Targeted Radionuclide Therapy of Prostate Cancer—From Basic Research to Clinical Perspectives

 ,
,  ,
,
Abstract
1. Introduction
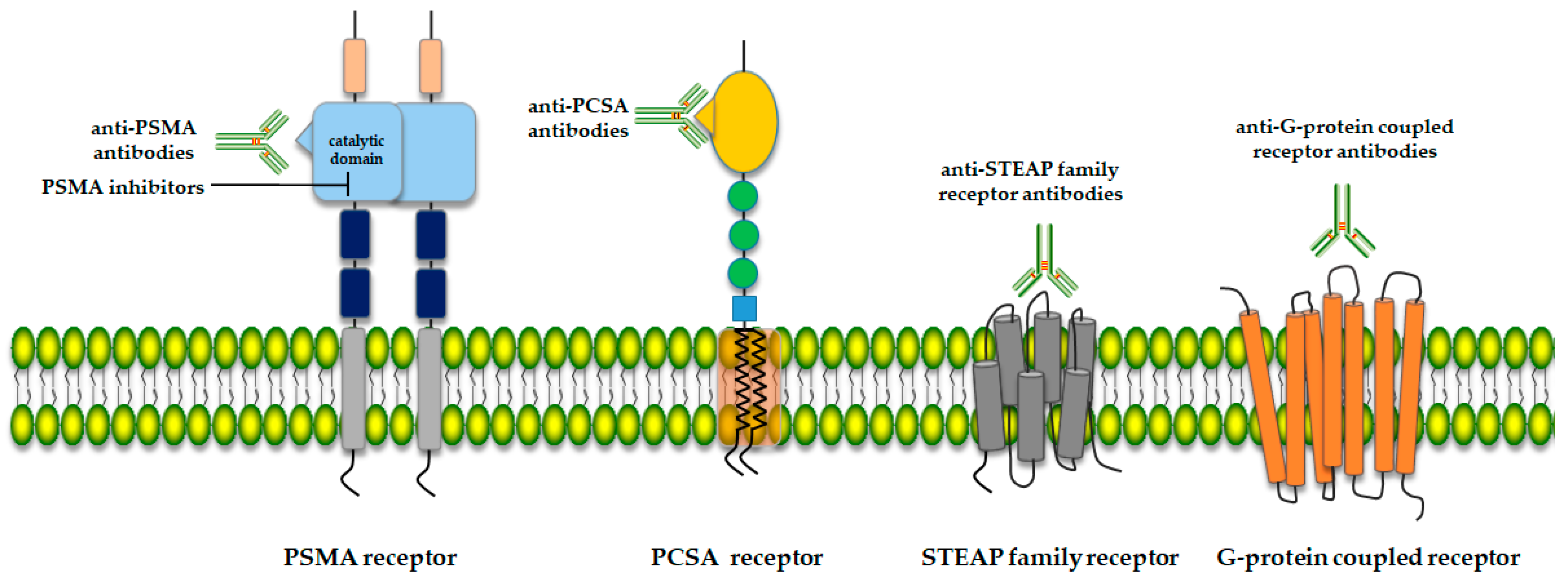
2. Potential Therapy Targets for Prostate Cancer
2.1. Prostate-Specific Membrane Antigen (PSMA)
2.2. Prostate Stem Cell Antigen (PSCA)
2.3. Six-Transmembrane Epithelial Antigen of the Prostate (STEAP)
2.4. G-Protein Coupled Receptor (5-oxoER)
3. PSMA Ligands for Targeted Radiotherapy
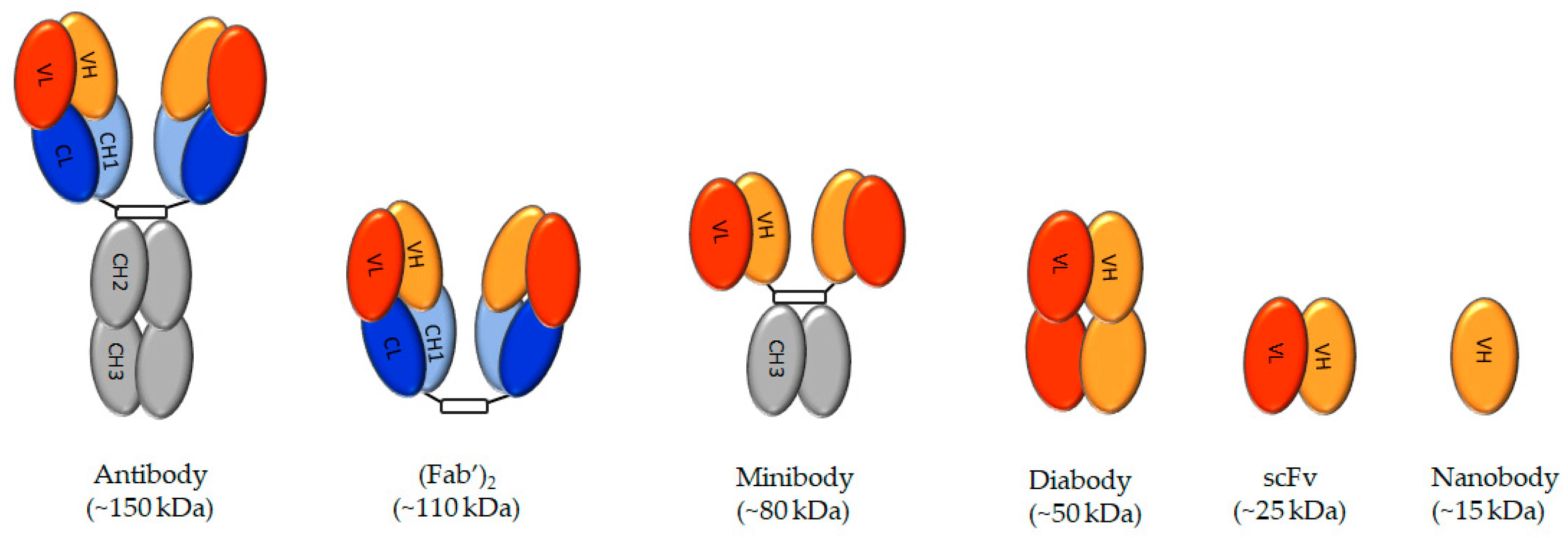
3.1. Antibodies
3.2. Minibodies, Diabodies, Single-Chain Variable Region Fragments, and Nanobodies
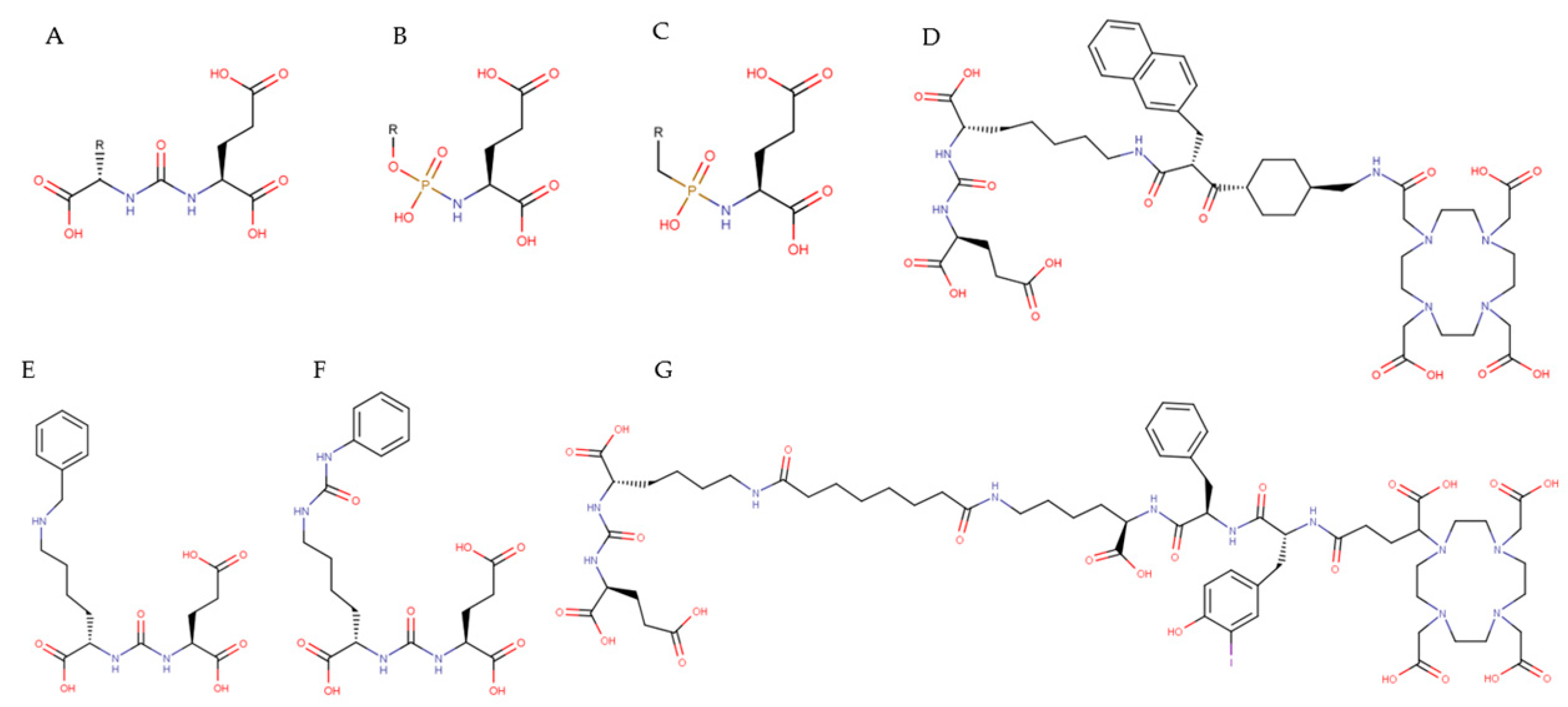
3.3. Small-Molecule PSMA Inhibitors
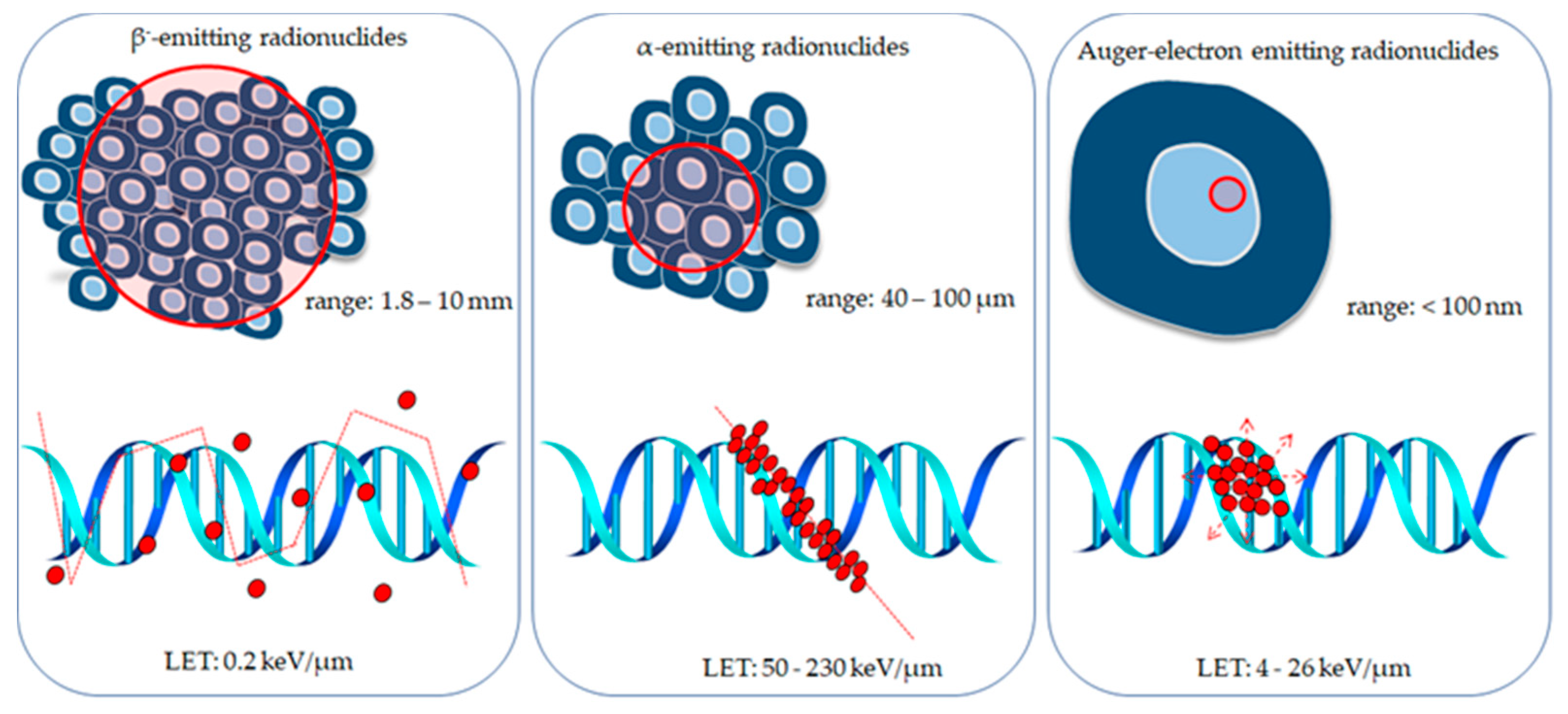
4. Radionuclides Investigated for Use in Targeted Prostate Cancer Therapy
4.1. β−-Emitting Radionuclides
4.2. α-Emitting Radionuclides
4.3. Auger Electron Emitters
5. PSMA-Targeting Ligands Labeled with Radionuclides for Targeted Prostate Cancer Therapy
5.1. PSMA-Targeting Antibody-Based Molecules Conjugated to β− Emitters
5.2. PSMA-Targeting Small Molecules Conjugated to β− Emitters
5.3. PSMA-Targeted Antibody-Based Molecules Conjugated to α-Emitters
5.4. PSMA-Targeted Small Molecules Conjugated to α-Emitters
5.5. PSMA-Targeting Ligands Conjugated to Auger Electron Emitters
6. PSMA-Targeted Nanoparticle-Based Radiopharmaceuticals
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef] [PubMed]
- Nuhn, P.; De Bono, J.S.; Fizazi, K.; Freedland, S.J.; Grilli, M.; Kantoff, P.W.; Sonpavde, G.; Sternberg, C.N.; Yegnasubramanian, S.; Antonarakis, E.S. Update on Systemic Prostate Cancer Therapies: Management of Metastatic Castration-resistant Prostate Cancer in the Era of Precision Oncology. Eur. Urol. 2019, 75, 88–99. [Google Scholar] [CrossRef]
- Nguyen-Nielsen, M.; Borre, M. Diagnostic and Therapeutic Strategies for Prostate Cancer. Semin. Nucl. Med. 2016, 46, 484–490. [Google Scholar] [CrossRef]
- Cattrini, C.; Castro, E.; Lozano, R.; Zanardi, E.; Rubagotti, A.; Boccardo, F.; Olmos, D. Current Treatment Options for Metastatic Hormone-Sensitive Prostate Cancer. Cancers 2019, 11, 1355. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, A.N.; Usman, A.; Morgans, A.; VanderWeele, D.J.; Sosman, J.; Wu, J.D. Past, Current, and Future of Immunotherapies for Prostate Cancer. Front. Oncol. 2019, 9, 884. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.M.; Howard, L.E.; Sourbeer, K.N.; Amarasekara, H.S.; Chow, L.C.; Cockrell, D.C.; Pratson, C.L.; Hanyok, B.T.; Aronson, W.J.; Kane, C.J.; et al. Predicting Time From Metastasis to Overall Survival in Castration-Resistant Prostate Cancer: Results From SEARCH. Clin. Genitourin. Cancer 2016, 15, 60–66.e2. [Google Scholar] [CrossRef]
- Logothetis, C.; Morris, M.J.; Den, R.; Coleman, R.E. Current perspectives on bone metastases in castrate-resistant prostate cancer. Cancer Metastasis Rev. 2018, 37, 189–196. [Google Scholar] [CrossRef]
- Elsasser-Beile, U.; Buhler, P.; Wolf, P. Targeted therapies for prostate cancer against the prostate specific membrane antigen. Curr. Drug Targets 2009, 10, 118–125. [Google Scholar] [CrossRef]
- Slovin, S.F. Targeting castration-resistant prostate cancer with monoclonal antibodies and constructs. Immunotherapy 2013, 5, 1347–1355. [Google Scholar] [CrossRef]
- Barve, A.; Jin, W.; Cheng, K. Prostate cancer relevant antigens and enzymes for targeted drug delivery. J. Control. Release 2014, 187, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Diao, W.; Cai, H.; Chen, L.; Jin, X.; Liao, X.; Jia, Z. Recent Advances in Prostate-Specific Membrane Antigen-Based Radiopharmaceuticals. Curr. Top. Med. Chem. 2019, 19, 33–56. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Bennett, M.J.; Thomas, L.M.; Bjorkman, P.J. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc. Natl. Acad. Sci. USA 2005, 102, 5981–5986. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; O’Keefe, D.S.; Bacich, D.J.; Reuter, V.E.; Heston, W.D.; Gaudin, P.B. Prostate-specific membrane antigen is produced in tumor-associated neovasculature. Clin. Cancer Res. 1999, 5, 2674–2681. [Google Scholar]
- Sokoloff, R.; Norton, K.C.; Gasior, C.L.; Marker, K.M.; Grauer, L.S. A dual-monoclonal sandwich assay for prostate-specific membrane antigen: Levels in tissues, seminal fluid and urine. Prostate 2000, 43, 150–157. [Google Scholar] [CrossRef]
- Zaviacic, M.; Ruzicková, M.; Jakubovský, J.; Danihel, L.; Babál, P.; Blazeková, J. The significance of prostate markers in the orthology of the female prostate. Bratisl. Lek. Listy 1994, 95, 491–497. [Google Scholar]
- Castanares, M.A.; Mukherjee, A.; Chowdhury, W.; Liu, M.; Chen, Y.; Mease, R.C.; Wang, Y.; Rodriguez, R.; Lupold, S.E.; Pomper, M.G. Evaluation of prostate-specific membrane antigen as an imaging reporter. J. Nucl. Med. 2014, 55, 805–811. [Google Scholar] [CrossRef]
- Bouchelouche, K.; Turkbey, B.; Choyke, P.L. PSMA PET and Radionuclide Therapy in Prostate Cancer. Semin. Nucl. Med. 2016, 46, 522–535. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Babich, J.W.; Kratochwil, C.; Giesel, F.L.; Eisenhut, M.; Kopka, K.; Haberkorn, U. The Rise of PSMA Ligands for Diagnosis and Therapy of Prostate Cancer. J. Nucl. Med. 2016, 57, 79–89. [Google Scholar] [CrossRef]
- Haberkorn, U.; Eder, M.; Kopka, K.; Babich, J.W.; Eisenhut, M. New Strategies in Prostate Cancer: Prostate-Specific Membrane Antigen (PSMA) Ligands for Diagnosis and Therapy. Clin. Cancer Res. 2016, 22, 9–15. [Google Scholar] [CrossRef]
- Schwarzenboeck, S.M.; Rauscher, I.; Bluemel, C.; Fendler, W.P.; Rowe, S.P.; Pomper, M.G.; Asfhar-Oromieh, A.; Herrmann, K.; Eiber, M. PSMA Ligands for PET Imaging of Prostate Cancer. J. Nucl. Med. 2017, 58, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Eiber, M.; Fendler, W.P.; Rowe, S.P.; Calais, J.; Hofman, M.S.; Maurer, T.; Schwarzenboeck, S.M.; Kratowchil, C.; Herrmann, K.; Giesel, F.L. Prostate-Specific Membrane Antigen Ligands for Imaging and Therapy. J. Nucl. Med. 2017, 58, 67S–76S. [Google Scholar] [CrossRef] [PubMed]
- Umbricht, C.A.; Benešová, M.; Schibli, R.; Müller, C. Preclinical Development of Novel PSMA-Targeting Radioligands: Modulation of Albumin-Binding Properties to Improve Prostate Cancer Therapy. Mol. Pharm. 2018, 15, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.E.; Gu, Z.; Watabe, T.; Thomas, G.; Szigeti, K.; Davis, E.; Wahl, M.; Nisitani, S.; Yamashiro, J.; Le Beau, M.M.; et al. Prostate stem cell antigen: A cell surface marker overexpressed in prostate cancer. Proc. Natl. Acad. Sci. USA 1998, 95, 1735–1740. [Google Scholar] [CrossRef]
- Ross, S.; Spencer, S.D.; Holcomb, I.; Tan, C.; Hongo, J.; Devaux, B.; Rangell, L.; Keller, G.A.; Schow, P.; Steeves, R.M.; et al. Prostate stem cell antigen as therapy target: Tissue expression and in vivo efficacy of an immunoconjugate. Cancer Res. 2002, 62, 2546–2553. [Google Scholar]
- Ono, H.; Sakamoto, H.; Yoshida, T.; Saeki, N. Prostate stem cell antigen is expressed in normal and malignant human brain tissues. Oncol. Lett. 2017, 15, 3081–3084. [Google Scholar] [CrossRef]
- Lam., J.S.; Yamashiro., J.; Shintaku., I.P.; Vessella, R.L.; Jenkins, R.B.; Horvath, S.; Said, J.W.; Reiter, R.E. Prostate stem cell antige is overexpressed in prostate cancer metastases. Clin. Cancer Res. 2005, 11, 2591–2596. [Google Scholar] [CrossRef]
- Yang, X.-L.; Guo, Z.; Liu, Y.; Si, T.; Yu, H.; Li, B.; Tian, W. Prostate stem cell antigen and cancer risk, mechanisms and therapeutic implications. Expert Rev. Anticancer Ther. 2013, 14, 31–37. [Google Scholar] [CrossRef]
- Heinrich, M.-C.; Göbel, C.; Kluth, M.; Bernreuther, C.; Sauer, C.; Schroeder, C.; Koop, C.; Hube-Magg, C.; Lebok, P.; Burandt, E.; et al. PSCA expression is associated with favorable tumor features and reduced PSA recurrence in operated prostate cancer. BMC Cancer 2018, 18, 612. [Google Scholar] [CrossRef]
- Zhigang, Z.; Wenlu, S. The association of prostate stem cell antigen (PSCA) mRNA expression and subsequent prostate cancer risk in men with benign prostatic hyperplasia following transurethral resection of the prostate. Prostate 2007, 68, 190–199. [Google Scholar] [CrossRef]
- Raff, A.B.; Gray, A.; Kast, W.M. Prostate stem cel antigen: A prospective therapeutic and diagnostic target. Cancer Lett. 2009, 277, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Saffran, D.C.; Raitano, A.B.; Hubert, R.S.; Witte, O.N.; Reiter, R.E.; Jakobovits, A. Anti-PSCA mAbs inhibit tumor growth and metastasis formation and prolong the survival of mice bearing human prostate cancer xenografts. Proc. Natl. Acad. Sci. USA 2001, 98, 2658–2663. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Yamashiro, J.; Kono, E.; Reiter, R.E. Anti-prostate stem cell antigen monoclonal antibody 1G8 induces cell death in vitro inhibits tumor growth in vivo via a Fc-independent mechanism. Cancer Cell 2005, 65, 9495–9500. [Google Scholar]
- Ling, Y.; Wei, K.; Luo, Y.; Gao, X.; Zhong, S. Dual docetaxel/superparamagnetic iron oxide loaded nanoparticles for both targeting magnetic resonance imaging and cancer therapy. Biomaterials 2011, 32, 7139–7150. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Luo, Y.; Wang, Y.; Pang, J.; Liao, C.; Lu, H.; Fang, Y. Prostate stem cell antigen-targeted nanoparticles with dual functional properties: In vivo imaging and cancer chemotherapy. Int. J. Nanomed. 2012, 7, 4037–4051. [Google Scholar] [CrossRef]
- Morris, M.J.; Eisenberger, M.A.; Pili, R.; Denmeade, S.R.; Rathkopf, D.E.; Slovin, S.F.; Farrelly, J.; Chudow, J.J.; Vincent, M.; Scher, H.I.; et al. A phase I/IIA study of AGS-PSCA for castration-resistant prostate cancer. Ann. Oncol. 2012, 23, 2714–2719. [Google Scholar] [CrossRef] [PubMed]
- Leyton, J.V.; Olafsen, T.; Lepin, E.J.; Hahm, S.; Bauer, K.B.; Reiter, R.E.; Wu, A.M. Humanized radioiodinated minibody for imaging of prostate stem cell antigen-expressing tumors. Clin. Cancer Res. 2008, 14, 7488–7496. [Google Scholar] [CrossRef]
- Lepin, E.J.; Leyton, J.V.; Zhou, Y.; Olafsen, T.; Salazar, F.B.; McCabe, K.E.; Hahm, S.; Marks, J.D.; Reiter, R.E.; Wu, A.M. An affinity matured minibody for PET imaging of prostate stem cell antigen (PSCA)-expressing tumors. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1529–1538. [Google Scholar] [CrossRef]
- Knowles, S.M.; Wu, A.M. Advances in Immuno–Positron Emission Tomography: Antibodies for Molecular Imaging in Oncology. J. Clin. Oncol. 2012, 30, 3884–3892. [Google Scholar] [CrossRef]
- Knowles, S.M.; Zettlitz, K.A.; Tavaré, R.; Rochefort, M.M.; Salazar, F.B.; Stout, D.B.; Yazaki, P.J.; Reiter, R.E.; Wu, A.M. Quantitative ImmunoPET of Prostate Cancer Xenografts with 89Zr- and 124I-Labeled Anti-PSCA A11 Minibody. J. Nucl. Med. 2014, 55, 452–459. [Google Scholar] [CrossRef]
- Yu, S.; Feng, F.; Wang, K.; Men, C.; Lin, C.; Liu, Q.; Yang, D.; Gao, Z. The therapeutic efficacy of I131-PSCA-mAb in orthotopic mouse models of prostate cancer. Eur. J. Med. Res. 2013, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Maia, C.; Santos, C.R.A. STEAP Proteins: From Structure to Applications in Cancer Therapy. Mol. Cancer Res. 2012, 10, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Hubert, R.S.; Vivanco, I.; Chen, E.; Rastegar, S.; Leong, K.; Mitchell, S.C.; Madraswala, R.; Zhou, Y.; Kuo, J.; Raitano, A.B.; et al. STEAP: A prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 14523–14528. [Google Scholar] [CrossRef] [PubMed]
- Challita-Eid, P.; Morrison, K.J.; Etessami, S.; An, Z.; Morrison, K.J.; Perez-Villar, J.J.; Raitano, A.B.; Jia, X.-C.; Gudas, J.M.; Kanner, S.B.; et al. Monoclonal Antibodies to Six-Transmembrane Epithelial Antigen of the Prostate-1 Inhibit Intercellular Communication In vitro and Growth of Human Tumor Xenografts in vivo. Cancer Res. 2007, 67, 5798–5805. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, J.; Shen, Z.; Liu, W.; Chen, Z. Clinical significance of six-transmembrane epithelial antigen of the prostate expressed in prostatic carcinoma. Zhonghua Nan Ke Xue = Natl. J. Androl. 2004, 10, 351–354. [Google Scholar]
- Grünewald, T.G.P.; Bach, H.; Cossarizza, A.; Matsumoto, I. The STEAP protein family: Versatile oxidoreductases and targets for cancer immunotherapy with overlapping and distinct cellular functions. Boil. Cell 2012, 104, 641–657. [Google Scholar] [CrossRef]
- Danila, D.C.; Szmulewitz, R.Z.; Higano, C.S.; Gilbert, H.; Kahn, R.S.; Wood, K.; Agarwal, P.; Lin, K.; Kabbarah, O.; Fine, B.M.; et al. A phase I study of the safety and pharmacokinetics of DSTP3086S, an anti-STEAP1 antibody-drug conjugate (ADC), in patients (pts) with metastatic castration-resistant prostate cancer (CRPC). J. Clin. Oncol. 2013, 31, 5020. [Google Scholar] [CrossRef]
- Korkmaz, K.S.; Elbi, C.; Korkmaz, C.G.; Loda, M.; Hager, G.L.; Saatcioglu, F. Molecular Cloning and Characterization of STAMP1, a Highly Prostate-specific Six Transmembrane Protein that Is Overexpressed in Prostate Cancer. J. Boil. Chem. 2002, 277, 36689–36696. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef]
- Whiteland, H.; Spencer-Harty, S.; Morgan, C.; Kynaston, H.; Thomas, D.H.; Bose, P.; Fenn, N.; Lewis, P.; Jenkins, S.; Doak, S.H. A role for STEAP2 in prostate cancer progression. Clin. Exp. Metastasis 2014, 31, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.B.; Dits, N.F.; Erkens-Schulze, S.; Van Ijcken, W.F.; Van Weerden, W.M.; Jenster, G. Modulation of Androgen Receptor Signaling in Hormonal Therapy-Resistant Prostate Cancer Cell Lines. PLoS ONE 2011, 6, e23144. [Google Scholar] [CrossRef] [PubMed]
- Varisli, L.; Gonen-Korkmaz, C.; Syed, H.M.; Böğürcü-Seidel, N.; Debelec-Butuner, B.; Erbaykent-Tepedelen, B.; Korkmaz, K.S. Androgen regulated HN1 leads proteosomal degradation of androgen receptor (AR) and negatively influences AR mediated transactivation in prostate cells. Mol. Cell. Endocrinol. 2012, 350, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Burnell, S.E.A.; Spencer-Harty, S.; Howarth, S.; Bodger, O.; Kynaston, H.; Morgan, C.; Doak, S.H. STEAP2 Knockdown Reduces the Invasive Potential of Prostate Cancer Cells. Sci. Rep. 2018, 8, 6252. [Google Scholar] [CrossRef]
- Lespagnol, A.; Duflaut, D.; Beekman, C.; Blanc, L.; Fiucci, G.; Marine, J.-C.; Vidal, M.; Amson, R.; Telerman, A. Exosome secretion, including the DNA damage-induced p53-dependent secretory pathway, is severely compromised in TSAP6/Steap3-null mice. Cell Death Differ. 2008, 15, 1723–1733. [Google Scholar] [CrossRef]
- Han, M.; Xu, R.; Wang, S.; Yang, N.; Ni, S.; Zhang, Q.; Xu, Y.; Zhang, X.; Zhang, C.; Wei, Y.; et al. Six-Transmembrane Epithelial Antigen of Prostate 3 Predicts Poor Prognosis and Promotes Glioblastoma Growth and Invasion. Neoplasia 2018, 20, 543–554. [Google Scholar] [CrossRef]
- Korkmaz, C.G.; Korkmaz, K.S.; Kurys, P.; Elbi, C.; Wang, L.; Klokk, T.I.; Hammarstrom, C.; Troen, G.; Svindland, A.; Hager, G.L.; et al. Molecular cloning and characterization of STAMP2, an androgen-regulated six transmembrane protein that is overexpressed in prostate cancer. Oncogene 2005, 24, 4934–4945. [Google Scholar] [CrossRef]
- Scarl, R.T.; Lawrence, C.M.; Gordon, H.M.; Nunemaker, C.S. STEAP4: Its emerging role in metabolism and homeostasis of cellular iron and copper. J. Endocrinol. 2017, 234, R123–R134. [Google Scholar] [CrossRef]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2012, 53, 531–556. [Google Scholar] [CrossRef]
- Xu, L.L.; Stackhouse, B.G.; Florence, K.; Zhang, W.; Shanmugam, N.; A Sesterhenn, I.; Zou, Z.; Srikantan, V.; Augustus, M.; Roschke, V.; et al. PSGR, a novel prostate-specific gene with homology to a G protein-coupled receptor, is overexpressed in prostate cancer. Cancer Res. 2000, 60, 6568–6572. [Google Scholar]
- Xia, C.; Ma, W.; Wang, F.; Hua, S.-B.; Liu, M. Identification of a prostate-specific G-protein coupled receptor in prostate cancer. Oncogene 2001, 20, 5903–5907. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, S.; Ghosh, J. Expression of 5-oxoETE receptor in prostate cancer cells: Critical role in survival. Biochem. Biophys. Res. Commun. 2006, 339, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Sarveswaran, S.; Ghosh, J. OXER1, a G protein coupled oxoeicisatetraenoid receptor mediates the survival-promoting effects of arachidonate 5-lipoxygenase in prostate cancer cells. Cancer Lett. 2013, 336, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Kalyvianaki, K.; Gebhart, V.M.; Peroulis, N.; Panagiotopoulou, C.; Kiagiadaki, F.; Pediaditakis, I.; Aivaliotis, M.; Moustou, E.; Tzardi, M.; Notas, G.; et al. Antagonizing effects of membrane-acting androgens on the eicosanoid receptor OXER1 in prostate cancer. Sci. Rep. 2017, 7, 44418. [Google Scholar] [CrossRef]
- Kratochwil, C.; Afshar-Oromieh, A.; Kopka, K.; Haberkorn, U.; Giesel, F.L. Current Status of Prostate-Specific Membrane Antigen Targeting in Nuclear Medicine: Clinical Translation of Chelator Containing Prostate-Specific Membrane Antigen Ligands Into Diagnostics and Therapy for Prostate Cancer. Semin. Nucl. Med. 2016, 46, 405–418. [Google Scholar] [CrossRef]
- Teo, M.Y.; Morris, M.J. Prostate-Specific Membrane Antigen-Directed Therapy for MCRPC. Cancer J. 2016, 22, 347–352. [Google Scholar] [CrossRef]
- Pillai, M.; Nanabala, R.; Joy, A.; Sasikumar, A.; Knapp, F.F. (Russ) Radiolabeled enzyme inhibitors and binding agents targeting PSMA: Effective theranostic tools for imaging and therapy of prostate cancer. Nucl. Med. Boil. 2016, 43, 692–720. [Google Scholar] [CrossRef]
- Hanack, K.; Messerschmidt, K.; Listek, M. Antibodies and Selection of Monoclonal Antibodies. Res. Probl. Cell Differ. 2016, 917, 11–22. [Google Scholar]
- Horoszewicz, J.S.; Kawinski, E.; Murphy, G.P. Monoclonal antibodies to a new antigenic marker in epithelial prostatic cells and serum of prostatic cancer patients. Anticancer Res. 1987, 7, 927–935. [Google Scholar]
- Troyer, J.K.; Beckett, M.L.; Wright, G.L. Location of prostate-specific membrane antigen in the LNCaP prostate carcinoma cell line. Prostate 1997, 30, 232–242. [Google Scholar] [CrossRef]
- Liu, H.; Moy, P.; Kim, S.; Xia, Y.; Rajasekaran, A.; Navarro, V.; Knudsen, B.; Bander, N.H. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997, 57, 3629–3634. [Google Scholar] [PubMed]
- Bařinka, C.; Mlčochová, P.; Sacha, P.; Hilgert, I.; Majer, P.; Slusher, B.S.; Horejsi, V.; Konvalinka, J. Amino acids at the N- and C-termini of human glutamate carboxypeptidase II are required for enzymatic activity and proper folding. JBIC J. Boil. Inorg. Chem. 2004, 271, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Kuratsukuri, K.; Newman, N.; Rovito, P.M.; Kaumaya, P.T.P.; Wang, C.Y.; Haas, G.P. Targeting epitopes in prostate-specific membrane antigen for antibody therapy of prostate cancer. Prostate Cancer Prostatic Dis. 2005, 8, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, D.; Olson, W.C.; Heston, W.D. In Vitro and In Vivo Responses of Advanced Prostate Tumors to PSMA ADC, an Auristatin-Conjugated Antibody to Prostate-Specific Membrane Antigen. Mol. Cancer Ther. 2011, 10, 1728–1739. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Penet, M.-F.; Nimmagadda, S.; Li, C.; Banerjee, S.R.; Winnard, P.T.; Artemov, D.; Glunde, K.; Pomper, M.G.; Bhujwalla, Z.M. PSMA-Targeted Theranostic Nanoplex for Prostate Cancer Therapy. ACS Nano 2012, 6, 7752–7762. [Google Scholar] [CrossRef] [PubMed]
- Knedlik, T.; Navratil, V.; Vik, V.; Pacík, D.; Sacha, P.; Konvalinka, J. Detection and quantitation of glutamate carboxypeptidase II in human blood. Prostate 2014, 74, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Tykvart, J.; Navratil, V.; Sedlak, F.; Corey, E.; Colombatti, M.; Fracasso, G.; Koukolík, F.; Bařinka, C.; Sacha, P.; Konvalinka, J. Comparative analysis of monoclonal antibodies against prostate-specific membrane antigen (PSMA). Prostate 2014, 74, 1674–1690. [Google Scholar] [CrossRef]
- Bates, D.; Abraham, S.; Campbell, M.; Zehbe, I.; Curiel, L. Development and Characterization of an Antibody-Labeled Super-Paramagnetic Iron Oxide Contrast Agent Targeting Prostate Cancer Cells for Magnetic Resonance Imaging. PLoS ONE 2014, 9, e97220. [Google Scholar] [CrossRef]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef]
- Murphy, G.P. Review of phase II hormone refractory prostate cancer trials. Urology 1999, 54, 19–21. [Google Scholar] [CrossRef]
- Galsky, M.D.; Eisenberger, M.; Moore-Cooper, S.; Kelly, W.K.; Slovin, S.F.; DeLaCruz, A.; Lee, Y.; Webb, I.J.; Scher, H.I. Phase I trial of the prostate-specific membrane antigen-directed immunoconjugate MLN2704 in patients with progressive Mcrpc. J. Clin. Oncol. 2008, 26, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.M.; Poty, S.; Sharma, S.K.; Lewis, J.S. Preclinical optimization of antibody-based radiopharmaceuticals for cancer imaging and radionuclide therapy-Model, vector, and radionuclide selection. J. Label. Compd. Radiopharm. 2018, 61, 611–635. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Shively, L.; Raubitschek, A.; Sherman, M.; Williams, L.E.; Wong, J.Y.; E Shively, J.; Wu, A.M. Minibody: A novel engineered anti-carcinoembryonic antigen antibody fragment (single-chain Fv-CH3) which exhibits rapid, high-level targeting of xenografts. Cancer Res. 1996, 56, 3055–3061. [Google Scholar]
- Viola, N.; Sevak, K.K.; Carlin, S.; Doran, M.G.; Evans, H.W.; Bartlett, D.W.; Wu, A.M.; Lewis, J.S. Noninvasive Imaging of PSMA in Prostate Tumors with 89Zr-Labeled huJ591 Engineered Antibody Fragments: The Faster Alternatives. Mol. Pharm. 2014, 11, 3965–3973. [Google Scholar] [CrossRef] [PubMed]
- Pandit-Taskar, N.; O’Donoghue, J.A.; Ruan, S.; Lyashchenko, S.K.; Carrasquillo, J.; Heller, G.; Martinez, D.F.; Cheal, S.M.; Lewis, J.S.; Fleisher, M.; et al. First-in-Human Imaging with 89Zr-Df-IAB2M Anti-PSMA Minibody in Patients with Metastatic Prostate Cancer: Pharmacokinetics, Biodistribution, Dosimetry, and Lesion Uptake. J. Nucl. Med. 2016, 57, 1858–1864. [Google Scholar] [CrossRef] [PubMed]
- Joraku, A.; Hatano, K.; Kawai, K.; Kandori, S.; Kojima, T.; Fukumitsu, N.; Isobe, T.; Mori, Y.; Sakata, M.; Hara, T.; et al. Phase I/IIa PET imaging study with 89zirconium labeled anti-PSMA minibody for urological malignancies. Ann. Nucl. Med. 2018, 33, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Holliger, P.; Prospero, T.; Winter, G. “Diabodies”: Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef]
- Kampmeier, F.; Williams, J.D.; Maher, J.; Mullen, G.E.; Blower, P.J. Design and preclinical evaluation of a 99mTc-labelled diabody of mAb J591 for SPECT imaging of prostate-specific membrane antigen (PSMA). EJNMMI Res. 2014, 4, 13. [Google Scholar] [CrossRef]
- Wong, P.; Li, L.; Chea, J.; Delgado, M.K.; Crow, D.; Poku, E.; Szpikowska, B.; Bowles, N.; Channappa, D.; Colcher, D.; et al. PET imaging of 64Cu-DOTA-scFv-anti-PSMA lipid nanoparticles (LNPs): Enhanced tumor targeting over anti-PSMA scFv or untargeted LNPs. Nucl. Med. Boil. 2017, 47, 62–68. [Google Scholar] [CrossRef]
- Frigerio, B.; Fracasso, G.; Luison, E.; Cingarlini, S.; Mortarino, M.; Coliva, A.; Seregni, E.; Bombardieri, E.; Zuccolotto, G.; Rosato, A.; et al. A single-chain fragment against prostate specific membrane antigen as a tool to build theranostic reagents for prostate cancer. Eur. J. Cancer 2013, 49, 2223–2232. [Google Scholar] [CrossRef]
- Lütje, S.; Van Rij, C.M.; Franssen, G.M.; Fracasso, G.; Helfrich, W.; Eek, A.; Oyen, W.; Colombatti, M.; Boerman, O.C. Targeting human prostate cancer with111In-labeled D2B IgG, F(ab′)2and Fab fragments in nude mice with PSMA-expressing xenografts. Contrast Media Mol. Imaging 2014, 10, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, B.; Franssen, G.; Luison, E.; Satta, A.; Seregni, E.; Colombatti, M.; Fracasso, G.; Valdagni, R.; Mezzanzanica, D.; Boerman, O.; et al. Full preclinical validation of the 123I-labeled anti-PSMA antibody fragment ScFvD2B for prostate cancer imaging. Oncotarget 2016, 8, 10919–10930. [Google Scholar]
- Frigerio, B.; Morlino, S.; Luison, E.; Seregni, E.; Lorenzoni, A.; Satta, A.; Valdagni, R.; Bogni, A.; Chiesa, C.; Mira, M.; et al. Anti-PSMA 124I-scFvD2B as a new immuno-PET tool for prostate cancer: Preclinical proof of principle. J. Exp. Clin. Cancer Res. 2019, 38, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, S.; Mullen, G.E.; Blower, P.J.; Ballinger, J.R. A 99mTc-labelled scFv antibody fragment that binds to prostate-specific membrane antigen. Nucl. Med. Commun. 2017, 38, 666–671. [Google Scholar] [CrossRef]
- Nawaz, S.; Mullen, G.E.D.; Sunassee, K.; Bordoloi, J.; Blower, P.J.; Ballinger, J.R. Simple, mild, one-step labelling of proteins with gallium-68 using a tris(hydroxypyridinone) bifunctional chelator: A 68Ga-THP-scFv targeting the prostate-specific membrane antigen. EJNMMI Res. 2017, 7, 86. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Lecocq, Q.; De Vlaeminck, Y.; Hanssens, H.; D’Huyvetter, M.; Raes, G.; Goyvaerts, C.; Keyaerts, M.; Devoogdt, N.; Breckpot, K. Theranostics in immuno-oncology using nanobody derivatives. Theranostics 2019, 9, 7772–7791. [Google Scholar] [CrossRef]
- Zare, H.; Rajabibazl, M.; Rasooli, I.; Ebrahimizadeh, W.; Bakherad, H.; Ardakani, L.S.; Gargari, S.L.M. Production of Nanobodies against Prostate-Specific Membrane Antigen (PSMA) Recognizing LnCaP Cells. Int. J. Boil. Mark. 2014, 29, 169–179. [Google Scholar] [CrossRef]
- Evazalipour, M.; D’Huyvetter, M.; Tehrani, B.S.; Abolhassani, M.; Omidfar, K.; Abdoli, S.; Arezumand, R.; Morovvati, H.; Lahoutte, T.; Muyldermans, S.; et al. Generation and characterization of nanobodies targeting PSMA for molecular imaging of prostate cancer. Contrast Media Mol. Imaging 2014, 9, 211–220. [Google Scholar] [CrossRef]
- Chatalic, K.L.; Veldhoven-Zweistra, J.; Bolkestein, M.; Hoeben, S.; Koning, G.A.; Boerman, O.C.; de Jong, M.; van Weerden, W.M. A Novel 111In-Labeled Anti-Prostate-Specific Membrane Antigen Nanobody for Targeted SPECT/CT Imaging of Prostate Cancer. J. Nucl. Med. 2015, 56, 1094–1099. [Google Scholar] [CrossRef]
- Nonnekens, J.; Chatalic, K.L.; Molkenboer-Kuenen, J.D.; Beerens, C.E.; Bruchertseifer, F.; Morgenstern, A.; Veldhoven-Zweistra, J.; Schottelius, M.; Wester, H.-J.; Van Gent, D.C.; et al. 213Bi-Labeled Prostate-Specific Membrane Antigen-Targeting Agents Induce DNA Double-Strand Breaks in Prostate Cancer Xenografts. Cancer Biother. Radiopharm. 2017, 32, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Bailey, C.W.; Huang, X.; Koide, S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Boil. 1998, 284, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, C.D.; Veerapandian, B.; Dai, X.-P.; Hamlin, R.C.; Xuong, N.-H.; Ruoslahti, E.; Ely, K.R. Crystal structure of the tenth type III cell adhesion module of human fibronectin. J. Mol. Boil. 1994, 236, 1079–1092. [Google Scholar] [CrossRef]
- Sha, F.; Salzman, G.; Gupta, A.; Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924. [Google Scholar] [CrossRef] [PubMed]
- Bloom, L.; Calabro, V. FN3: A new protein scaffold reaches the clinic. Drug Discov. Today 2009, 14, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Park, S.; Kim, D.-Y.; Pyo, A.; Kimura, R.H.; Sathirachinda, A.; Choy, H.E.; Min, J.-J.; Gambhir, S.S.; Hong, Y. Isolation and Characterization of a Monobody with a Fibronectin Domain III Scaffold That Specifically Binds EphA2. PLoS ONE 2015, 10, e0132976. [Google Scholar] [CrossRef]
- Kiess, A.; Minn, I.; Chen, Y.; Hobbs, R.; Sgouros, G.; Mease, R.C.; Pullambhatla, U.; Shen, C.J.; Foss, C.A.; Pomper, M.G. Auger Radiopharmaceutical Therapy Targeting Prostate-Specific Membrane Antigen. J. Nucl. Med. 2015, 56, 1401–1407. [Google Scholar] [CrossRef]
- Jackson, P.F.; Cole, D.C.; Slusher, B.S.; Stetz, S.L.; Ross, L.E.; Donzanti, B.A.; Trainor, D.A. Design, Synthesis, and Biological Activity of a Potent Inhibitor of the Neuropeptidase N-Acetylated α-Linked Acidic Dipeptidase. J. Med. Chem. 1996, 39, 619–622. [Google Scholar] [CrossRef]
- Liu, T.; Wu, L.Y.; Kazak, M.; Berkman, C.E. Cell-Surface labeling and internalization by a fluorescent inhibitor of prostate-specific membrane antigen. Prostate 2008, 68, 955–964. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Nan, F.; Conti, P.; Zhang, J.; Ramadan, E.; Bzdega, T.; Wroblewska, B.; Neale, J.H.; Pshenichkin, S.; Wroblewski, J.T.; et al. Design of Remarkably Simple, Yet Potent Urea-Based Inhibitors of Glutamate Carboxypeptidase II (NAALADase). J. Med. Chem. 2001, 44, 298–301. [Google Scholar] [CrossRef]
- Bodei, L.; Kassis, A.I.; Adelstein, S.J.; Mariani, G. Radionuclide Therapy with Iodine-125 and Other Auger–Electron-Emitting Radionuclides: Experimental Models and Clinical Applications. Cancer Biother. Radiopharm. 2003, 18, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Kiess, A.P.; Minn, I.; Vaidyanathan, G.; Hobbs, R.F.; Josefsson, A.; Shen, C.; Brummet, M.; Chen, Y.; Choi, J.; Koumarianou, E.; et al. (2S)-2-(3-(1-Carboxy-5-(4-211At-Astatobenzamido)Pentyl)Ureido)-Pentanedioic Acid for PSMA-Targeted α-Particle Radiopharmaceutical Therapy. J. Nucl. Med. 2016, 57, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Hillier, S.M.; Maresca, K.P.; Femia, F.J.; Marquis, J.C.; Foss, C.A.; Nguyen, N.; Zimmerman, C.N.; Barrett, J.A.; Eckelman, W.C.; Pomper, M.G.; et al. Preclinical evaluation of novel glutamateurea-lysine analogues that target prostate-specific membrane antigen as molecular imaging pharmaceuticals for prostate cancer. Cancer Res. 2009, 69, 6932–6940. [Google Scholar] [CrossRef] [PubMed]
- Maresca, K.P.; Hillier, S.M.; Femia, F.J.; Keith, D.; Barone, C.; Joya, J.L.; Zimmerman, C.N.; Kozikowski, A.P.; Barrett, J.A.; Eckelman, W.C.; et al. A series of halogenated heterodimeric inhibitors of prostate specific membrane antigen (PSMA) as radiolabeled probes for targeting prostate cancer. J. Med. Chem. 2009, 52, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Kopka, K.; Benešová, M.; Bařinka, C.; Haberkorn, U.; Babich, J. Glu-Ureido-Based Inhibitors of Prostate-Specific Membrane Antigen: Lessons Learned During the Development of a Novel Class of Low-Molecular-Weight Theranostic Radiotracers. J. Nucl. Med. 2017, 58, 17S–26S. [Google Scholar] [CrossRef]
- Yeong, C.H.; Cheng, M.H.; Ng, K.H. Therapeutic radionuclides in nuclear medicine: Current and future prospects. J. Zhejiang Univ. Sci. B 2014, 15, 845–863. [Google Scholar] [CrossRef] [PubMed]
- Kassis, A.I. Therapeutic radionuclides: Biophysical and radiobiologic principles. Semin. Nucl. Med. 2008, 38, 358–366. [Google Scholar] [CrossRef]
- Westcott, M.A.; Coldwell, D.M.; Liu, D.M.; Zikria, J.F. The development, commercialization, and clinical context of yttrium-90 radiolabeled resin and glass microspheres. Adv. Radiat. Oncol. 2016, 1, 351–364. [Google Scholar] [CrossRef]
- Chakravarty, R.; Dash, A.; Pillai, M.R. Availability of yttrium-90 from strontium-90: A nuclear medicine perspective. Cancer Biother. Radiopharm. 2012, 27, 621–641. [Google Scholar] [CrossRef]
- Das, T.; Banerjee, S. Theranostic Applications of Lutetium-177 in Radionuclide Therapy. Curr. Radiopharm. 2016, 9, 94–101. [Google Scholar] [CrossRef]
- Dash, A.; Pillai, M.R.; Knapp, F.F., Jr. Production of 177Lu for Targeted Radionuclide Therapy: Available Options. Nucl. Med. Mol. Imaging 2015, 49, 85–107. [Google Scholar] [CrossRef] [PubMed]
- Mettler, F.A.; Guiberteau, M.J. Essentials of Nuclear Medicine and Molecular Imaging, 7th ed.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Müller, C.; Umbricht, C.A.; Gracheva, N.; Tschan, V.J.; Pellegrini, G.; Bernhardt, P.; Zeevaart, J.R.; Köster, U.; Schibli, R.; van der Meulen, N.P. Terbium-161 for PSMA-targeted radionuclide therapy of prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Wulbrand, C.; Seidl, C.; Gaertner, F.C.; Bruchertseifer, F.; Morgenstern, A.; Essler, M.; Senekowitsch-Schmidtke, R. Alpha-particle emitting 213Bi-anti-EGFR immunoconjugates eradicate tumor cells independent of oxygenation. PLoS ONE 2013, 8, e64730. [Google Scholar] [CrossRef] [PubMed]
- Kozempel, J.; Mokhodoeva, O.; Vlk, M. Progress in targeted alpha-particle therapy. What we learned about recoils release from in vivo generators. Molecules 2018, 23, 581. [Google Scholar] [CrossRef]
- de Kruijff, R.M.; Wolterbeek, H.T.; Denkova, A.G. A critical review of alpha radionuclide therapy-how to deal with recoiling daughters? Pharmaceuticals 2015, 8, 321–336. [Google Scholar] [CrossRef]
- Aghevlian, S.; Boyle, A.J.; Reilly, R.M. Radioimmunotherapy of cancer with high linear energy transfer (LET) radiation delivered by radionuclides emitting α-particles or Auger electrons. Adv. Drug Deliv. Rev. 2017, 109, 102–118. [Google Scholar] [CrossRef]
- Schwartz, J.; Jaggi, J.S.; O’Donoghue, J.A.; Ruan, S.; McDevitt, M.; Larson, S.M.; Scheinberg, D.A.; Humm, J.L. Renal uptake of bismuth-213 and its contribution to kidney radiation dose following administration of actinium-225-labeled antibody. Phys. Med. Biol. 2011, 56, 721–733. [Google Scholar] [CrossRef]
- Nedrow, J.R.; Josefsson, A.; Park, S.; Hobbs, R.F.; Bruchertseifer, F.; Morgenstern, A.; Sgouros, G. Reducing renal uptake of free 213Bi associated with the decay of 225Ac-labeled radiopharmaceuticals. In Proceedings of the 10th International Symposium on Targeted Alpha Therapy, Kanazawa, Japan, 30 May–1 June 2017; p. 67. [Google Scholar]
- Morgenstern, A.; Bruchertseifer, F.; Apostolidis, C. Bismuth-213 and actinium-225 generator performance and evolving therapeutic applications of two generator-derived alpha-emitting radioisotopes. Curr. Radiopharm. 2012, 5, 221–227. [Google Scholar] [CrossRef]
- Dekempeneer, Y.; Keyaerts, M.; Krasniqi, A.; Puttemans, J.; Muyldermans, S.; Lahoutte, T.; D’huyvetter, M.; Devoogdt, N. Targeted alpha therapy using short-lived alpha-particles and the promise of nanobodies as targeting vehicle. Expert Opin. Biol. Ther. 2016, 16, 1035–1047. [Google Scholar] [CrossRef]
- Griswold, J.R.; Medvedev, D.G.; Engle, J.W.; Copping, R.; Fitzsimmons, J.M.; Radchenko, V.; Cooley, J.C.; Fassbender, M.E.; Denton, D.L.; Murphy, K.E.; et al. Large scale accelerator production of 225Ac: Effective cross sections for 78-192MeV protons incident on 232Th targets. Appl. Radiat. Isot. 2016, 118, 366–374. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Pruszynski, M. Astatine-211: Production and availability. Curr. Radiopharm. 2011, 4, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Eckerman, K.; Endo, A. ICRP Publication 107. Nuclear decay data for dosimetric calculations. Ann. ICRP 2008, 38, 7–96. [Google Scholar] [PubMed]
- Volterrani, D.; Erba, P.A.; Carrió, I.H.; Strauss, W.; Mariani, G. Nuclear Medicine Textbook: Methodology and Clinical Applications; Volterrani, D., Erba, P.A., Carrio, I., Strauss, H.W., Mariani, G., Eds.; Springer: New York, NY, USA, 2019. [Google Scholar]
- Henriksen, G.; Hoff, P.; Larsen, R.H. Evaluation of potential chelating agents for radium. Appl. Radiat. Isot. 2002, 56, 667–671. [Google Scholar] [CrossRef]
- Henriksen, G.; Schoultz, B.W.; Michaelsen, T.E.; Bruland, Ø.S.; Larsen, R.H. Sterically stabilized liposomes as a carrier for alpha-emitting radium and actinium radionuclides. Nucl. Med. Biol. 2004, 31, 441–449. [Google Scholar] [CrossRef]
- Piotrowska, A.; Leszczuk, E.; Bruchertseifer, F.; Morgenstern, A.; Bilewicz, A. Functionalized NaA nanozeolites labeled with 224,225Ra for targeted alpha therapy. J. Nanopart. Res. 2013, 15, 2082. [Google Scholar] [CrossRef]
- Mokhodoeva, O.; Vlk, M.; Málková, E.; Kukleva, E.; Mičolová, P.; Štamberg, K.; Šlouf, M.; Dzhenloda, R.; Kozempel, J. Study of 223Ra uptake mechanism by Fe3O4 nanoparticles: Towards new prospective theranostic SPIONs. J. Nanopart. Res. 2016, 18, 301–313. [Google Scholar] [CrossRef]
- Gott, M.; Yang, P.; Kortz, U.; Stephan, H.; Pietzsch, H.J.; Mamat, C. A 224Ra-labeled polyoxopalladate as a putative radiopharmaceutical. Chem. Commun. 2019, 55, 7631–7634. [Google Scholar] [CrossRef]
- Kozempel, J.; Vlk, M.; Málková, E.; Bajzíková, A.; Bárta, J.; Santos-Oliveira, R.; Malta Rossi, A. Prospective carriers of 223Ra for targeted alpha particle therapy. J. Radioanal. Nucl. Chem. 2015, 304, 443–447. [Google Scholar] [CrossRef]
- Suchánková, P.; Kukleva, E.; Štamberg, K.; Nykl, P.; Vlk, M.; Kozempel, J. Study of 223Ra uptake mechanism on hydroxyapatite and titanium dioxide nanoparticles as a function of pH. RSC Adv. 2020, 10, 3659–3666. [Google Scholar] [CrossRef]
- Hogle, S.; Boll, R.A.; Murphy, K.; Denton, D.; Owens, A.; Haverlock, T.J.; Garland, M.; Mirzadeh, S. Reactor production of Thorium-229. Appl. Radiat. Isot. 2016, 114, 19–27. [Google Scholar] [CrossRef]
- Banerjee, S.R.; Minn, I.; Kumar, V.; Josefsson, A.; Lisok, A.; Brummet, M.; Chen, J.; Kiess, A.P.; Baidoo, K.; Brayton, C.; et al. Preclinical Evaluation of 203/212Pb-Labeled Low-Molecular-Weight Compounds for Targeted Radiopharmaceutical Therapy of Prostate Cancer. J. Nucl. Med. 2020, 61, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Kassis, A.I. Cancer therapy with Auger electrons: Are we almost there? J. Nucl. Med. 2003, 44, 1479–1481. [Google Scholar] [PubMed]
- Ku, A.; Facca, V.J.; Cai, Z.; Reilly, R.M. Auger electrons for cancer therapy—A review. EJNMMI Radiopharm. Chem. 2019, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Kahn, D.; Williams, R.D.; Seldin, D.W.; Libertino, J.A.; Hirschhorn, M.; Dreicer, R.; Weiner, G.J.; Bushnell, D.; Gulfo, J. Radioimmunoscintigraphy with 111 indium labeled CYT-356 for the detection of occult prostate cancer recurrence. J. Urol. 1994, 152, 1490–1495. [Google Scholar] [CrossRef]
- Deb, N.; Goris, M.; Trisler, K.; Fowler, S.; Saal, J.; Ning, S.; Becker, M.; Marquez, C.; Knox, S. Treatment of hormone-refractory prostate cancer with 90Y-CYT-356 monoclonal antibody. Clin. Cancer Res. 1996, 2, 1289–1297. [Google Scholar] [PubMed]
- Smith-Jones, P.M.; Vallabahajosula, S.; Goldsmith, S.J.; Navarro, V.; Hunter, C.J.; Bastidas, D.; Bander, N.H. In Vitro characterization of radiolabeled monoclonal antibodies specific for the extracellular domain of prostate-specific membrane antigen. Cancer Res. 2000, 60, 5237–5243. [Google Scholar]
- Smith-Jones, P.M.; Vallabhajosula, S.; St. Omer, S.; Navarro, V.; Goldsmith, S.J.; Bander, N.H. 177Lu-DOTA-HuJ591: A new radiolabeled monoclonal antibody (mAb) for targeted therapy of prostate cancer. J. Label. Compds. Radiopharm. 2001, 44, 90–92. [Google Scholar] [CrossRef]
- Smith-Jones, P.M.; Vallabhajosula, S.; Navarro, V.; Bastidas, D.; Goldsmith, S.J.; Bander, N.H. Radiolabeled monoclonal antibodies specific to the extracellular domain of prostate-specific membrane antigen: Preclinical studies in nude mice bearing LNCaP human prostate tumor. J. Nucl. Med. 2003, 44, 610–617. [Google Scholar]
- Vallabhajosula, S.; Smith-Jones, P.M.; Navarro, V.; Goldsmith, S.J.; Bander, N.H. Radioimmunotherapy of Prostate cancer in human xenografts using monoclonal antibodies specific to prostate specific membrane antigen: Studies in nude mice. Prostate 2004, 58, 145–155. [Google Scholar] [CrossRef]
- Regino, C.A.; Wong, K.J.; Milenic, D.E.; Holmes, E.H.; Garmestani, K.; Choyke, P.L.; Brechbiel, M.W. Preclinical evaluation of a monoclonal antibody (3C6) specific for prostate-specific membrane antigen. Curr. Radiopharm. 2009, 2, 9–17. [Google Scholar] [CrossRef]
- Behe, M.; Alt, K.; Deininger, F.; Buhler, P.; Wetterauer, U.; Weber, W.A.; Elsässer-Beile, U.; Wolf, P. In Vivo testing of 177Lu-labelled anti-PSMA antibody as a new radioimmunotherapeutic agent against prostate cancer. In Vivo 2011, 25, 55–59. [Google Scholar] [PubMed]
- Bander, N.H.; Trabulsi, E.J.; Kostakoglu, L.; Yao, D.; Vallabhajosula, S.; Smith-Jones, P.; Joyce, M.A.; Milowsky, M.; Nanus, D.M.; Goldsmith, S.J. Targeting metastatic prostate cancer with radiolabeled monoclonal antibody J591 to the extracellular domain of prostate specific membrane antigen. J. Urol. 2003, 170, 171. [Google Scholar] [CrossRef] [PubMed]
- Milowsky, M.I.; Nanus, D.M.; Kostakoglu, L.; Vallabhajosula, S.; Goldsmith, S.J.; Bander, N.H. Phase I trial of 90Y-labeled anti-prostate specific membrane antigen monoclonal antibody J591 for androgen-independent prostate cancer. J. Clin. Oncol. 2004, 22, 2522–2531. [Google Scholar] [CrossRef] [PubMed]
- Bander, N.H.; Milowsky, M.I.; Nanus, D.M.; Kostakoglu, L.; Vallabhajosula, S.; Goldsmith, S.J. Phase I trial of 177lutetiumlabeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J. Clin. Oncol. 2005, 23, 4591–4601. [Google Scholar] [CrossRef] [PubMed]
- Vallabhajosula, S.; Goldsmith, S.J.; Kostakoglu, L.; Milowsky, M.I.; Nanus, D.M.; Bander, N.H. Radioimmunotherapy of Prostate Cancer Using 90Y- and 177Lu-Labeled J591 monoclonal antibodies: Effect of multiple treatments on myelotoxicity. Clin. Cancer Res. 2005, 11, 7195s–7200s. [Google Scholar] [CrossRef] [PubMed]
- Vallabhajosula, S.; Goldsmith, S.J.; Hamacher, K.A.; Kostakoglu, L.; Konishi, S.; Milowski, M.I.; Nanus, D.M.; Bander, N.H. Prediction of myelotoxicity based on bone marrow radiation-absorbed dose: Radioimmunotherapy studies using 90Y- and 177Lu-labeled J591 antibodies specific for prostate-specific membrane antigen. J. Nucl. Med. 2005, 46, 850–858. [Google Scholar]
- Vallabhajosula, S.; Kuji, I.; Hamacher, A.; Konishi, S.; Kostakoglu, L.; Kothari, P.A.; Milowski, M.I.; Nanus, D.M.; Bander, N.H.; Goldsmith, S.J. Pharmacokinetics and biodistribution of 111In- and 177Lu-labeled J591 antibody specific for prostate-specific membrane antigen: Prediction of 90Y-J591 radiation dosimetry based on 111In or 177Lu? J. Nucl. Med. 2005, 46, 634–641. [Google Scholar]
- Tagawa, S.T.; Milowsky, M.I.; Morris, M.J.; Vallabhajosula, S.; Goldsmith, S.; Matulich, D.; Nanus, D.M.; Kaplan, J.; Berger, F.; Scher, H.I.; et al. Phase II trial of 177Lutetium radiolabeled anti-prostate-specific membrane antigen (PSMA) monoclonal antibody J591 (177Lu-J591) in patients with metastatic castrate resistent prostate cancer. J. Clin. Oncol. 2008, 26, 5140. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Milowsky, M.I.; Morris, M.; Vallabhajosula, S.; Christos, P.; Akhtar, N.H.; Osborne, J.; Goldsmith, S.J.; Larson, S.; Taskar, N.P.; et al. Phase II study of lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for mCRPC. Clin. Cancer Res. 2013, 19, 5182–5191. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Vallabhajosula, S.; Osborne, J.; Goldsmith, S.J.; Petrillo, K.; Tyrell, L.; Dhillon, G.S.; Beltran, H.; Bander, N.H.; Nanus, D.M. Phase I trial of fractionated-dose 177-lutetium radiolabeled anti-prostatespecific membrane antigen (PSMA) monoclonal antibody J591 (177Lu-J591) in patients (pts) with metastatic castration resistant prostate cancer (metCRPC). J. Clin. Oncol. 2010, 28, 4667. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Akhtar, N.H.; Nikolopoulou, A.; Kaur, G.; Robinson, B.; Kahn, R.; Vallabhajosula, S.; Goldsmith, S.J.; Nanus, D.M.; Bander, N.H. Bone marrow recovery and subsequent chemotherapy following radiolabeled anti-prostate-specific membrane antigen monoclonal antibody J591 in men with mCRPC. Front. Oncol. 2013, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Whang, Y.E.; Kaur, K.; Vallabhajosula, S.; Paul, J.C.; Nikolopoulou, A.; Jhanwar, Y.; Sheikh, A.; Ireland, A.; Garcias-Espana, C.; et al. Phase I trial of docetaxel/prednisone plus fractionated dose radiolabeled anti-prostate specific membrane antigen (PSMA) monoclonal antibody 177lu-J591 in patients with metastatic, castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2014. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Vallabhajosula, S.; Christos, P.J.; Jhanwar, Y.S.; Batra, J.S.; Lam, L.; Osborne, J.; Beltran, H.; Molina, A.M.; Goldsmith, S.J.; et al. Phase 1/2 study of fractionated dose lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 (177Lu-J591) for mCRPC. Cancer 2019, 125, 2561–2569. [Google Scholar] [PubMed]
- Weineisen, M.; Schottelius, M.; Simecek, J.; Eiber, M.; Schwaiger, M.; Wester, H. Development and first in human evaluation of PSMA I&T—A ligand for diagnostic imaging and endoradiotherapy of prostate cancer. J. Nucl. Med. 2014, 55, 1083. [Google Scholar]
- Weineisen, M.; Schottelius, M.; Simecek, J.; Baum, R.P.; Yildiz, A.; Beykan, S.; Kulkarni, H.R.; Lassmann, M.; Klette, I.; Eiber, M.; et al. 68Ga- and 177Lu-labeled PSMA I&T: Optimization of a PSMA-targeted theranostic concept and first proof-of-concept human studies. J. Nucl. Med. 2015, 56, 1169–1176. [Google Scholar] [PubMed]
- Kratochwil, C.; Giesel, F.L.; Eder, M.; Afshar-Oromieh, A.; Benešová, M.; Mier, W.; Kopka, K.; Haberkorn, U. [177Lu]lutetium-labelled PSMA ligand induced remission in a patient with metastatic prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 987–988. [Google Scholar] [CrossRef]
- Zechmann, C.M.; Afshar-Oromieh, A.; Armor, T.; Stubbs, J.; Mier, W.; Hadaschik, B.; Joyal, J.; Kopka, K.; Debus, J.; Babich, J.W.; et al. Radiation dosimetry and first therapy results with a 124I/131I-labeled small molecule (MIP-1095) targeting PSMA for prostate cancer therapy. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1280–1292. [Google Scholar] [CrossRef]
- Okamoto, S.; Thieme, A.; Allmann, J.; D’Alessandria, C.; Maurer, T.; Retz, M.; Tauber, R.; Heck, M.M.; Wester, H.J.; Tamaki, N.; et al. Radiation dosimetry for 177Lu-PSMA I&T in mCRPC: Absorbed dose in normal organs and tumor lesions. J. Nucl. Med. 2017, 58, 445–450. [Google Scholar]
- Baum, R.P.; Kulkarni, H.R.; Schuchardt, C.; Singh, A.; Wirtz, M.; Wiessalla, S.; Schottelius, M.; Mueller, D.; Klette, I.; Wester, H.J. 177Lu-labeled prostate-specific membrane antigen radioligand therapy of mCRPC: Safety and efficacy. J. Nucl. Med. 2016, 57, 1006–1013. [Google Scholar] [CrossRef]
- Kulkarni, H.R.; Singh, A.; Schuchardt, C.; Niepsch, K.; Sayeg, M.; Leshch, Y.; Wester, H.J.; Baum, R.P. PSMA-Based Radioligand Therapy for MCRPC: The Bad Berka Experience Since 2013. J. Nucl. Med. 2016, 57, 97S–104S. [Google Scholar] [CrossRef]
- Ahmadzadehfar, H.; Rahbar, K.; Kurpig, S.; Bogemann, M.; Claesener, M.; Eppard, E.; Gartner, F.; Rogenhofer, S.; Schafers, M.; Essler, M. Early side effects and first results of radioligand therapy with (177)Lu-DKFZ-617 PSMA of castrate-resistant metastatic prostate cancer: A two-centre study. EJNMMI Res. 2015, 5, 114. [Google Scholar] [CrossRef]
- Heck, M.M.; Retz, M.; D’Alessandria, C.; Rauscher, I.; Scheidhauer, K.; Storz, E.; Janssen, F.; Schottelius, M.; Wester, H.J.; Gschwend, J.E.; et al. Systemic Radioligand Therapy with (177)Lu Labeled Prostate Specific Membrane Antigen Ligand for Imaging and Therapy in Patients with Metastatic Castration Resistant Prostate Cancer. J. Urol. 2016, 196, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadehfar, H.; Eppard, E.; Kurpig, S.; Fimmers, R.; Yordanova, A.; Schlenkhoff, C.D.; Gärtner, F.; Rogenhofer, S.; Essler, M. Therapeutic response and side effects of repeated radioligand therapy with 177Lu-PSMA-DKFZ-617 of castrate-resistant metastatic prostate cancer. Oncotarget 2016, 7, 12477–12488. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, K.; Schmidt, M.; Heinzel, A.; Eppard, E.; Bode, A.; Yordanova, A.; Claesener, M.; Ahmadzadehfar, H. Response and tolerability of a single dose of 177Lu-PSMA-617 in patients with mCRPC: A multicenter retrospective analysis. J. Nucl. Med. 2016, 57, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Hohberg, M.; Eschner, W.; Schmidt, M.; Dietlein, M.; Kobe, C.; Fischer, T.; Drzezga, A.; Wild, M. Lacrimal Glands May Represent Organs at Risk for Radionuclide Therapy of Prostate Cancer with [(177)Lu]DKFZ-PSMA-617. Mol. Imaging Biol. 2016, 18, 437–445. [Google Scholar] [CrossRef]
- Delker, A.; Fendler, W.P.; Kratochwil, C.; Brunegraf, A.; Gosewisch, A.; Gildehaus, F.J.; Tritschler, S.; Stief, C.G.; Kopka, K.; Haberkorn, U.; et al. Dosimetry for (177)Lu-DKFZ-PSMA-617: A new radiopharmaceutical for the treatment of metastatic prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 42–51. [Google Scholar] [CrossRef]
- Fendler, W.P.; Reinhardt, S.; Ilhan, H.; Delker, A.; Böning, G.; Gildehaus, F.J.; Stief, C.; Bartenstein, P.; Gratzke, C.; Lehner, S.; et al. Preliminary experience with dosimetry, response and patient reported outcome after 177Lu-PSMA-617 therapy for mCRPC. Oncotarget 2017, 8, 3581–3590. [Google Scholar] [CrossRef]
- Yadav, M.P.; Ballal, S.; Tripathi, M.; Damle, N.A.; Sahoo, R.K.; Seth, A.; Bal, C. Post-therapeutic dosimetry of 177Lu-DKFZ-PSMA-617 in the treatment of patients with mCRPC. Nucl. Med. Commun. 2017, 38, 91–98. [Google Scholar] [CrossRef]
- Scarpa, L.; Buxbaum, S.; Kendler, D.; Fink, K.; Bektic, J.; Gruber, L.; Decristoforo, C.; Uprimny, C.; Lukas, P.; Horninger, W.; et al. The 68Ga/177Lu theragnostic concept in PSMA targeting of castration-resistant prostate cancer: Correlation of SUVmax values and absorbed dose estimates. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 788–800. [Google Scholar] [CrossRef]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Thang, S.P.; Akhurst, T.; Iravani, A.; Kong, G.; Ravi Kumar, A.; Murphy, D.G.; et al. [177Lu] PSMA radionuclide treatment in patients with mCRPC (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Edler von Eyben, F.; Singh, A.; Zhang, J.; Nipsch, K.; Meyrick, D.; Lenzo, N.; Kairemo, K.; Joensuu, T.; Virgolini, I.; Soydal, C.; et al. 177Lu-PSMA radioligand therapy of predominant lymph node metastatic prostate cancer. Oncotarget 2019, 10, 2451–2461. [Google Scholar] [PubMed]
- Rahbar, K.; Ahmadzadehfar, H.; Kratochwil, C.; Haberkorn, U.; Schäfers, M.; Essler, M.; Baum, R.P.; Kulkarni, H.R.; Schmidt, M.; Drzezga, A.; et al. German multicenter study investigating 177Lu-PSMA-617 radioligand therapy in advanced prostate cancer patients. J. Nucl. Med. 2017, 58, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Giesel, F.L.; Stefanova, M.; Benesova, M.; Bronzel, M.; Afshar-Oromieh, A.; Mier, W.; Eder, M.; Kopka, K.; Haberkorn, U. PSMA-Targeted Radionuclide Therapy of MCRPC with 177Lu-Labeled PSMA-617. J. Nucl. Med. 2016, 57, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, M.R.; Barendswaard, E.; Ma, D.; Lai, L.; Curcio, M.J.; Sgouros, G.; Ballangrud, A.M.; Yang, W.H.; Finn, R.D.; Pellegrini, V.; et al. An alpha-particle emitting antibody 213Bi-J591 for radioimmuno-therapy of prostate cancer. Cancer Res. 2000, 60, 6095–6100. [Google Scholar] [PubMed]
- Ballangrud, A.M.; Yang, W.H.; Charlton, D.E.; McDevitt, M.R.; Hamacher, K.A.; Panageas, K.S.; Ma, D.; Bander, N.H.; Scheinberg, D.A.; Sgouros, G. Response of LNCaP spheroids after treatment with an alpha-particle emitter (213Bi)-labeled anti-prostate-specific membrane antigen antibody (J591). Cancer Res. 2001, 61, 2008–2014. [Google Scholar] [PubMed]
- Li, Y.; Tian, Z.; Rizv, S.M.; Bander, N.H.; Allen, B.J. In Vitro and preclinical targeted alpha therapy of human prostate cancer with Bi-213 labeled J59 antibody against the prostate specific membrane antigen. Prost. Cancer Prostat. Dis. 2002, 5, 36–46. [Google Scholar] [CrossRef]
- Hammer, S.; Larssen, A.; Ellingsen, C.; Geraudie, S.; Grant, D.; Indrevoll, B.; von Ahsen, O.; Kristian, A.; Hagemann, U.B.; Karlsson, J.; et al. Preclinical pharmacology of the PSMA-targeted thorium-227 conjugate PSMA-TTC: A novel targeted alpha therapeutic for the treatment of prostate cancer. In Proceedings of the American Association for Cancer Research Annual Meeting 2017, Washington, DC, USA, 1–5 April 2017. [Google Scholar]
- Hammer, S.; Hagemann, U.B.; Zitzmann-Kolbe, S.; Larsen, A.; Ellingsen, C.; von Ahsen, O.; Karlsson, J.; Bjerke, R.M.; Ryan, O.B.; Lejeune, P.; et al. Preclinical activity of PSMA-TTC, a targeted alpha therapeutic in patient-derived prostate cancer models. In Proceedings of the American Association for Cancer Research Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.L.; Weis, M.; Verburg, F.A.; Mottaghy, F.; Kopka, K.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. 225-Ac-PSMA-617 for PSMA-Targeted α-Radiation Therapy of Metastatic Castration-Resistant Prostate Cancer. J. Nucl. Med. 2016, 57, 1941–1944. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Bronzel, M.; Apostolidis, C.; Weichert, W.; Haberkorn, U.; Giesel, F.L.; Morgenstern, A. Targeted α-therapy of metastatic castration resistant prostate cancer with 225Ac-PSMA-617: Dosimetry estimate and empiric dose finding. J. Nucl. Med. 2017, 58, 1624–1631. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Hohenfellner, M.; Giesel, F.L.; Haberkorn, U.; Morgenstern, A. Targeted α-therapy of metastatic castration resistant prostate cancer with 225Ac-PSMA-617: Swimmer-plot analysis suggests efficacy regarding duration of tumor control. J. Nucl. Med. 2018, 59, 795–802. [Google Scholar] [CrossRef]
- Khreish, F.; Ebert, N.; Ries, M.; Maus, S.; Rosar, F.; Bohnenberger, H.; Stemler, T.; Saar, M.; Bartholomä, M.; Ezziddin, S. 225Ac-PSMA-617/177Lu-PSMA-617 tandem therapy of mCRPC: Pilot experience. Eur. J. Nucl. Med. Mol. Imaging 2019. [Google Scholar] [CrossRef]
- Sathekge, M.; Bruchertseifer, F.; Knoesen, O.; Reyneke, F.; Lawal, I.; Lengana, T.; Davis, C.; Mahapane, J.; Corbett, C.; Vorster, M.; et al. 225Ac-PSMA-617 in chemotherapy-naive patients with advanced prostate cancer: A pilot study. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 129–138. [Google Scholar] [PubMed]
- Sathekge, M.; Bruchertseifer, F.; Vorster, M.; Lawal, I.; Knoesen, O.; Mahapane, J.; Davis, C.; Reyneke, F.; Maes, A.; Kratochwil, C.; et al. Predictors of overall and disease free survival in metastatic castration-resistant prostate cancer patients receiving 225Ac-PSMA-617 radioligand therapy. J. Nucl. Med. 2020, 61, 62–69. [Google Scholar] [PubMed]
- Sathekge, M.; Knoesen, O.; Meckel, M.; Modiselle, M.; Vorster, M.; Marx, S. 213Bi-PSMA-617 targeted alpha-radionuclide therap in mCRPC. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1099–1100. [Google Scholar] [PubMed]
- Kratochwil, C.; Schmidt, K.; Afshar-Oromieh, A.; Bruchertseifer, F.; Rathke, H.; Morgenstern, A.; Haberkorn, U.; Giesel, F.L. Targeted alpha therapy of mCRPC: Dosimetry estimate of 213Bismuth-PSMA-617. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 31–37. [Google Scholar]
- Müller, C.; Singh, A.; Umbricht, C.A.; Kulkarni, H.R.; Johnston, K.; Benešová, M.; Senftleben, S.; Müller, D.; Vermeulen, C.; Schibli, R.; et al. Preclinical investigations and first-in-human application of 152Tb-PSMA-617 for PET/CT imaging of prostate cancer. EJNMMI Res. 2019, 9, 68. [Google Scholar]
- Karsten, B.B.; Liu, T.; Nedrow-Byers, J.R.; Benny, P.D.; Berkman, C.E. Targeting prostate cancer cells with PSMA inhibitor-guided gold nanoparticles. Bioorg. Med. Chem. Lett. 2013, 23, 565–568. [Google Scholar]
- Lu, W.; Singh, A.K.; Khan, S.A.; Senapati, D.; Yu, H.; Ray, P.C. Gold nano-popcorn-based targeted diagnosis, nanotherapy treatment, and in situ monitoring of photothermal therapy response of prostate cancer cells using surface-enhanced Raman spectroscopy. J. Am. Chem. Soc. 2010, 132, 18103–18114. [Google Scholar]
- Flores, O.; Santra, S.; Kaittanis, C.; Bassiouni, R.; Khaled, A.S.; Khaled, A.; Grimm, J.; Perez, J.M. PSMA-Targeted Theranostic Nanocarrier for Prostate Cancer. Theranostics 2017, 7, 2477–2494. [Google Scholar]
- Mangadlao, J.D.; Wang, X.; McCleese, C.; Escamilla, M.; Ramamurthy, G.; Wang, Z.; Govande, M.; Basilion, J.P.; Burda, C. Prostate-Specific Membrane Antigen Targeted Gold Nanoparticles for Theranostics of Prostate Cancer. ACS Nano 2018, 12, 3714–3725. [Google Scholar]
- Bandekar, A.; Zhu, C.; Jindal, R.; Bruchertseifer, F.; Morgenstern, A.; Sofou, S. Anti-prostate-specific membrane antigen liposomes loaded with 225Ac for potential targeted antivascular α-particle therapy of cancer. J. Nucl. Med. 2014, 55, 107–114. [Google Scholar]
- Zhu, C.; Bandekar, A.; Sempkowski, M.; Banerjee, S.R.; Pomper, M.G.; Bruchertseifer, F.; Morgenstern, A.; Sofou, S. Nanoconjugation of PSMA-Targeting Ligands Enhances Perinuclear Localization and Improves Efficacy of Delivered Alpha-Particle Emitters against Tumor Endothelial Analogues. Mol. Cancer Ther. 2016, 15, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Miyahira, A.K.; Pienta, K.J.; Morris, M.J.; Bander, N.H.; Baum, R.P.; Fendler, W.P.; Goeckeler, W.; Gorin, M.A.; Hennekes, H.; Pomper, M.G.; et al. Meeting report from the Prostate Cancer Foundation PSMA-directed radionuclide scientific working group. Prostate 2018, 78, 775–789. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radionuclide | Emitted Particle | Half-Life | Maximum Range in Tissue [mm] | Energy of Emitted Particle [MeV] | Production Mode | Availability | References |
|---|---|---|---|---|---|---|---|
| Yttrium-90 | β− | 2.67 days | 12 | 2.28 | 90Sr/90Y generator | Commercially available (low price) | [118,119] |
| Lutetium-177 | β− | 6,65 days | 1.6 | 0.49 | 176Lu(n,γ)177Lu | Commercially available (low price) | [120,121] |
| 176Yb(n,γ) 177Yb 177Lu | Commercially available (high price) | ||||||
| Iodine-131 | β−/γ | 8.02 days | 2.3 | 0.97 | natTe(n,γ)131I | Commercially available (low price) | [122] |
| Terbium-161 | β−/Auger and CE | 6.89 days | 0.03 | 0.15 | 160Gd(n,γ)161Gd 161Tb | Low availability, difficult production | [123] |
| Bismuth-213 | α/β− | 45.6 min. | 0.084 | 8.38 | 225Ac/213Bi generator | Moderate availability | [130,131] |
| Actinium-225 | α | 10.0 days | 0.061 | 28 | 229Th/225Ac 226Ra(p,2n)225Ac 232Th(p,spall.) 225Ac | Moderate availability | [125,132] |
| Astatine-211 | α | 7.2 days | 0.067 | 5.87 | natBi(α,2n)211At | Moderate availability | [133] |
| Radium-223 | α | 11.43 days | 0.08 | 28.2 | 227Ac/223Ra generator | Commercially available | [134,135] |
| Thorium-227 | α | 18.7 days | 0.1 | 6.14 | 227Ac/227Th generator | Moderate availability | [136] |
| Lead-212 | β−/αdecays to 212Bi | 10.6 h | 0.08 | 6.05 | 228Th/224Ra/212Pb generator. | Commercially available | [137] |
| Iodine-125 | Auger and CE | 59.4 days | 0.0001 | 0.035 | 124Xe(n,γ)125Xe 125I. | Commercially available (moderate price) | [111] |
| Agent | Administered Activity | Animals Cell Type | Main Findings | Ref. |
|---|---|---|---|---|
| 131I-J415, 131I-J533 131I-J591, 111In-J415 111In-J533,111In-J591 | 350 MBq/mg | LNCaP cells | J415 and J591 mAbs competed for binding to PSMA antigen, J533 did not interfered with J415, rapid elimination of 131I from the cell and high retention of 111In | [149] |
| 177Lu-huJ591 mAb | 10 µCi 100–400 µCi | BALB/c nude mice bearing LNCaP tumor xenografts | high uptake of 177Lu-huJ591 in tumors, lack of bone radioactivity, high stability of conjugate, dose-dependent tumor remission | [150] |
| 131I-J415, 131I-J533 131I-J591, 131I-1I-7E11 111In-J415, 111In-J533, 111In-J591, 111In 1I-7E11 | 80 kBq | BALB/c nude mice bearing LNCaP tumor xenografts | 131I-J533 showed lower tumor localization and reduced tumor/blood and tumor/muscle ratios than 131I-J415 or 131I-J591, better tumor uptake of 111In-labeled mAbs compared to 131I labeled mAbs | [151] |
| 131I-huJ591 mAb 90Y-huJ591 mAb | 3.7–11.1 MBq 131I-huJ591; 1.11–7.4 MBq 90Y-huJ591 | BALB/c nude mice bearing LNCaP tumor xenografts | anti-tumor effects dependent on a size of tumor, radionuclide used and dose, median survival time increased, total dose, and dose rate equally important for bone marrow toxicity | [152] |
| 111In-CHX-A-3C6 mAb | 7.5 μCi 100 μCi | LNCaP, 22Rv, DU145 cells and BALB/c nude mice bearing LNCaP, 22Rv1, or DU145 tumor xenografts | high ability to bind to LNCaP and 22Rv1, but not to DU145 cells, excellent in vivo PSMA-targeting of 111In-CHX-A′′-3C6 in mice bearing LNCaP, 22Rv1 xenografts | [153] |
| 177Lu-DOTA-3/F11 | 300 kBq0.5, 1, or 2 MBq | SCID mice bearing C4-2 tumor xenografts | increasing tumor uptake over time. Single dose of 1 MBq 177Lu-DOTA-3/F11 inhibited tumor growth and prolonged survival | [154] |
| 213Bi-J591 | 0.06–6.4 mCi/mg | 5E4 LNCaP cells, spheroids, nude mice bearing LNCaP tumors | efficient binding and internationalization of 213Bi-J591, improved median tumor-free survival, PSA decline | [187] |
| 213Bi-J591 | 0.9 and 1.8 MBq/ml | LNCaP-LN3 spheroids | reduction in spheroid volume with increasing radioactivity | [188] |
| 213Bi-J591 | 1–100 mCi | LNCaP-LN3 cells, nude mice bearing LNCaP tumors | in vitro very high cytotoxicity with increasing activity, inhibition of tumor growth, no side effects | [189] |
| 227Th-PSMA IgG1 | 100–500 kBq/kg | nude mice bearing LNCaP-luc, C4-2 or 22Rv1tumors | selective anti-tumor efficacy at 250 and 500 kBq/kg, prevention of tumor growth at 100 kBq/kg | [190] |
| 227Th-PSMA IgG1 | 75–500 kBq/kg | nude mice bearing PDx or KuCap1 tumors | dose-dependent tumor growth inhibition, stable disease or tumor regression at 300 kBq/kg, partial or complete regression of tumor growth at 250 or 500 kBq/kg in the PDx model | [191] |
| 211At-6 | 3.7 kBq/100 μL 37–740 kBq | PC3 PIP and PC3-flu cells, nude mice bearing PC3 tumors | efficient cellular uptake in PC3 PIP tumors and kidneys, tumor growth delay in PC3 PIP xenograft model and improved survival in micrometastatic PC model | [112] |
| 213Bi-PSMA I&T, 213Bi-JVZ-008 | 0.3 MBq5.4–6.6 MBq 213Bi-PSMA I&T 4.5–5.4 MBq 213Bi-JVZ-008 | LNCaP cells, BALB/c nude mice bearing LNCaP tumors | induced DSBs in vitro, 2x higher tumor uptake of 213Bi-PSMA I&T than 213Bi-JVZ-008, 213Bi-PSMA I&T more potent to induce DNA damage in the tumor, possible nephrotoxicity | [101] |
| 125I-DCIBzL | 3.7–370 kBq/mL 3.7–111 MBq | PC3 cells, nude mice bearing PC3 tumors | increased DNA damage in vitro, significant tumor growth delay | [107] |
| 161Tb-PSMA-617 177Lu-PSMA-617 | 0.01–20 MBq/mL 5 or 10 MBq | PC3 cells, nude mice bearing PC3 tumors | enhanced therapeutic effects of 161Tb compared to 177Lu, equal pharmacokinetics of 161Tb-PSMA-617 and 177Lu-PSMA617 | [123] |
| Agent | Treatment Dose | Number of Patients | Main Findings | Ref. |
|---|---|---|---|---|
| 90Y-huJ591 | 0.185–0.740 GBq/m2 | 29 | DLT—0.74 MBq/m2, MTD—0.65 MBq/m2, myelosuppression was the DLT, excellent targeting of prostate cancer metastases | [155,156,157,158] |
| 177Lu-huJ591 | 0.370 to 2.775 GBq/m2 | 24 | myelosuppression was the DLT, excellent targeting of prostate cancer metastases | [159,160] |
| 177Lu-huJ591 | 2.4 GBq/m2 2.6 GBq/m2 | 47 | dose–response relationship in PSA decline (71% vs. 46% of patients), MTD—2.59 MBq/m2, acceptable mielosuppresion, MOS—11.9 months with 2.4 MBq/m2 and 21.8 months with 2.6 MBq/m2 | [161,162] |
| 177Lu-huJ591 | 3.3 GBq/m2, fractionated doses: 0.74 GBq/m2 escalation dose: 0.18 GBq/m2 | 28 | MTD—1.48 MBq/m2, fractionated dosing allowed higher cumulative doses of 177Lu-J591 with less toxicity | [163] |
| 90Y-huJ591 177Lu-huJ591 | 0.185–0.740 GBq/m2 0.740–3.3 GBq/m2 | 121 29 | myelosuppression was the DLT, patients could tolerate subsequent therapies after RIT | [164] |
| docetaxel/ 177Lu-J591 | docetaxel (75mg/m2) with escalation doses of 177Lu-J591 (0.74–1.48 GBq/m2) | 15 | PSA decline in 80% (>50% in 73% of patients (and >50% in 73%), toxicity limited to reversible myelosuppression | [165] |
| 177Lu-J591 | 0.74–1.67 GBq/m2 | 49 | fractionated dosing allowed higher doses of 177Lu-J591 with less toxicity, myelosuppression was the DLT | [166] |
| 131I-MIP-1095 | 8 GBq (range: 2–7.2 GBq) | 28 | PSA decline in 61% of patients, PD (14%), high doses received by the salivary glands, liver, and kidneys, reduction of pain | [170] |
| 177Lu-PSMA I&T | 7.4 GBq/cycle | 18 | the kidneys and glandular tissue were the critical organs, with a mean absorbed dose of 0.72 Gy/GBq and 3.8 Gy/GBq, respectively | [171] |
| 177Lu-PSMA I&T | 5.8 GBq/cycle | 56 | PSA decline in 80% of patients, PD (36%), no long-term side effects | [172] |
| 177Lu-PSMA I&T | 6.0 GBq/cycle (range: 2 to 9.7 GBq) | 119 | PSA decline in 76.3% of patients (>50% decline in PSA in 57.5%), no long-term side effects | [173] |
| 177Lu-PSMA-617 | 5.6 GBq (range: 4.1–6.1 GBq) | 10 | PSA decline in 70% of patients, no long-term side effects | [174] |
| 177Lu-PSMA-617 | 6 GBq/cycle | 22 | PSA decline in 80% of patients (>50% decline in PSA in 45%), PD (32%) | [175] |
| 177Lu-PSMA-617 | 4.8 GBq/cycle | 30 | PSA decline in 70% of patients (>50% decline in PSA in 43.3%), PD (21%) | [176] |
| 177Lu-PSMA-617 | 5.9 GB/cycles | 82 | PSA decline in 53% of patients, PD (21%), therapy was well tolerated | [177] |
| 177Lu-PSMA-617 | 5.52 GBq (range: 5.28 to 5.77 GBq) | 9 | lacrimal glands represent the dose-limiting organs | [178] |
| 177Lu-PSMA-617 | 3.6 GBq (range: 3.4 to 3.9 GBq) | 5 | salivary glands represent the dose-limiting organs | [179] |
| 177Lu-PSMA-617 | 6.0 GBq | 10 | PSA decline in 47% of patients, PD (33%), no acute events for salivary gland or kidney, | [180] |
| 177Lu-PSMA-617 | 1.11–5.55 GBq | 31 | PSA decline by 75% (mean for all patients), PD (20%), PFS—12 months, MOS—15 months | [181] |
| 177Lu-PSMA-617 | 6.1 GBq (range: 5.4 to 6.5 GBq) | 10 | PSA decline in 50% patients, PD (30%), no side effects, xerostomia observed only in 1 patient | [182] |
| 177Lu-PSMA-617 | 4–8 GBq | 30 | PSA decline in 97% patients (>50% decline in PSA in 57%) PFS—7.6 months, MOS—13.5 months | [183] |
| 177Lu-PSMA I&T177Lu-PSMA-617 | ≥14.8 GBq | 45 | PFS better with higher dose, re-challenge therapy at progression decreased tumor grade, mild and transitory adverse effects | [184] |
| 177Lu-PSMA-617 | 2–8 GBq | 145 | PSA decline > 50% in 45% patients, mild side effects | [185] |
| 225Ac-PSMA-617 | 100 kBq/kg b.w. | 40 | PSA decline > 50% in 63% patients, xerostomia was the main side effect, PFS—7 months, MOS > 12 months | [194] |
| 225Ac-PSMA-617177Lu-PSMA-617tandem therapy | 5.3 MBq (range: 1.5 to 7.9 MBq) 6.9 GBq (range: 5.0–11.6 GBq) | 20 | PSA decline > 50% in 65% patients, PFS—19 weeks, OS—48 weeks tandem therapy enhanced response to therapy and decreased xerostomia severity | [195] |
| 225Ac-PSMA-617 | 4–8 MBq | 17 | PSA decline in 94% of patients (>90% decline in PSA in 82%), grade 1/2 xerostomia in all patients | [196] |
| 225Ac-PSMA-617 | 4–8 MBq | 73 | PSA decline in 83% (>50% decline in PSA in 70%), PD (32%), PFS—15.2 months, OS—18 months, xerostomia in 85% of patients | [197] |
| Trial Identification | Agent | Phase | Study Arms | Estimated Enrolment | Primary Endpoints |
|---|---|---|---|---|---|
| NCT03828838 | 177Lu-PSMA-617 | I/II | two cycles (3 GBq and 3–6 GBq) | 10 | doses delivered to the tumors and organs at risk |
| NCT 03511664 | 177Lu-PSMA-617 | III | 7.4 GBq (±10%) 177Lu-PSMA-617 every 6 weeks for a maximum of 6 cycles + Best supportive/best standard of care (BS/BSOC) | 750 | overall survival |
| NCT03392428 | 177Lu-PSMA-617 | II | 8.5 GBq (0.5 GBq per cycle) every 6 weeks | 200 | PSA response |
| NCT03780075 | 177Lu-EB-PSMA-617 | I | 1.11GBq (30 mCi) of 177Lu-EB-PSMA-617 1.85GBq (50 mCi) of 177Lu-EB-PSMA-617 3.70GBq (100 mCi) of 177Lu-EB-PSMA-617 | 30 | PSA response |
| NCT00859781 | 177Lu-J591 + ketoconazole 111In-J591+ ketoconazole | II | group-1: ketoconazole + hydrocortisone followed by a single dose of 177Lu-J591 (70 mCi/m2) group-2: ketoconazole + hydrocortisone followed by a single dose of 111In-J591 (5 mCi) | 140 | proportion free of radiographically evident metastases at 18 months by CT and/or MRI scan of the abdomen and pelvis, chest X-ray, or CT scan of the chest and bone scan |
| NCT03545165 | 177Lu−J591 177Lu−PSMA−617 | I/II | cumulative 2.7 GBq/m2 dose of 177Lu−J591 and the cumulative 177Lu−PSMA−617 dose from 3.7 GBq to 18.5 GBq. Dose escalation in 6 different dose levels (3+3 dose−escalation study/de-escalation design) | 48 | dose-limiting toxicity (DLT), cumulative maximum tolerated dose (MTD), PSA response |
| NCT03276572 | 225Ac−J591 | I | a single dose of 225Ac−J591 (13.3 KBq/Kg–93.3 KBq/Kg or 0.36 μCi/Kg–2.52 μCi/Kg) | 42 | dose-limiting toxicity (DLT) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czerwińska, M.; Bilewicz, A.; Kruszewski, M.; Wegierek-Ciuk, A.; Lankoff, A. Targeted Radionuclide Therapy of Prostate Cancer—From Basic Research to Clinical Perspectives. Molecules 2020, 25, 1743. https://doi.org/10.3390/molecules25071743
Czerwińska M, Bilewicz A, Kruszewski M, Wegierek-Ciuk A, Lankoff A. Targeted Radionuclide Therapy of Prostate Cancer—From Basic Research to Clinical Perspectives. Molecules. 2020; 25(7):1743. https://doi.org/10.3390/molecules25071743
Chicago/Turabian StyleCzerwińska, Malwina, Aleksander Bilewicz, Marcin Kruszewski, Aneta Wegierek-Ciuk, and Anna Lankoff. 2020. "Targeted Radionuclide Therapy of Prostate Cancer—From Basic Research to Clinical Perspectives" Molecules 25, no. 7: 1743. https://doi.org/10.3390/molecules25071743
APA StyleCzerwińska, M., Bilewicz, A., Kruszewski, M., Wegierek-Ciuk, A., & Lankoff, A. (2020). Targeted Radionuclide Therapy of Prostate Cancer—From Basic Research to Clinical Perspectives. Molecules, 25(7), 1743. https://doi.org/10.3390/molecules25071743