discov. 2020, 19, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Kurrikoff, K.; Aphkhazava, D.; Langel, Ü. The future of peptides in cancer treatment. Curr. Opin. Pharmacol. 2019, 47, 27–32. [Google Scholar] [CrossRef]
- Lammi, C.; Zanoni, C.; Aiello, G.; Arnoldi, A.; Grazioso, G. Lupin Peptides Modulate the Protein-Protein Interaction of PCSK9 with the Low Density Lipoprotein Receptor in HepG2 Cells. Sci. Rep. 2016. [Google Scholar] [CrossRef]
- Geng, H.; Chen, F.; Ye, J.; Jiang, F. Applications of Molecular Dynamics Simulation in Structure Prediction of Peptides and Proteins. Comput. Struct. Biotechnol. J. 2019, 17, 1162–1170. [Google Scholar] [CrossRef]
- Jiang, F.; Geng, H. Computational Methods for Studying Conformational Behaviors of Cyclic Peptides. Methods Mol. Biol. 2019, 2001, 61–71. [Google Scholar] [CrossRef]
- Lavecchia, A. Deep learning in drug discovery: Opportunities, challenges and future prospects. Drug. Discov. Today 2019, 24, 2017–2032. [Google Scholar] [CrossRef]
- Zhao, H.; Caflisch, A. Molecular dynamics in drug design. Eur. J. Med. Chem. 2015, 91, 4–14. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Williams-Noonan, B.J.; Yuriev, E.; Chalmers, D.K. Free Energy Methods in Drug Design: Prospects of “Alchemical Perturbation” in Medicinal Chemistry. J. Med. Chem. 2018, 61, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Damiati, S.A.; Alaofi, A.L.; Dhar, P.; Alhakamy, N.A. Novel machine learning application for prediction of membrane insertion potential of cell-penetrating peptides. Int. J. Pharm. 2019. [Google Scholar] [CrossRef]
- Neuhaus, C.S.; Gabernet, G.; Steuer, C.; Root, K.; Hiss, J.A.; Zenobi, R.; Schneider, G. Simulated Molecular Evolution for Anticancer Peptide Design. Angew. Chem. Int. Ed. Engl. 2019, 58, 1674–1678. [Google Scholar] [CrossRef]
- Gabernet, G.; Gautschi, D.; Muller, A.T.; Neuhaus, C.S.; Armbrecht, L.; Dittrich, P.S.; Hiss, J.A.; Schneider, G. In silico design and optimization of selective membranolytic anticancer peptides. Sci. Rep. 2019. [Google Scholar] [CrossRef]
- Maltarollo, V.G.; Gertrudes, J.C.; Oliveira, P.R.; Honorio, K.M. Applying machine learning techniques for ADME-Tox prediction: A review. Expert Opin. Drug. Metab. Toxicol. 2015, 11, 259–271. [Google Scholar] [CrossRef]
- Ghaemi, Z.; Alberga, D.; Carloni, P.; Laio, A.; Lattanzi, G. Permeability Coefficients of Lipophilic Compounds Estimated by Computer Simulations. J. Chem. Theory Comput. 2016, 12, 4093–4099. [Google Scholar] [CrossRef]
- Bocci, G.; Carosati, E.; Vayer, P.; Arrault, A.; Lozano, S.; Cruciani, G. ADME-Space: A new tool for medicinal chemists to explore ADME properties. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Lee, A.C.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019. [Google Scholar] [CrossRef]
- Van Montfort, R.L.M.; Workman, P. Structure-based drug design: Aiming for a perfect fit. Essays Biochem. 2017, 61, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Griffith, R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, 395–401. [Google Scholar] [CrossRef]
- Sgrignani, J.; Novati, B.; Colombo, G.; Grazioso, G. Covalent docking of selected boron-based serine beta-lactamase inhibitors. J. Comput. Aided Mol. Des. 2015, 29, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef]
- Magistrato, A.; Sgrignani, J.; Krause, R.; Cavalli, A. Single or Multiple Access Channels to the CYP450s Active Site? An Answer from Free Energy Simulations of the Human Aromatase Enzyme. J. Phys. Chem. Lett. 2017, 8, 2036–2042. [Google Scholar] [CrossRef]
- Do, P.-C.; Lee, E.H.; Le, L. Steered Molecular Dynamics Simulation in Rational Drug Design. J. Chem. Inf. Mod. 2018, 58, 1473–1482. [Google Scholar] [CrossRef]
- Cavalli, A.; Spitaleri, A.; Saladino, G.; Gervasio, F.L. Investigating Drug–Target Association and Dissociation Mechanisms Using Metadynamics-Based Algorithms. Acc. Chem. Res. 2015, 48, 277–285. [Google Scholar] [CrossRef]
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. Exploring Protein-Peptide Recognition Pathways Using a Supervised Molecular Dynamics Approach. Structure 2017, 25, 655–662. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Chessari, G.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Nissink, J.W.; Taylor, R.D.; Taylor, R. Modeling water molecules in protein-ligand docking using GOLD. J. Med. Chem. 2005, 48, 6504–6515. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, A.; Deganutti, G.; Salmaso, V.; Sturlese, M.; Moro, S. AquaMMapS: An Alternative Tool to Monitor the Role of Water Molecules During Protein-Ligand Association. ChemMedChem 2018, 13, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Sgrignani, J.; De Luca, F.; Torosyan, H.; Docquier, J.D.; Duan, D.; Novati, B.; Prati, F.; Colombo, G.; Grazioso, G. Structure-based approach for identification of novel phenylboronic acids as serine-beta-lactamase inhibitors. J. Comput. Aided Mol. Des. 2016, 30, 851–861. [Google Scholar] [CrossRef]
- Catto, C.; Grazioso, G.; Dell’Orto, S.; Gelain, A.; Villa, S.; Marzano, V.; Vitali, A.; Villa, F.; Cappitelli, F.; Forlani, F. The response of Escherichia coli biofilm to salicylic acid. Biofouling 2017, 33, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Sgrignani, J.; Bonaccini, C.; Grazioso, G.; Chioccioli, M.; Cavalli, A.; Gratteri, P. Insights into docking and scoring neuronal alpha4beta2 nicotinic receptor agonists using molecular dynamics simulations and QM/MM calculations. J. Comput. Chem. 2009, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Chaskar, P.; Zoete, V.; Rohrig, U.F. On-the-Fly QM/MM Docking with Attracting Cavities. J. Chem. Inf. Model. 2017, 57, 73–84. [Google Scholar] [CrossRef]
- Rastelli, G.; Pinzi, L. Refinement and Rescoring of Virtual Screening Results. Front. Chem. 2019. [Google Scholar] [CrossRef]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Almlof, M.; Brandsdal, B.O.; Aqvist, J. Binding affinity prediction with different force fields: Examination of the linear interaction energy method. J. Comput. Chem. 2004, 25, 1242–1254. [Google Scholar] [CrossRef]
- Brandsdal, B.O.; Osterberg, F.; Almlof, M.; Feierberg, I.; Luzhkov, V.B.; Aqvist, J. Free energy calculations and ligand binding. Adv. Protein Chem. 2003, 66, 123–158. [Google Scholar]
- Aqvist, J.; Marelius, J. The linear interaction energy method for predicting ligand binding free energies. Comb. Chem. High. Throughput Screen. 2001, 4, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Gagic, Z.; Ruzic, D.; Djokovic, N.; Djikic, T.; Nikolic, K. In silico Methods for Design of Kinase Inhibitors as Anticancer Drugs. Front. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Krishna, S.; Siddiqi, M.I. Virtual screening strategies: Recent advances in the identification and design of anti-cancer agents. Methods 2015, 71, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Sgrignani, J.; Magistrato, A. Influence of the membrane lipophilic environment on the structure and on the substrate access/egress routes of the human aromatase enzyme. A computational study. J. Chem. Inf. Model. 2012, 52, 1595–1606. [Google Scholar] [CrossRef]
- Ritacco, I.; Saltalamacchia, A.; Spinello, A.; Ippoliti, E.; Magistrato, A. All-Atom Simulations Disclose How Cytochrome Reductase Reshapes the Substrate Access/Egress Routes of Its Partner CYP450s. J. Phys. Chem. Lett. 2020, 11, 1189–1193. [Google Scholar] [CrossRef]
- Gobbi, S.; Rampa, A.; Belluti, F.; Bisi, A. Nonsteroidal aromatase inhibitors for the treatment of breast cancer: An update. Anticancer Agents Med. Chem. 2014, 14, 54–65. [Google Scholar] [CrossRef]
- Favia, A.D.; Nicolotti, O.; Stefanachi, A.; Leonetti, F.; Carotti, A. Computational methods for the design of potent aromatase inhibitors. Expert. Opin. Drug. Discov. 2013, 8, 395–409. [Google Scholar] [CrossRef]
- Liu, J.; Flockhart, P.J.; Lu, D.; Lv, W.; Lu, W.J.; Han, X.; Cushman, M.; Flockhart, D.A. Inhibition of cytochrome p450 enzymes by the e- and z-isomers of norendoxifen. Drug Metab. Dispos. 2013, 41, 1715–1720. [Google Scholar] [CrossRef]
- Lu, W.J.; Desta, Z.; Flockhart, D.A. Tamoxifen metabolites as active inhibitors of aromatase in the treatment of breast cancer. Breast. Cancer Res. Treat. 2011, 131, 473–481. [Google Scholar] [CrossRef]
- Lv, W.; Liu, J.; Lu, D.; Flockhart, D.A.; Cushman, M. Synthesis of Mixed (E,Z)-, (E)-, and (Z)-Norendoxifen with Dual Aromatase Inhibitory and Estrogen Receptor Modulatory Activities. J. Med. Chem. 2013, 56, 4611–4618. [Google Scholar] [CrossRef]
- Sgrignani, J.; Bon, M.; Colombo, G.; Magistrato, A. Computational approaches elucidate the allosteric mechanism of human aromatase inhibition: A novel possible route to Small-molecule regulation of CYP450s activities? J. Chem. Inf. Model. 2014, 54, 2856–2868. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T. New Method for Fast and Accurate Binding-site Identification and Analysis. Chem. Biol. Drug. Des. 2007, 69, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Mod. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Martini, S.; Berti, F.; Pennati, M.; Pavlin, M.; Sgrignani, J.; Grazioso, G.; Colombo, G.; Zaffaroni, N.; Magistrato, A. Rational design of allosteric modulators of the aromatase enzyme: An unprecedented therapeutic strategy to fight breast cancer. Eur. J. Med. Chem. 2019, 168, 253–262. [Google Scholar] [CrossRef]
- Caciolla, J.; Spinello, A.; Martini, S.; Bisi, A.; Zaffaroni, N.; Gobbi, S.; Magistrato, A. Targeting Orthosteric and Allosteric Pockets of Aromatase via Dual-Mode Novel Azole Inhibitors. ACS Med. Chem. Lett. 2020. [Google Scholar] [CrossRef]
- Regan, M.C.; Horanyi, P.S.; Pryor, E.E., Jr.; Sarver, J.L.; Cafiso, D.S.; Bushweller, J.H. Structural and dynamic studies of the transcription factor ERG reveal DNA binding is allosterically autoinhibited. Proc. Natl. Acad. Sci. USA 2013, 110, 13374–13379. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, S.B.; Johnson, A.P. eHiTS: A new fast, exhaustive flexible ligand docking system. J. Mol. Graph. Model. 2007, 26, 198–212. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Ciemny, M.; Kurcinski, M.; Kamel, K.; Kolinski, A.; Alam, N.; Schueler-Furman, O.; Kmiecik, S. Protein–peptide docking: Opportunities and challenges. Drug Discov. Today 2018, 23, 1530–1537. [Google Scholar] [CrossRef]
- Lammi, C.; Sgrignani, J.; Arnoldi, A.; Grazioso, G. Biological Characterization of Computationally Designed Analogs of peptide TVFTSWEEYLDWV (Pep2-8) with Increased PCSK9 Antagonistic Activity. Sci. Rep. 2019. [Google Scholar] [CrossRef]
- Lammi, C.; Sgrignani, J.; Roda, G.; Arnoldi, A.; Grazioso, G. Inhibition of PCSK9(D374Y)/LDLR Protein-Protein Interaction by Computationally Designed T9 Lupin Peptide. ACS Med. Chem. Lett. 2019, 10, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Garton, M.; Corbi-Verge, C.; Hu, Y.; Nim, S.; Tarasova, N.; Sherborne, B.; Kim, P.M. Rapid and accurate structure-based therapeutic peptide design using GPU accelerated thermodynamic integration. Proteins 2019, 87, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Bhachoo, J.; Beuming, T. Investigating Protein-Peptide Interactions Using the Schrodinger Computational Suite. Methods Mol. Biol. 2017, 1561, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, F.S.; Tomassi, S.; Di Maro, S.; Reichart, F.; Notni, J.; Dangi, A.; Marelli, U.K.; Brancaccio, D.; Merlino, F.; Wester, H.J.; et al. From a Helix to a Small Cycle: Metadynamics-Inspired alphavbeta6 Integrin Selective Ligands. Angew. Chem. Int. Ed. Engl. 2018, 57, 14645–14649. [Google Scholar] [CrossRef]
- Besker, N.; Gervasio, F.L. Using metadynamics and path collective variables to study ligand binding and induced conformational transitions. Methods Mol. Biol. 2012, 819, 501–513. [Google Scholar] [CrossRef]
- Wang, C.; Greene, D.A.; Xiao, L.; Qi, R.; Luo, R. Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci. 2018, 4, 87. [Google Scholar] [CrossRef]
- Geng, L.; Wang, Z.; Yang, X.; Li, D.; Lian, W.; Xiang, Z.; Wang, W.; Bu, X.; Lai, W.; Hu, Z.; et al. Structure-based Design of Peptides with High Affinity and Specificity to HER2 Positive Tumors. Theranostics 2015, 5, 1154–1165. [Google Scholar] [CrossRef]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef]
- Mermelstein, D.J.; Lin, C.; Nelson, G.; Kretsch, R.; McCammon, J.A.; Walker, R.C. Fast and flexible gpu accelerated binding free energy calculations within the amber molecular dynamics package. J. Comput. Chem. 2018, 39, 1354–1358. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Spodzieja, M.; Lach, S.; Iwaszkiewicz, J.; Cesson, V.; Kalejta, K.; Olive, D.; Michielin, O.; Speiser, D.E.; Zoete, V.; Derré, L.; et al. Design of short peptides to block BTLA/HVEM interactions for promoting anticancer T-cell responses. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Wu, H.C.; Tseng, Y.L.; Lin, C.T. A novel peptide specifically binding to nasopharyngeal carcinoma for targeted drug delivery. Cancer. Res. 2004, 64, 8002–8008. [Google Scholar] [CrossRef] [PubMed]
- Das, A.A.; Sharma, O.P.; Kumar, M.S.; Krishna, R.; Mathur, P.P. PepBind: A Comprehensive Database and Computational Tool for Analysis of Protein–peptide Interactions. Genom. Proteom. Bioinform. 2013, 11, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Lee, A.C.; Chen, I.J.; Chang, N.C.; Wu, H.C.; Yu, H.M.; Chang, Y.J.; Lee, T.W.; Yu, J.C.; Yu, A.L.; et al. Structure-based optimization of GRP78-binding peptides that enhances efficacy in cancer imaging and therapy. Biomaterials 2016, 94, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, D.T.; Lang, P.T.; Pegg, S.; Pettersen, E.; Kuntz, I.D.; Brooijmans, N.; Rizzo, R.C. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput. Aided Mol. Des. 2006, 20, 601–619. [Google Scholar] [CrossRef]
- Wang, S.-H.; Wu, Y.-T.; Kuo, S.-C.; Yu, J. HotLig: A Molecular Surface-Directed Approach to Scoring Protein–Ligand Interactions. J. Chem. Inf. Mod. 2013, 53, 2181–2195. [Google Scholar] [CrossRef]
- Warden, B.A.; Fazio, S.; Shapiro, M.D. The PCSK9 revolution: Current status, controversies, and future directions. Trends Cardiovasc. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Grazioso, G.; Bollati, C.; Sgrignani, J.; Arnoldi, A.; Lammi, C. First Food-Derived Peptide Inhibitor of the Protein-Protein Interaction between Gain-of-Function PCSK9(D374Y) and the Low-Density Lipoprotein Receptor. J. Agric. Food Chem. 2018, 66, 10552–10557. [Google Scholar] [CrossRef]
- Ylilauri, M.; Pentikainen, O.T. MMGBSA as a tool to understand the binding affinities of filamin-peptide interactions. J. Chem. Inf. Model. 2013, 53, 2626–2633. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef]
- Postupalenko, V.; Desplancq, D.; Orlov, I.; Arntz, Y.; Spehner, D.; Mely, Y.; Klaholz, B.P.; Schultz, P.; Weiss, E.; Zuber, G. Protein Delivery System Containing a Nickel-Immobilized Polymer for Multimerization of Affinity-Purified His-Tagged Proteins Enhances Cytosolic Transfer. Angew. Chem. Int. Ed. Engl. 2015, 54, 10583–10586. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Vassilev, L.; Ranki, T.; Hemminki, A.; Karioja-Kallio, A.; Levalampi, O.; Vuolanto, A.; Cerullo, V.; Pesonen, S. Toxicological and bio-distribution profile of a GM-CSF-expressing, double-targeted, chimeric oncolytic adenovirus ONCOS-102—Support for clinical studies on advanced cancer treatment. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Magarkar, A.; Cervera-Carrascon, V.; Fusciello, M.; Feola, S.; Muller, M.; Garofalo, M.; Kuryk, L.; Tähtinen, S.; Pastore, L.; et al. A novel in silico framework to improve MHC-I epitopes and break the tolerance to melanoma. OncoImmunology 2017. [Google Scholar] [CrossRef]
- Koski, A.; Rajecki, M.; Guse, K.; Kanerva, A.; Ristimaki, A.; Pesonen, S.; Escutenaire, S.; Hemminki, A. Systemic adenoviral gene delivery to orthotopic murine breast tumors with ablation of coagulation factors, thrombocytes and Kupffer cells. J. Gene. Med. 2009, 11, 966–977. [Google Scholar] [CrossRef]
- Freytag, S.O.; Stricker, H.; Movsas, B.; Kim, J.H. Prostate cancer gene therapy clinical trials. Mol. Ther. 2007, 15, 1042–1052. [Google Scholar] [CrossRef]
- Lubaroff, D.M.; Konety, B.R.; Link, B.; Gerstbrein, J.; Madsen, T.; Shannon, M.; Howard, J.; Paisley, J.; Boeglin, D.; Ratliff, T.L.; et al. Phase I clinical trial of an adenovirus/prostate-specific antigen vaccine for prostate cancer: Safety and immunologic results. Clin. Cancer Res. 2009, 15, 7375–7380. [Google Scholar] [CrossRef]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. OncoImmunology 2016. [Google Scholar] [CrossRef]
- Kuryk, L.; Haavisto, E.; Garofalo, M.; Capasso, C.; Hirvinen, M.; Pesonen, S.; Ranki, T.; Vassilev, L.; Cerullo, V. Synergistic anti-tumor efficacy of immunogenic adenovirus ONCOS-102 (Ad5/3-D24-GM-CSF) and standard of care chemotherapy in preclinical mesothelioma model. Int. J. Cancer 2016, 139, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Moller, A.W.; Jaderberg, M. Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model. OncoImmunology 2019. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Møller, A.-S.W.; Garofalo, M.; Cerullo, V.; Pesonen, S.; Alemany, R.; Jaderberg, M. Antitumor-specific T-cell responses induced by oncolytic adenovirus ONCOS-102 (AdV5/3-D24-GM-CSF) in peritoneal mesothelioma mouse model. J. Med. Virol. 2018, 90, 1669–1673. [Google Scholar] [CrossRef] [PubMed]
- Cerullo, V.; Diaconu, I.; Romano, V.; Hirvinen, M.; Ugolini, M.; Escutenaire, S.; Holm, S.L.; Kipar, A.; Kanerva, A.; Hemminki, A. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol. Ther. 2012, 20, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Garofalo, M.; Hirvinen, M.; Cerullo, V. The evolution of adenoviral vectors through genetic and chemical surface modifications. Viruses 2014, 6, 832–855. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Møller, A.-S.W.; Vuolanto, A.; Pesonen, S.; Garofalo, M.; Cerullo, V.; Jaderberg, M. Optimization of Early Steps in Oncolytic Adenovirus ONCOS-401 Production in T-175 and HYPERFlasks. Int. J. Mol. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fountzilas, C.; Patel, S.; Mahalingam, D. Review: Oncolytic virotherapy, updates and future directions. Oncotarget 2017, 8, 102617–102639. [Google Scholar] [CrossRef]
- Martin, N.T.; Bell, J.C. Oncolytic Virus Combination Therapy: Killing One Bird with Two Stones. Mol. Ther. 2018, 26, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Iovine, B.; Oliviero, G.; Garofalo, M.; Orefice, M.; Nocella, F.; Borbone, N.; Piccialli, V.; Centore, R.; Mazzone, M.; Piccialli, G.; et al. The anti-proliferative effect of L-carnosine correlates with a decreased expression of hypoxia inducible factor 1 alpha in human colon cancer cells. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Iovine, B.; Kuryk, L.; Capasso, C.; Hirvinen, M.; Vitale, A.; Yliperttula, M.; Bevilacqua, M.A.; Cerullo, V. Oncolytic Adenovirus Loaded with L-carnosine as Novel Strategy to Enhance the Antitumor Activity. Mol. Cancer Ther. 2016, 15, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Ottolino-Perry, K.; Diallo, J.S.; Lichty, B.D.; Bell, J.C.; McCart, J.A. Intelligent design: Combination therapy with oncolytic viruses. Mol. Ther. 2010, 18, 251–263. [Google Scholar] [CrossRef]
- Nguyen, A.; Ho, L.; Wan, Y. Chemotherapy and Oncolytic Virotherapy: Advanced Tactics in the War against Cancer. Front. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Møller, A.-S.W.; Jaderberg, M. Quantification and functional evaluation of CD40L production from the adenovirus vector ONCOS-401. Cancer Gene Ther. 2019, 26, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Diaconu, I.; Cerullo, V.; Hirvinen, M.L.; Escutenaire, S.; Ugolini, M.; Pesonen, S.K.; Bramante, S.; Parviainen, S.; Kanerva, A.; Loskog, A.S.; et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer. Res. 2012, 72, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Galluzzi, L.; Martins, I.; Schlemmer, F.; Adjemian, S.; Michaud, M.; Sukkurwala, A.Q.; Menger, L.; Zitvogel, L.; Kroemer, G. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev. 2011, 30, 61–69. [Google Scholar] [CrossRef]
- Wong, D.Y.; Ong, W.W.; Ang, W.H. Induction of Immunogenic Cell Death by Chemotherapeutic Platinum Complexes. Angew. Chem. Int. Ed. Engl. 2015. [Google Scholar] [CrossRef]
- Siurala, M.; Bramante, S.; Vassilev, L.; Hirvinen, M.; Parviainen, S.; Tahtinen, S.; Guse, K.; Cerullo, V.; Kanerva, A.; Kipar, A.; et al. Oncolytic adenovirus and doxorubicin-based chemotherapy results in synergistic antitumor activity against soft-tissue sarcoma. Int. J. Cancer 2015, 136, 945–954. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Gilboa, E. How tumors escape immune destruction and what we can do about it. Cancer Immunol. Immunother. 1999, 48, 382–385. [Google Scholar] [CrossRef]
- Liikanen, I.; Ahtiainen, L.; Hirvinen, M.L.; Bramante, S.; Cerullo, V.; Nokisalmi, P.; Hemminki, O.; Diaconu, I.; Pesonen, S.; Koski, A.; et al. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol. Ther. 2013, 21, 1212–1223. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Khair, D.O.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Azoury, S.C.; Straughan, D.M.; Shukla, V. Immune Checkpoint Inhibitors for Cancer Therapy: Clinical Efficacy and Safety. Curr. Cancer Drug Targets 2015, 15, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Redman, J.M.; Gibney, G.T.; Atkins, M.B. Advances in immunotherapy for melanoma. BMC Med. 2016. [Google Scholar] [CrossRef]
- Ai, M.; Curran, M.A. Immune checkpoint combinations from mouse to man. Cancer Immunol. Immunother. 2015, 64, 885–892. [Google Scholar] [CrossRef]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014. [Google Scholar] [CrossRef]
- Vile, R.G. How to train your oncolytic virus: The immunological sequel. Mol. Ther. 2014, 22, 1881–1884. [Google Scholar] [CrossRef]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef]
- Gregoriadis, G. Engineering liposomes for drug delivery: Progress and problems. Trends Biotechnol. 1995, 13, 527–537. [Google Scholar] [CrossRef]
- Allen, T.M. Liposomal drug formulations. Rationale for development and what we can expect for the future. Drugs 1998, 56, 747–756. [Google Scholar] [CrossRef]
- Gregoriadis, G. Drug entrapment in liposomes. FEBS Lett. 1973, 36, 292–296. [Google Scholar] [CrossRef]
- Park, Y.S. Tumor-directed targeting of liposomes. Biosci. Rep. 2002, 22, 267–281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sudimack, J.; Lee, R.J. Targeted drug delivery via the folate receptor. Adv. Drug Deliv. Rev. 2000, 41, 147–162. [Google Scholar] [CrossRef]
- Mori, A.; Klibanov, A.L.; Torchilin, V.P.; Huang, L. Influence of the steric barrier activity of amphipathic poly(ethyleneglycol) and ganglioside GM1 on the circulation time of liposomes and on the target binding of immunoliposomes in vivo. FEBS Lett. 1991, 284, 263–266. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug. Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Jhaveri, A.; Deshpande, P.; Pattni, B.; Torchilin, V. Transferrin-targeted, resveratrol-loaded liposomes for the treatment of glioblastoma. J. Control. Release 2018, 277, 89–101. [Google Scholar] [CrossRef]
- Garofalo, M.; Villa, A.; Crescenti, D.; Marzagalli, M.; Kuryk, L.; Limonta, P.; Mazzaferro, V.; Ciana, P. Heterologous and cross-species tropism of cancer-derived extracellular vesicles. Theranostics 2019, 9, 5681–5693. [Google Scholar] [CrossRef]
- Stremersch, S.; Marro, M.; Pinchasik, B.E.; Baatsen, P.; Hendrix, A.; De Smedt, S.C.; Loza-Alvarez, P.; Skirtach, A.G.; Raemdonck, K.; Braeckmans, K. Identification of Individual Exosome-Like Vesicles by Surface Enhanced Raman Spectroscopy. Small 2016, 12, 3292–3301. [Google Scholar] [CrossRef]
- Saleh, A.F.; Lazaro-Ibanez, E.; Forsgard, M.A.; Shatnyeva, O.; Osteikoetxea, X.; Karlsson, F.; Heath, N.; Ingelsten, M.; Rose, J.; Harris, J.; et al. Extracellular vesicles induce minimal hepatotoxicity and immunogenicity. Nanoscale 2019, 11, 6990–7001. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef]
- Stremersch, S.; De Smedt, S.C.; Raemdonck, K. Therapeutic and diagnostic applications of extracellular vesicles. J. Control. Release 2016, 244, 167–183. [Google Scholar] [CrossRef]
- Ker, D.F.E.; Wang, D.; Behn, A.W.; Wang, E.T.H.; Zhang, X.; Zhou, B.Y.; Mercado-Pagan, A.E.; Kim, S.; Kleimeyer, J.; Gharaibeh, B.; et al. Functionally Graded, Bone- and Tendon-Like Polyurethane for Rotator Cuff Repair. Adv. Funct. Mater. 2018. [Google Scholar] [CrossRef] [PubMed]
- Vader, P.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Emerging targets for cancer therapy. Trends Mol. Med. 2014, 20, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Saari, H.; Lazaro-Ibanez, E.; Viitala, T.; Vuorimaa-Laukkanen, E.; Siljander, P.; Yliperttula, M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J. Control. Release 2015, 220, 727–737. [Google Scholar] [CrossRef]
- Garofalo, M.; Saari, H.; Somersalo, P.; Crescenti, D.; Kuryk, L.; Aksela, L.; Capasso, C.; Madetoja, M.; Koskinen, K.; Oksanen, T.; et al. Antitumor effect of oncolytic virus and paclitaxel encapsulated in extracellular vesicles for lung cancer treatment. J. Control. Release 2018, 283, 223–234. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Stremersch, S.; Vandenbroucke, R.E.; Van Wonterghem, E.; Hendrix, A.; De Smedt, S.C.; Raemdonck, K. Comparing exosome-like vesicles with liposomes for the functional cellular delivery of small RNAs. J. Control. Release 2016, 232, 51–61. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Gangadaran, P.; Li, X.J.; Kalimuthu, S.K.; Min, O.J.; Hong, C.M.; Rajendran, R.L.; Lee, H.W.; Zhu, L.; Baek, S.H.; Jeong, S.Y.; et al. New Optical Imaging Reporter-labeled Anaplastic Thyroid Cancer-Derived Extracellular Vesicles as a Platform for In Vivo Tumor Targeting in a Mouse Model. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control. Release 2015, 199, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, L.; Saunders, D.N.; Ranson, M.; Thurecht, K.J.; Storm, G.; Vine, K.L. Towards clinical translation of ligand-functionalized liposomes in targeted cancer therapy: Challenges and opportunities. J. Control. Release 2018, 277, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018. [Google Scholar] [CrossRef]
- He, C.; Zheng, S.; Luo, Y.; Wang, B. Exosome Theranostics: Biology and Translational Medicine. Theranostics 2018, 8, 237–255. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Garofalo, M.; Villa, A.; Rizzi, N.; Kuryk, L.; Rinner, B.; Cerullo, V.; Yliperttula, M.; Mazzaferro, V.; Ciana, P. Extracellular vesicles enhance the targeted delivery of immunogenic oncolytic adenovirus and paclitaxel in immunocompetent mice. J. Control. Release 2019, 294, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Villa, A.; Rizzi, N.; Kuryk, L.; Mazzaferro, V.; Ciana, P. Systemic Administration and Targeted Delivery of Immunogenic Oncolytic Adenovirus Encapsulated in Extracellular Vesicles for Cancer Therapies. Viruses 2018. [Google Scholar] [CrossRef] [PubMed]
- Ornes, S. Antibody-drug conjugates. Proc. Natl. Acad. Sci. USA 2013. [Google Scholar] [CrossRef] [PubMed]
- Mathe, G.; Tran Ba, L.O.C.; Bernard, J. Effect on mouse leukemia 1210 of a combination by diazo-reaction of amethopterin and gamma-globulins from hamsters inoculated with such leukemia by heterografts. C. R. Acad. Sci. 1958, 246, 1626–1628. [Google Scholar]
- Ford, C.H.; Newman, C.E.; Johnson, J.R.; Woodhouse, C.S.; Reeder, T.A.; Rowland, G.F.; Simmonds, R.G. Localisation and toxicity study of a vindesine-anti-CEA conjugate in patients with advanced cancer. Br. J. Cancer 1983, 47, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Morris, C.Q. Antibody-Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef] [PubMed]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, A.; Dhillon, S. Trastuzumab emtansine: First global approval. Drugs 2013, 73, 755–765. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Moore, K.N.; O’Malley, D.M.; Vergote, I.; Martin, L.P.; Gonzalez-Martin, A.; Malek, K.; Birrer, M.J. Safety and activity findings from a phase 1b escalation study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with carboplatin in patients with platinum-sensitive ovarian cancer. Gynecol. Oncol. 2018, 151, 46–52. [Google Scholar] [CrossRef]
- Müller, P.; Kreuzaler, M.; Khan, T.; Thommen, D.S.; Martin, K.; Glatz, K.; Savic, S.; Harbeck, N.; Nitz, U.; Gluz, O.; et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci. Transl. Med. 2015. [Google Scholar] [CrossRef]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef]
- Widdison, W.C.; Ponte, J.F.; Coccia, J.A.; Lanieri, L.; Setiady, Y.; Dong, L.; Skaletskaya, A.; Hong, E.E.; Wu, R.; Qiu, Q.; et al. Development of Anilino-Maytansinoid ADCs that Efficiently Release Cytotoxic Metabolites in Cancer Cells and Induce High Levels of Bystander Killing. Bioconjug. Chem. 2015, 26, 2261–2278. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Berkenblit, A. Antibody-Drug Conjugates for Cancer Treatment. Ann. Rev. Med. 2018, 69, 191–207. [Google Scholar] [CrossRef]
- Ghaemi, Z.; Minozzi, M.; Carloni, P.; Laio, A. A novel approach to the investigation of passive molecular permeation through lipid bilayers from atomistic simulations. J. Phys. Chem. B 2012, 116, 8714–8721. [Google Scholar] [CrossRef] [PubMed]
- Minozzi, M.; Lattanzi, G.; Benz, R.; Costi, M.P.; Venturelli, A.; Carloni, P. Permeation through the cell membrane of a boron-based beta-lactamase inhibitor. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, J.; Ying, S.; Ren, H.; Dai, J.; Zhang, L.; Liang, L.; Wang, Q.; Shen, Q.; Shen, J.W. Molecular dynamics study on the encapsulation and release of anti-cancer drug doxorubicin by chitosan. Int. J. Pharm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Melo, R.; Lemos, A.; Preto, A.J.; Almeida, J.G.; Correia, J.D.G.; Sensoy, O.; Moreira, I.S. Computational Approaches in Antibody-drug Conjugate Optimization for Targeted Cancer Therapy. Curr. Top. Med. Chem. 2018, 18, 1091–1109. [Google Scholar] [CrossRef]
- Norman, R.A.; Ambrosetti, F.; Bonvin, A.M.J.J.; Colwell, L.J.; Kelm, S.; Kumar, S.; Krawczyk, K. Computational approaches to therapeutic antibody design: Established methods and emerging trends. Brief. Bioinform. 2019. [Google Scholar] [CrossRef]
- De Leo, F.; Sgrignani, J.; Bonifazi, D.; Magistrato, A. Structural and dynamic properties of monoclonal antibodies immobilized on CNTs: A computational study. Chemistry 2013, 19, 12281–12293. [Google Scholar] [CrossRef]
- Hashemzadeh, H.; Javadi, H.; Darvishi, M.H. Study of Structural stability and formation mechanisms in DSPC and DPSM liposomes: A coarse-grained molecular dynamics simulation. Sci. Rep. 2020. [Google Scholar] [CrossRef]
- Perilla, J.R.; Hadden, J.A.; Goh, B.C.; Mayne, C.G.; Schulten, K. All-Atom Molecular Dynamics of Virus Capsids as Drug Targets. J. Phys. Chem. Lett. 2016, 7, 1836–1844. [Google Scholar] [CrossRef]
- Durrant, J.D.; Kochanek, S.E.; Casalino, L.; Ieong, P.U.; Dommer, A.C.; Amaro, R.E. Mesoscale All-Atom Influenza Virus Simulations Suggest New Substrate Binding Mechanism. ACS Cent. Sci. 2020, 6, 189–196. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garofalo, M.; Grazioso, G.; Cavalli, A.; Sgrignani, J. How Computational Chemistry and Drug Delivery Techniques Can Support the Development of New Anticancer Drugs. Molecules 2020, 25, 1756. https://doi.org/10.3390/molecules25071756
Garofalo M, Grazioso G, Cavalli A, Sgrignani J. How Computational Chemistry and Drug Delivery Techniques Can Support the Development of New Anticancer Drugs. Molecules. 2020; 25(7):1756. https://doi.org/10.3390/molecules25071756
Chicago/Turabian StyleGarofalo, Mariangela, Giovanni Grazioso, Andrea Cavalli, and Jacopo Sgrignani. 2020. "How Computational Chemistry and Drug Delivery Techniques Can Support the Development of New Anticancer Drugs" Molecules 25, no. 7: 1756. https://doi.org/10.3390/molecules25071756
APA StyleGarofalo, M., Grazioso, G., Cavalli, A., & Sgrignani, J. (2020). How Computational Chemistry and Drug Delivery Techniques Can Support the Development of New Anticancer Drugs. Molecules, 25(7), 1756. https://doi.org/10.3390/molecules25071756