
Antibacterial Prodrugs to Overcome Bacterial Resistance




Abstract
1. Introduction
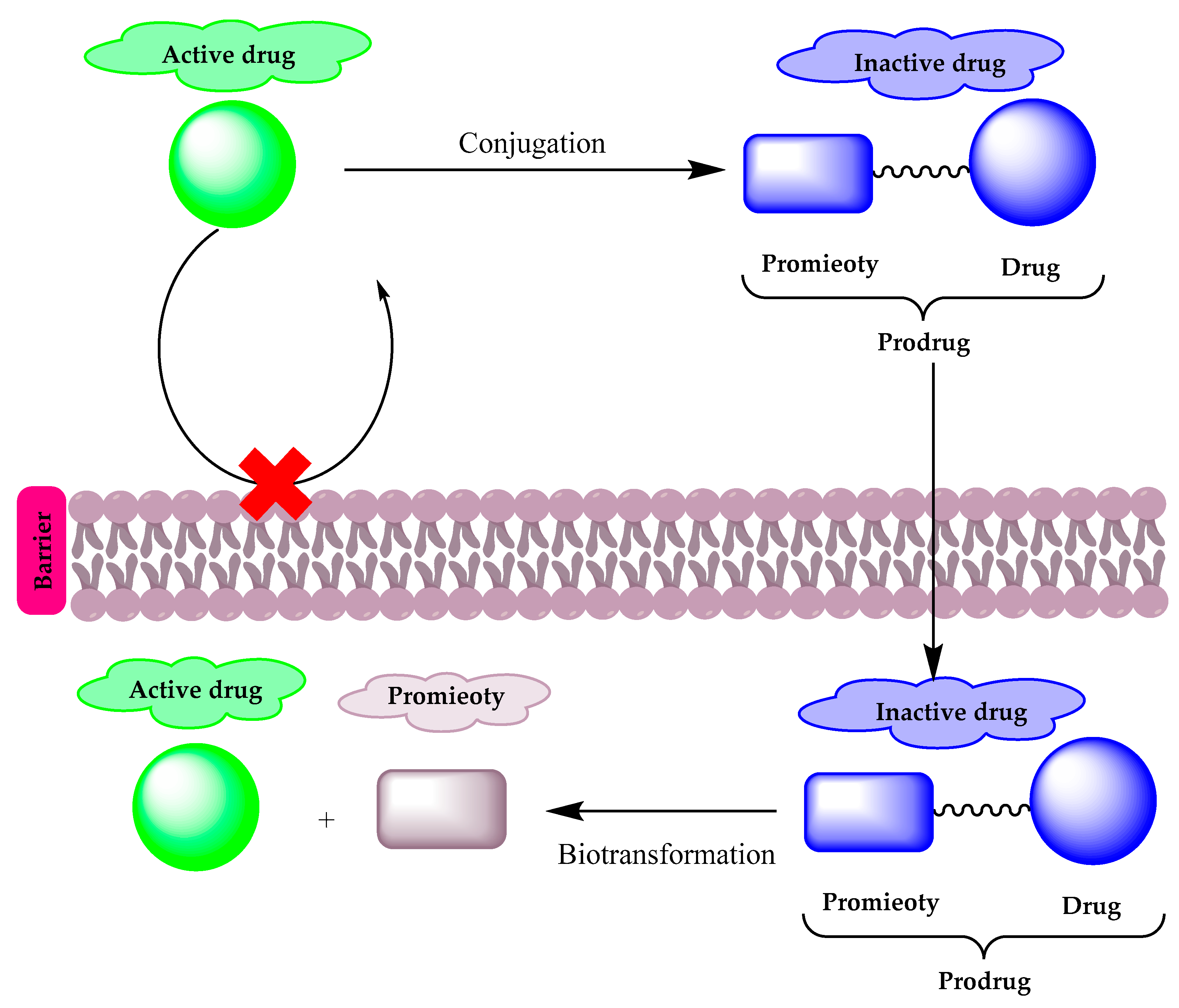
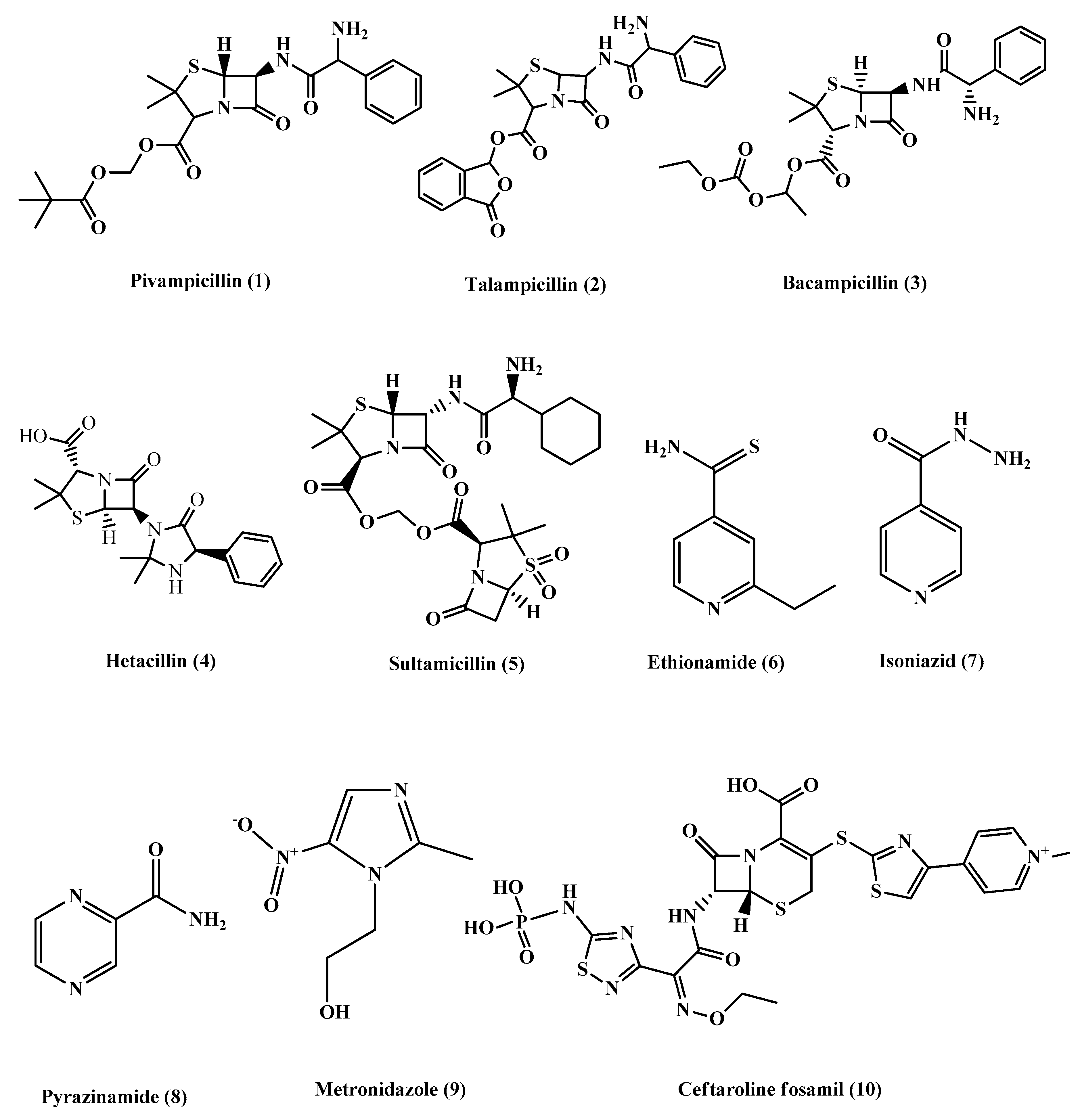
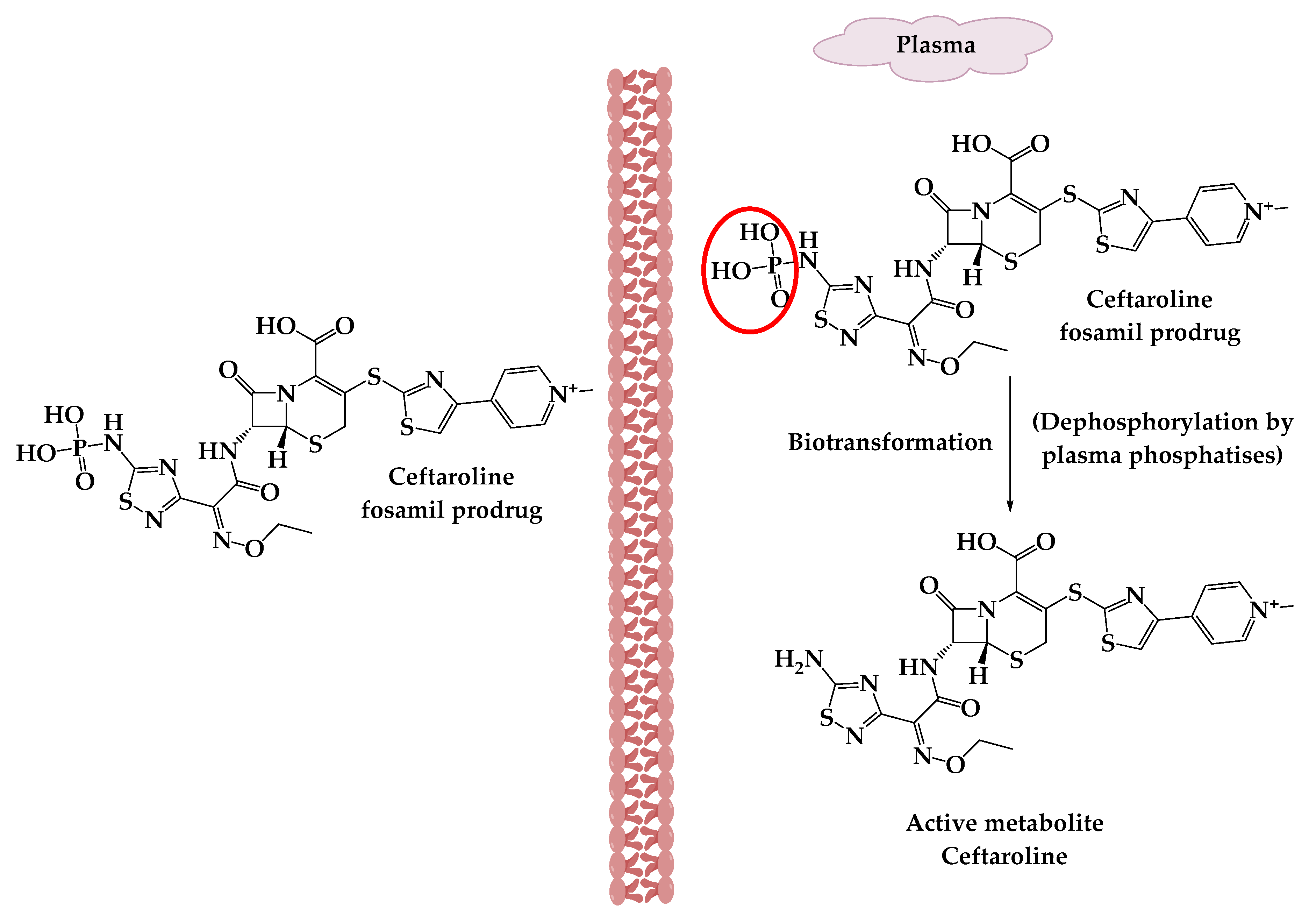
2. Prodrugs
3. Prodrug Applications against Resistant Bacterial Pathogens
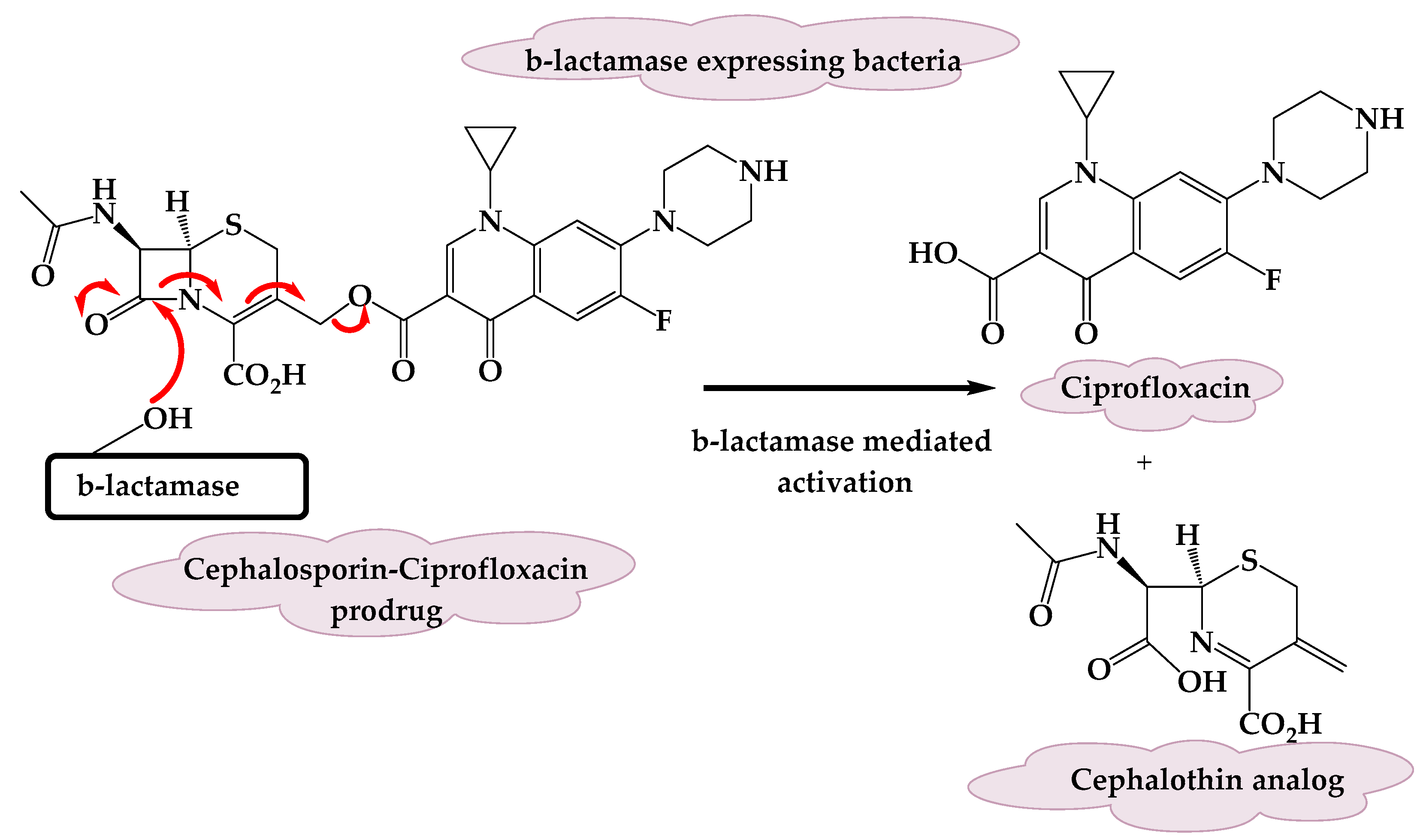
3.1. A β-Lactamase-Activated Ciprofloxacin Prodrug
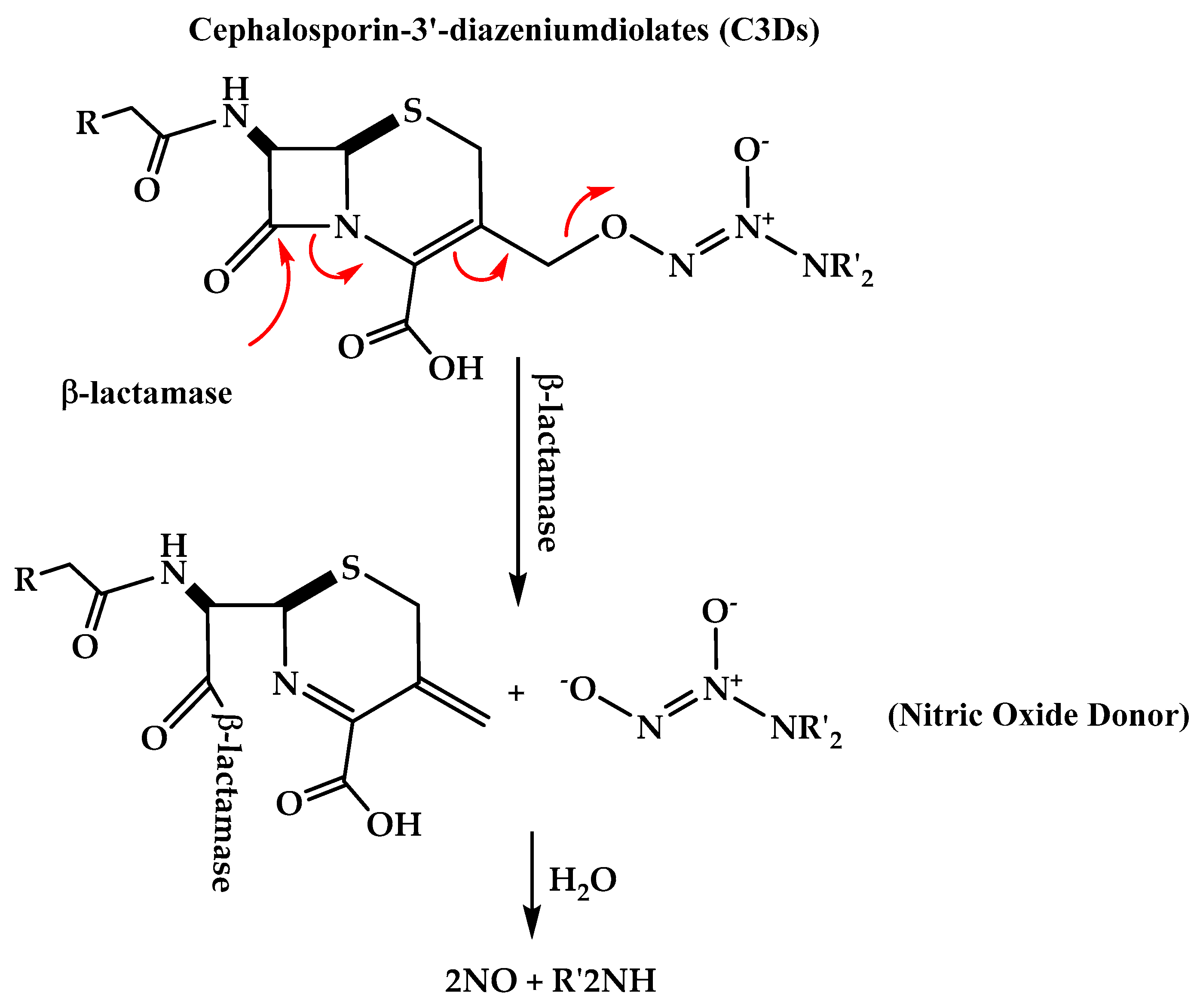
3.2. Cephalosporin-3-Diazeniumdiolate
3.3. Triclosan Glycoside Prodrugs
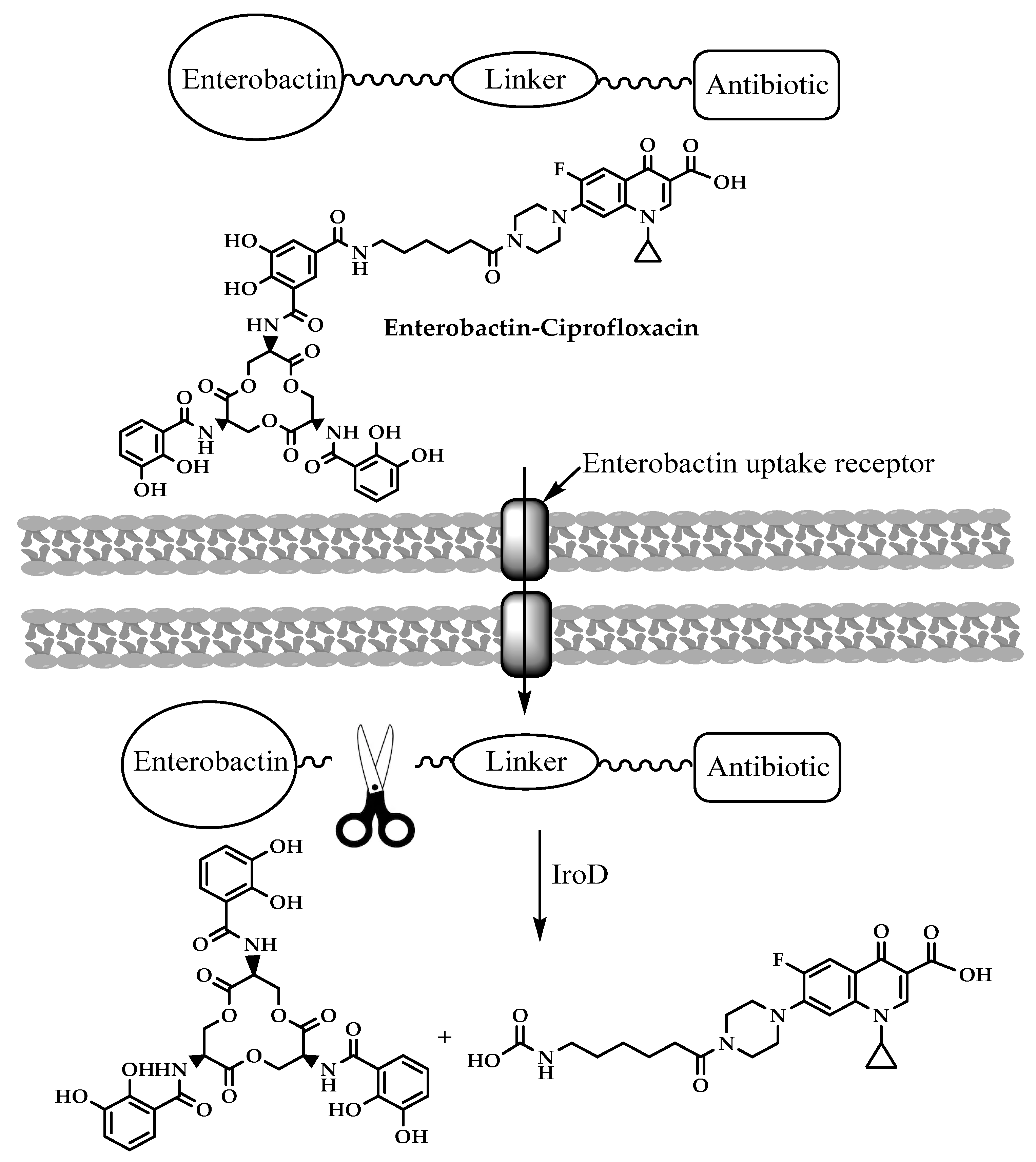
3.4. Enterobactin-Antibiotic Conjugates
3.5. Antimicrobial Peptide Prodrugs
3.6. Prodrugs of 5-Modified 2ʹ-Deoxyuridines
3.7. The Tebipenempivoxil Prodrug
3.8. Avibactam Prodrugs
3.9. Tedizolid Phosphate (TR701)
3.10. FtsZ-Targeting Benzamide Prodrugs
3.11. Carvacrol Prodrugs
3.12. ADC111, ADC112 and ADC113
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Gajdacs, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Zaman, S.B.; Hussain, M.A.; Nye, R.; Mehta, V.; Mamun, K.T.; Hossain, N. A Review on Antibiotic Resistance: Alarm Bells are Ringing. Cureus 2017, 9, e1403. [Google Scholar] [CrossRef]
- Karaman, R. Using predrugs to optimize drug candidates. Expert Opin. Drug Discov. 2014, 9, 1405–1419. [Google Scholar] [CrossRef] [PubMed]
- Najjar, A.; Karaman, R. Successes, failures, and future prospects of prodrugs and their clinical impact. Expert Opin. Drug Discov. 2019, 14, 199–220. [Google Scholar] [CrossRef]
- Karaman, R. Prodrugs design based on inter- and intramolecular chemical processes. Chem. Biol. Drug Des. 2013, 82, 643–668. [Google Scholar] [CrossRef] [PubMed]
- Najjar, A.; Karaman, R. The Prodrug Approach in the Era of Drug Design; Taylor & Francis: Abingdon-on-Thames, UK, 2019. [Google Scholar]
- Amly, W.; Karaman, R. Recent updates in utilizing prodrugs in drug delivery (2013–2015). Expert Opin. Drug Deliv. 2016, 13, 571–591. [Google Scholar] [CrossRef]
- Dahan, A.; Khamis, M.; Agbaria, R.; Karaman, R. Targeted prodrugs in oral drug delivery: The modern molecular biopharmaceutical approach. Expert Opin. Drug Deliv. 2012, 9, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Monserrat-Martinez, A.; Gambin, Y.; Sierecki, E. Thinking Outside the Bug: Molecular Targets and Strategies to Overcome Antibiotic Resistance. Int. J. Mol. Sci. 2019, 20, 1255. [Google Scholar] [CrossRef]
- Smyth, R.D.; Pfeffer, M.; Van Harken, D.R.; Cohen, A.; Hottendorf, G.H. Human pharmacokinetics and disposition of sarmoxicillin, a lipophilic amoxicillin prodrug. Antimicrob. Agents Chemother. 1981, 19, 1004–1012. [Google Scholar] [CrossRef]
- Jusko, W.J.; Lewis, G.P.; Schmitt, G.W. Ampicillin and hetacillin pharmacokinetics in normal and anephric subjects. Clin. Pharmacol. Ther. 1973, 14, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.H. Bioavailability of talampicillin. Br. Med. J. 1977, 2, 232–233. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soares GM, S.; Figueiredo, L.C.; Faveri, M.; Cortelli, S.C.; Duarte, P.M.; Feres, M. Mechanisms of action of systemic antibiotics used in periodontal treatment and mechanisms of bacterial resistance to these drugs. J. Appl. Oral Sci. 2012, 20, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Shirley, D.A.; Heil, E.L.; Johnson, J.K. Ceftaroline fosamil: A brief clinical review. Infect Dis. Ther. 2013, 2, 95–110. [Google Scholar] [CrossRef]
- Kong, K.F.; Schneper, L.; Mathee, K. Beta-lactam antibiotics: From antibiosis to resistance and bacteriology. Apmis 2010, 118, 1–36. [Google Scholar] [CrossRef]
- Bush, K. Proliferation and significance of clinically relevant β-lactamases. Ann. N. Y. Acad. Sci. 2013, 1277, 84–90. [Google Scholar] [CrossRef]
- Cantón, R.; González-Alba, J.M.; Galán, J.C. CTX-M enzymes: Origin and diffusion. Front. Microbiol. 2012, 3, 110. [Google Scholar] [CrossRef]
- Scheld, W.M. Maintaining fluoroquinolone class efficacy: Review of influencing factors. Emerg. Infect. Dis. 2003, 9, 1. [Google Scholar] [CrossRef]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-induced changes in the intestinal microbiota and disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef]
- Stewardson, A.J.; Gaïa, N.; Francois, P.; Malhotra-Kumar, S.; Delemont, C.; de Tejada, B.M.; Schrenzel, J.; Harbarth, S.; Lazarevic, V.; Wp, S. Collateral damage from oral ciprofloxacin versus nitrofurantoin in outpatients with urinary tract infections: A culture-free analysis of gut microbiota. Clin. Microbiol. Infect. 2015, 21, 344. [Google Scholar] [CrossRef]
- Evans, L.E.; Krishna, A.; Ma, Y.; Webb, T.E.; Marshall, D.C.; Tooke, C.L.; Spencer, J.; Clarke, T.B.; Armstrong, A.; Edwards, A.M. Exploitation of antibiotic resistance as a novel drug target: Development of a β-lactamase-activated antibacterial prodrug. J. Med. Chem. 2019, 62, 4411–4425. [Google Scholar] [CrossRef] [PubMed]
- Allan, R.N.; Kelso, M.J.; Rineh, A.; Yepuri, N.R.; Feelisch, M.; Soren, O.; Brito-Mutunayagam, S.; Salib, R.J.; Stoodley, P.; Clarke, S.C. Cephalosporin-NO-donor prodrug PYRRO-C3D shows β-lactam-mediated activity against Streptococcus pneumoniae biofilms. Nitric Oxide 2017, 65, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Barraud, N.; Kardak, B.G.; Yepuri, N.R.; Howlin, R.P.; Webb, J.S.; Faust, S.N.; Kjelleberg, S.; Rice, S.A.; Kelso, M.J. Cephalosporin-3′-diazeniumdiolates: Targeted NO-Donor Prodrugs for Dispersing Bacterial Biofilms. Angew. Chem. Int. Ed. 2012, 51, 9057–9060. [Google Scholar] [CrossRef] [PubMed]
- Soren, O.; Rineh, A.; Silva, D.G.; Cai, Y.; Howlin, R.P.; Allan, R.N.; Feelisch, M.; Davies, J.C.; Connett, G.J.; Faust, S.N. Cephalosporin nitric oxide-donor prodrug DEA-C3D disperses biofilms formed by clinical cystic fibrosis isolates of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2019, 75, 117–125. [Google Scholar] [CrossRef]
- Collins, S.A.; Kelso, M.J.; Rineh, A.; Yepuri, N.R.; Coles, J.; Jackson, C.L.; Halladay, G.D.; Walker, W.T.; Webb, J.S.; Hall-Stoodley, L. Cephalosporin-3′-Diazeniumdiolate NO Donor Prodrug PYRRO-C3D Enhances Azithromycin Susceptibility of Nontypeable Haemophilus influenzae Biofilms. Antimicrob. Agents Chemother. 2017, 61, e02086. [Google Scholar]
- Kämpfer, P.; Rauhoff, O.; Dott, W. Glycosidase profiles of members of the family Enterobacteriaceae. J. Clin. Microbiol. 1991, 29, 2877–2879. [Google Scholar] [CrossRef]
- Levy, C.W.; Roujeinikova, A.; Sedelnikova, S.; Baker, P.J.; Stuitje, A.R.; Slabas, A.R.; Slabas, A.R.; Rice, D.W.; Rafferty, J.B. Molecular basis of triclosan activity. Nature 1999, 398, 383–384. [Google Scholar] [CrossRef]
- Howse, G.L.; Bovill, R.A.; Stephens, P.J.; Osborn, H.M. Synthesis and antibacterial profiles of targeted triclosan derivatives. Eur. J. Med. Chem. 2019, 162, 51–58. [Google Scholar] [CrossRef]
- Neumann, W.; Sassone-Corsi, M.; Raffatellu, M.; Nolan, E.M. Esterase-catalyzed siderophore hydrolysis activates an enterobactin–ciprofloxacin conjugate and confers targeted antibacterial activity. J. Am. Chem. Soc. 2018, 140, 5193–5201. [Google Scholar] [CrossRef]
- Zheng, T.; Nolan, E.M. Enterobactin-mediated delivery of β-lactam antibiotics enhances antibacterial activity against pathogenic Escherichia coli. J. Am. Chem. Soc. 2014, 136, 9677–9691. [Google Scholar] [CrossRef]
- Chairatana, P.; Zheng, T.; Nolan, E.M. Targeting virulence: Salmochelin modification tunes the antibacterial activity spectrum of β-lactams for pathogen-selective killing of Escherichia coli. Chem. Sci. 2015, 6, 4458–4471. [Google Scholar] [CrossRef]
- Lai, Y.; Gallo, R.L. AMPed up immunity: How antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009, 30, 131–141. [Google Scholar] [CrossRef]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Sahl, H.-G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 2006, 4, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.Y.; Khanum, R. Antimicrobial peptides as potential anti-biofilm agents against multidrug-resistant bacteria. J. Microbiol. Immunol. Infect. 2017, 50, 405–410. [Google Scholar] [CrossRef]
- Zhou, Y.; Peng, Y. Synergistic effect of clinically used antibiotics and peptide antibiotics against Gram-positive and Gram-negative bacteria. Exp. Ther. Med. 2013, 6, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Samy, R.P.; Stiles, B.G.; Franco, O.L.; Sethi, G.; Lim, L.H. Animal venoms as antimicrobial agents. Biochem. Pharmacol. 2017, 134, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yan, J.; Chen, R.; Dang, W.; Zhang, B.; Zhang, W.; Song, J.; Wang, R. Membrane-active action mode of polybia-CP, a novel antimicrobial peptide isolated from the venom of Polybia paulista. Antimicrob. Agents Chemother. 2012, 56, 3318–3323. [Google Scholar] [CrossRef]
- das Neves, R.C.; Mortari, M.R.; Schwartz, E.F.; Kipnis, A.; Junqueira-Kipnis, A.P. Antimicrobial and antibiofilm effects of peptides from venom of social Wasp and scorpion on multidrug-resistant Acinetobacter baumannii. Toxins 2019, 11, 216. [Google Scholar] [CrossRef]
- Forde, É.; Shafiy, G.; Fitzgerald-Hughes, D.; Strömstedt, A.A.; Devocelle, M. Action of antimicrobial peptides and their prodrugs on model and biological membranes. J. Pept. Sci. 2018, 24, e3086. [Google Scholar] [CrossRef] [PubMed]
- Hilpert, K.; Volkmer-Engert, R.; Walter, T.; Hancock, R.E. High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotechnol. 2005, 23, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Desgranges, S.; Ruddle, C.C.; Burke, L.P.; McFadden, T.M.; O’Brien, J.E.; Fitzgerald-Hughes, D.; Humphreys, H.; Smyth, T.P.; Devocelle, M. β-Lactam-host defence peptide conjugates as antibiotic prodrug candidates targeting resistant bacteria. RSC Adv. 2012, 2, 2480–2492. [Google Scholar] [CrossRef]
- Ferrari, V.; Serpi, M. Nucleoside analogs and tuberculosis: New weapons against an old enemy. Future Med. Chem. 2015, 7, 291–314. [Google Scholar] [CrossRef]
- Serpi, M.; Ferrari, V.; Pertusati, F. Nucleoside derived antibiotics to fight microbial drug resistance: New utilities for an established class of drugs? J. Med. Chem. 2016, 59, 10343–10382. [Google Scholar] [CrossRef]
- Khandazhinskaya, A.L.; Alexandrova, L.A.; Matyugina, E.S.; Solyev, P.N.; Efremenkova, O.V.; Buckheit, K.W.; Wilkinson, M.; Buckheit, R.W.; Chernousova, L.N.; Smirnova, T.G. Novel 5′-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents. Molecules 2018, 23, 3069. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Solyev, P.N.; Karpenko, I.L.; Efremenkova, O.V.; Vasilyeva, B.F.; Sumarukova, I.G.; Kochetkov, S.N.; Alexandrova, L.A. Synthesis of water-soluble prodrugs of 5-modified 2′-deoxyuridines and their antibacterial activity. J. Antibiot. 2020, 2020, 1–11. [Google Scholar] [CrossRef]
- McEntee, L.; Johnson, A.; Farrington, N.; Unsworth, J.; Dane, A.; Jain, A.; Cotroneo, N.; Critchley, I.; Melnick, D.; Parr, T. Pharmacodynamics of Tebipenem: New Options for Oral Treatment of Multidrug-Resistant Gram-Negative Infections. Antimicrob. Agents Chemother. 2019, 63, e00603. [Google Scholar] [CrossRef]
- Gordon, E.M.; Duncton, M.A.; Gallop, M.A. Orally absorbed derivatives of the β-lactamase inhibitor avibactam. Design of novel prodrugs of sulfate containing drugs. J. Med. Chem. 2018, 61, 10340–10344. [Google Scholar] [CrossRef]
- Moellering, R., Jr. Linezolid: The first oxazolidinone antimicrobial. Ann. Intern. Med. 2003, 138, 135. [Google Scholar] [CrossRef]
- Kanafani, Z.A.; Corey, G.R. Tedizolid (TR-701): A new oxazolidinone with enhanced potency. Expert Opin. Investig. Drugs 2012, 21, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Barber, K.E.; Smith, J.R.; Raut, A.; Rybak, M.J. Evaluation of tedizolid against Staphylococcus aureus and enterococci with reduced susceptibility to vancomycin, daptomycin or linezolid. J. Antimicrob. Chemother. 2015, 71, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.J.; Poppe, S.; Schaadt, R.; Brown-Driver, V.; Finn, J.; Pillar, C.M.; Shinabarger, D.; Zurenko, G. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob. Agents Chemother. 2008, 52, 4442–4447. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. An FtsZ-targeting prodrug with oral antistaphylococcal efficacy in vivo. Antimicrob. Agents Chemother. 2013, 57, 5860–5869. [Google Scholar] [CrossRef]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; Lyu, Y.L.; Pawlak, J.; Saravolatz, S.; Saravolatz, L.D.; Weinstein, M.P.; LaVoie, E.J. TXA709, an FtsZ-targeting benzamide prodrug with improved pharmacokinetics and enhanced in vivo efficacy against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2015, 59, 4845–4855. [Google Scholar] [CrossRef]
- Marinelli, L.; Di Stefano, A.; Cacciatore, I. Carvacrol and its derivatives as antibacterial agents. Phytochem. Rev. 2018, 17, 903–921. [Google Scholar] [CrossRef]
- Marinelli, L.; Fornasari, E.; Eusepi, P.; Ciulla, M.; Genovese, S.; Epifano, F.; Fiorito, S.; Turkez, H.; Rtc, S.; Mingoia, M. Carvacrol prodrugs as novel antimicrobial agents. Eur. J. Med. Chem. 2019, 178, 515–529. [Google Scholar] [CrossRef]
- Fleck, L.E.; North, E.J.; Lee, R.E.; Mulcahy, L.R.; Casadei, G.; Lewis, K. A screen for and validation of prodrug antimicrobials. Antimicrob. Agents Chemother. 2014, 58, 1410–1419. [Google Scholar] [CrossRef]
- Guay, D.R. An update on the role of nitrofurans in the management of urinary tract infections. Drugs 2001, 61, 353–364. [Google Scholar] [CrossRef]
- Gershon, H.; Parmegiani, R. Antimicrobial activity of 8-quinolinol, its salts with salicylic acid and 3-hydroxy-2-naphthoic acid, and the respective copper (II) chelates in liquid culture. Appl. Environ. Microbiol. 1963, 11, 62–65. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotics | Date Introduced | Date of Resistance Development |
|---|---|---|
| Penicillin | Discovery-1928 Mass production-1942 | 1940 |
| Tetracycline | 1950 | 1959 |
| Erythromycin | 1953 | 1968 |
| Methicillin | 1960 | 1962 |
| Gentamicin | 1967 | 1979 |
| Vancomycin | 1972 | 1988/2002 |
| Imipenem/Ceftazidime | 1985 | 1998/1987 |
| Levofloxacin | 1996 | 1996 |
| Linezolid | 2000 | 2001 |
| Daptomycin | 2003 | 2014 |
| Ceftaroline | 2010 | 2011 |
| Telavancin | 2013 | 2019 |
| Tedizolid phosphate/Dalbavancin | 2014 | 2016/2015 |
| Delafloxacin | 2017 | 2019 |
| Omadacycline | 2018 | - |
| Iclaprim | 2019 | - |
| Cefiderocol | 2020 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jubeh, B.; Breijyeh, Z.; Karaman, R. Antibacterial Prodrugs to Overcome Bacterial Resistance. Molecules 2020, 25, 1543. https://doi.org/10.3390/molecules25071543
Jubeh B, Breijyeh Z, Karaman R. Antibacterial Prodrugs to Overcome Bacterial Resistance. Molecules. 2020; 25(7):1543. https://doi.org/10.3390/molecules25071543
Chicago/Turabian StyleJubeh, Buthaina, Zeinab Breijyeh, and Rafik Karaman. 2020. "Antibacterial Prodrugs to Overcome Bacterial Resistance" Molecules 25, no. 7: 1543. https://doi.org/10.3390/molecules25071543
APA StyleJubeh, B., Breijyeh, Z., & Karaman, R. (2020). Antibacterial Prodrugs to Overcome Bacterial Resistance. Molecules, 25(7), 1543. https://doi.org/10.3390/molecules25071543