Probing the Interfacial Behavior of Type IIIa Binary Mixtures Along the Three-Phase Line Employing Molecular Thermodynamics



Abstract
1. Introduction
2. Theory
2.1. Square Gradient Theory for Mixtures
2.2. The Statistical Associating Fluid Theory Model
2.3. The Three-Phase Equilibrium from SAFT-VR Mie EoS
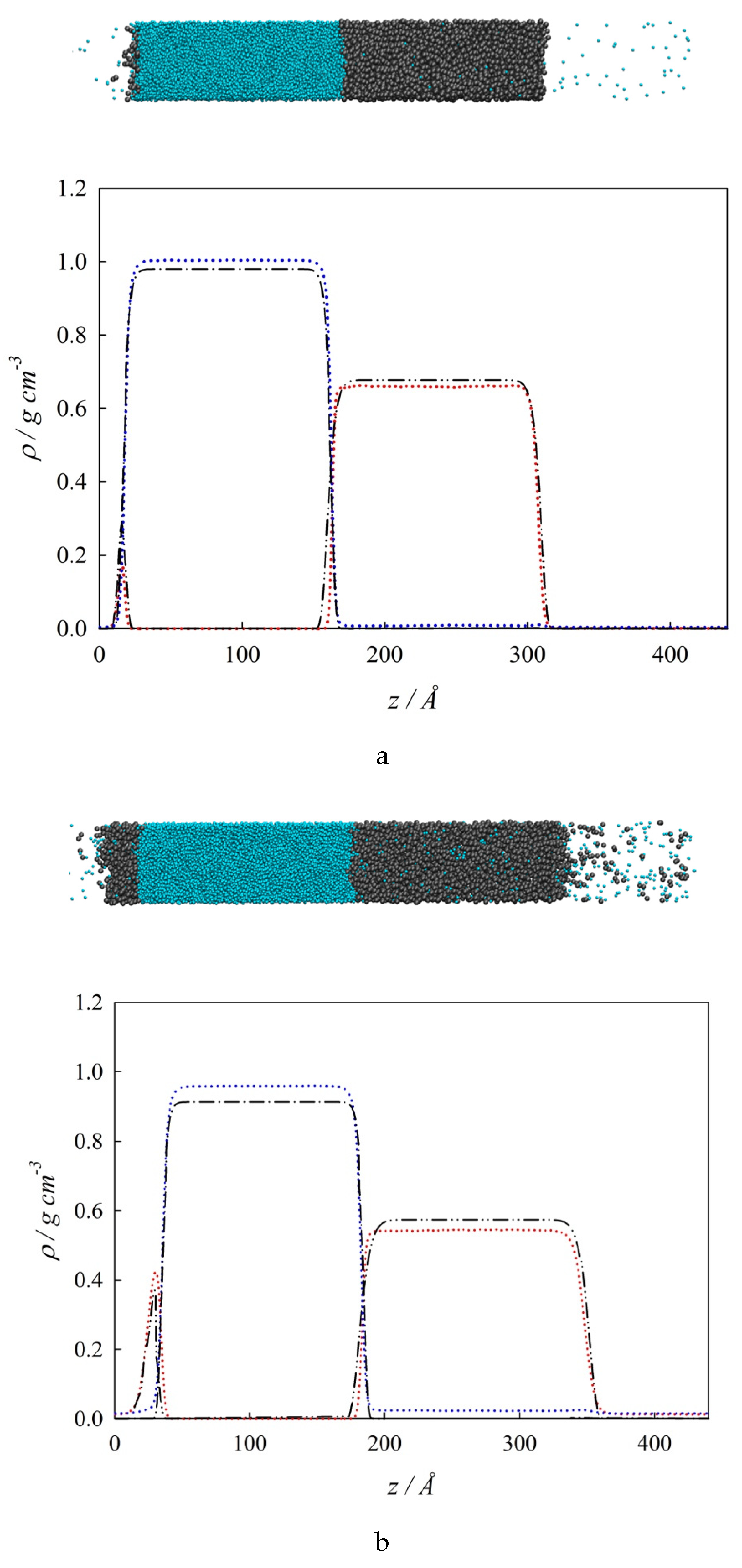
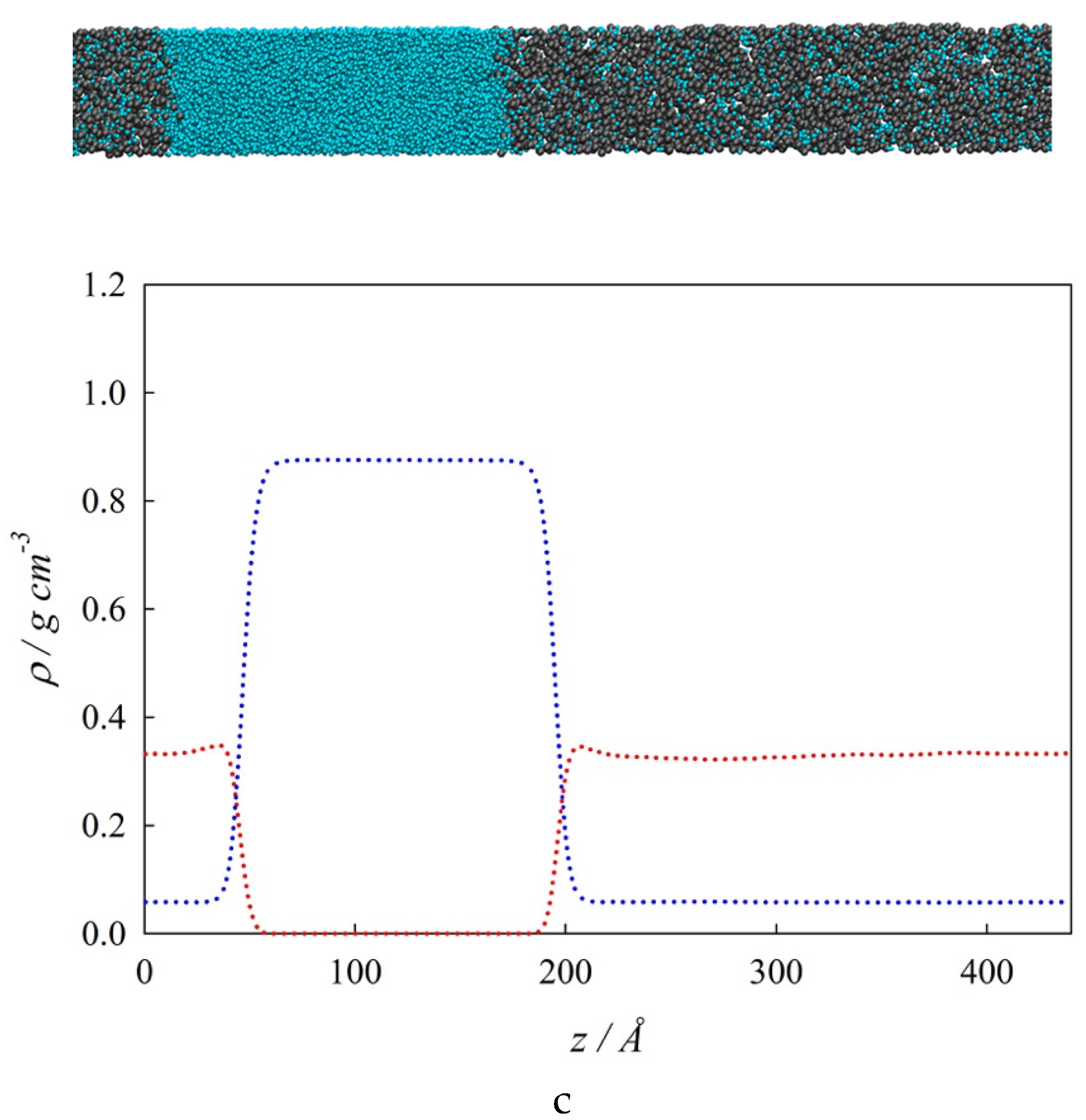
3. Molecular Dynamics Simulations
4. Results and Discussions
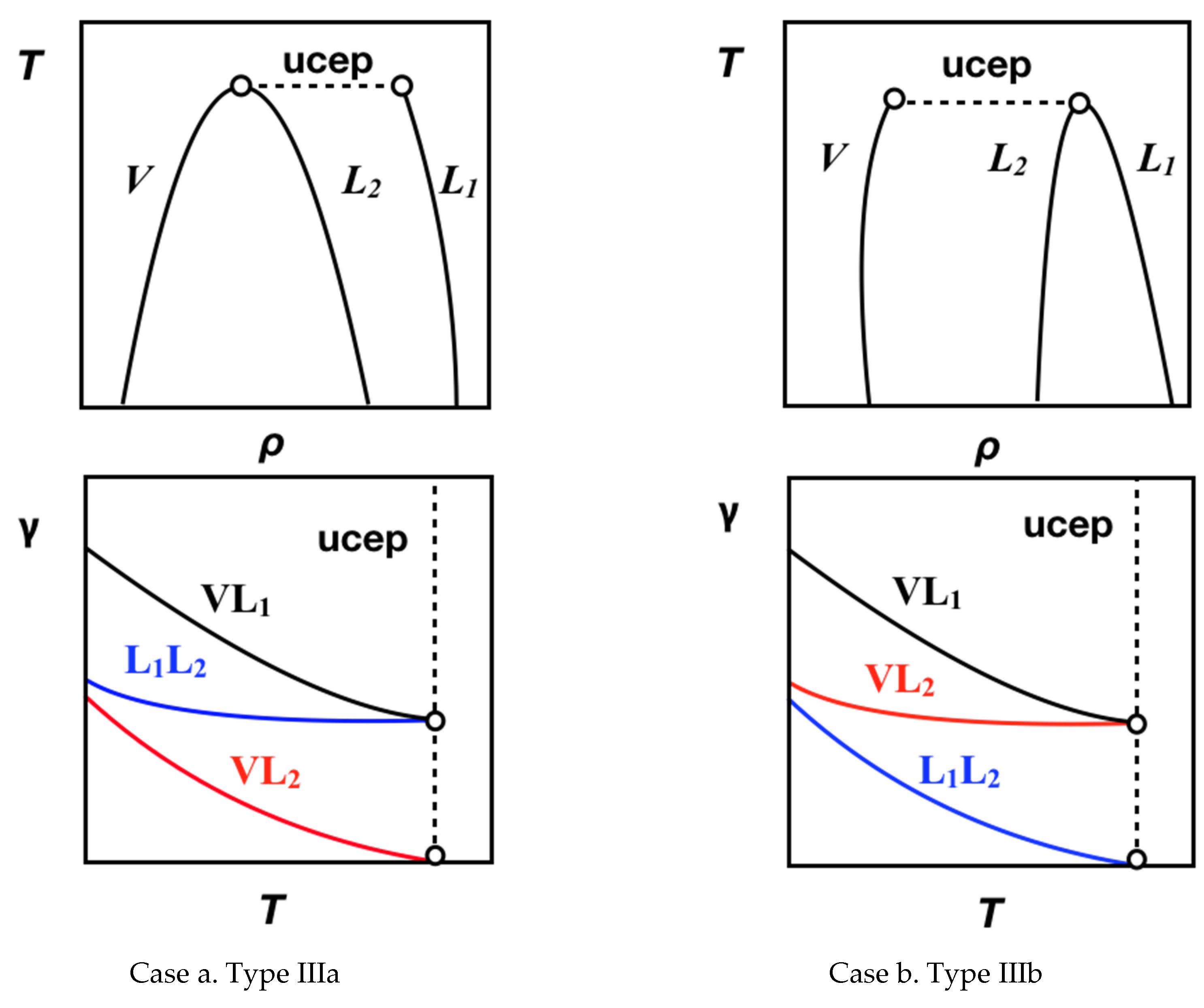
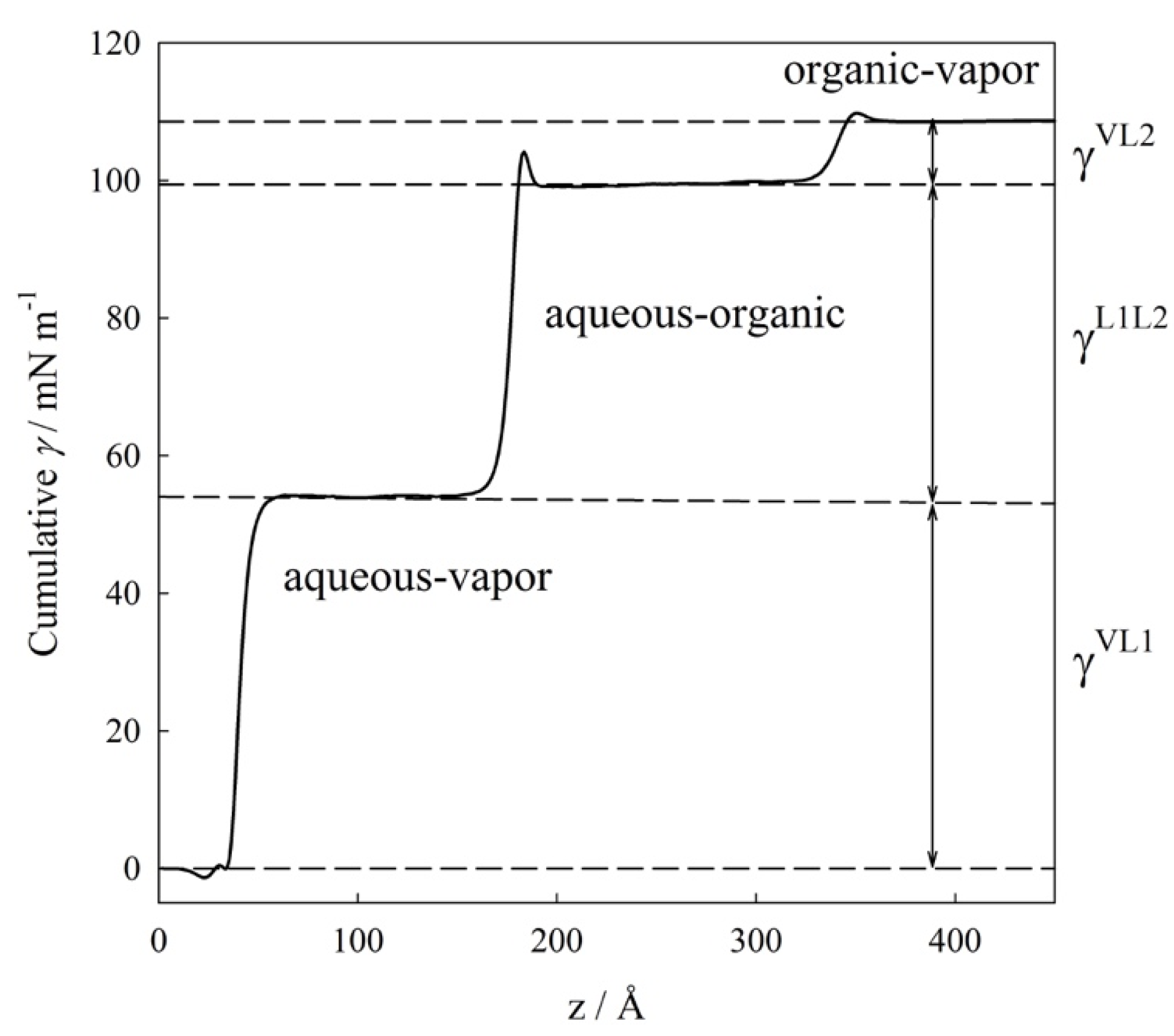
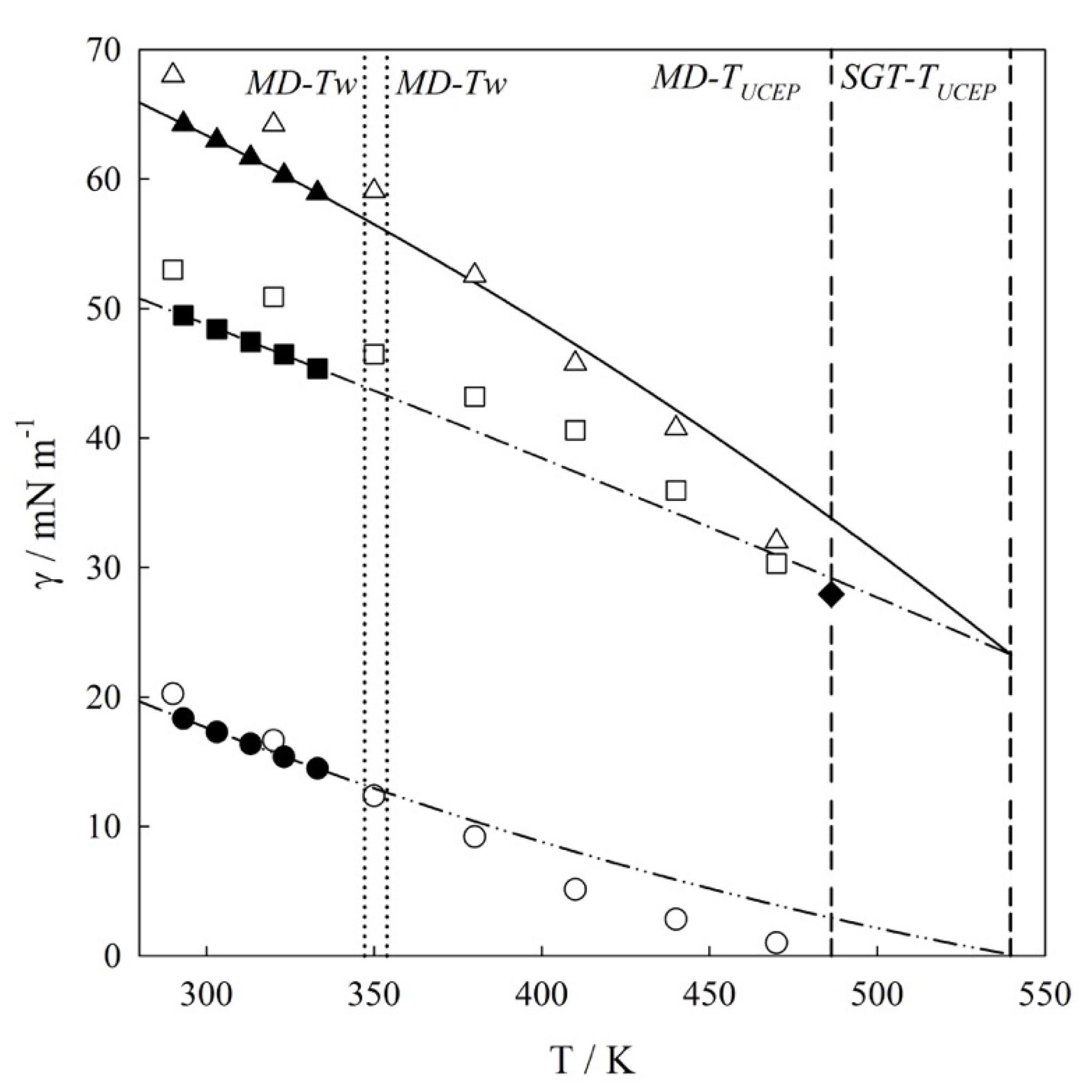
4.1. Interfacial Tension Along a Three-Phase Equilibrium
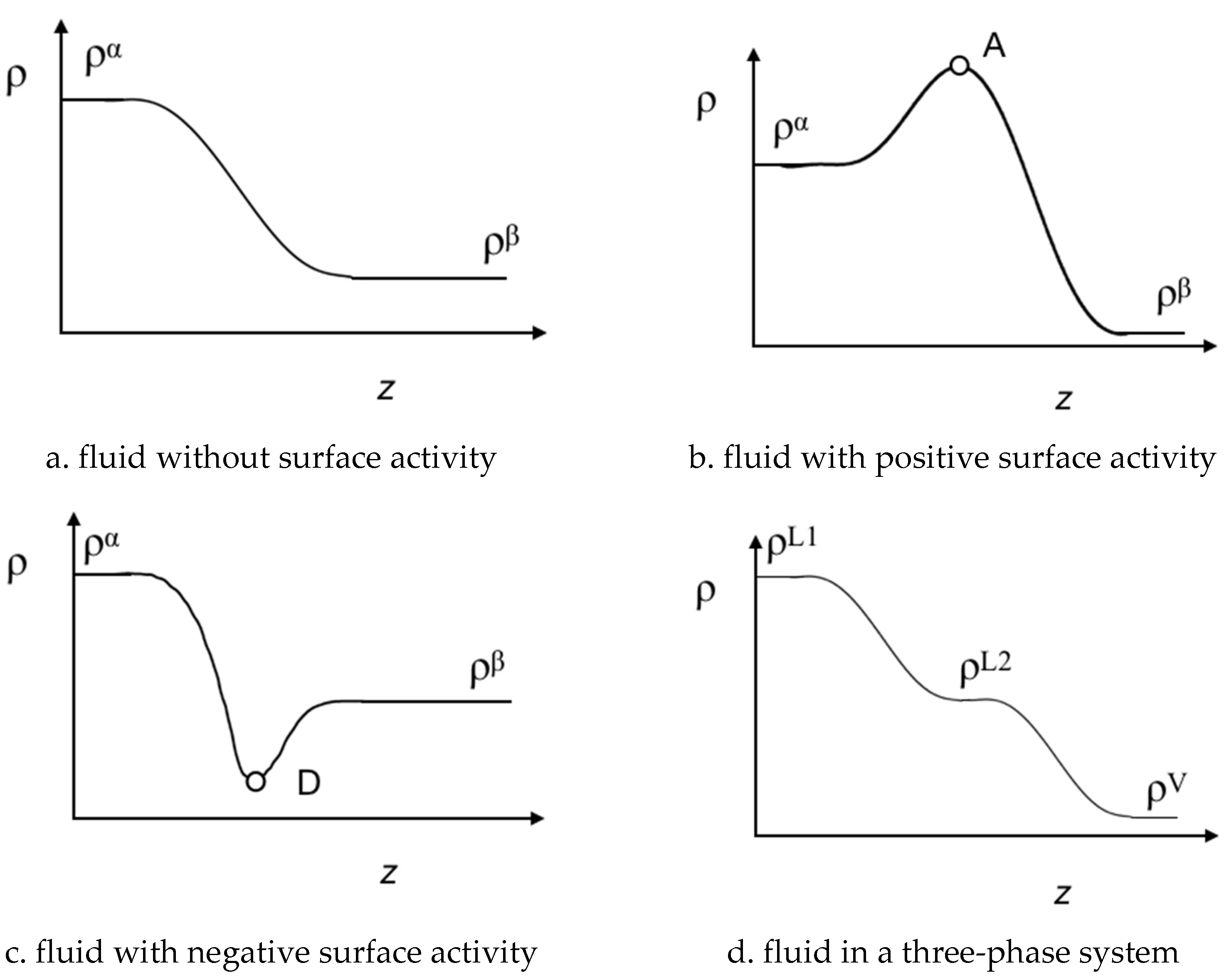
4.2. Bulk Densities and Interfacial Concentration Profiles Along a Three-Phase Equilibrium
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Van Konynenburg, P.; Scott, R. Critical lines and phase equilibria in binary van der Waals mixtures. Philos. Trans. R. Soc. 1980, 298, 495–539. [Google Scholar]
- Bidart, C.; Segura, H.; Wisniak, J. Phase equilibrium behavior in water (1) + n-alkane (2) mixtures. Ind. Eng. Chem. Res. 2007, 46, 947–954. [Google Scholar] [CrossRef]
- Virnau, P.; Müller, M.; MacDowell, L.G.; Binder, K. Phase behavior of n-alkanes in supercritical solution: A Monte Carlo study. J. Chem. Phys. 2004, 121, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; MacDowell, L.G.; Virnau, P.; Binder, K. Interface properties and bubble nucleation in compressible mixtures containing polymers. J. Chem. Phys. 2002, 117, 5480–5496. [Google Scholar] [CrossRef]
- Rowlinson, J.S.; Widom, B. Molecular Theory of Capillarity; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Cahn, J.W. Critical point wetting. J. Chem. Phys. 1977, 66, 3667–3672. [Google Scholar] [CrossRef]
- Dietrich, S.; Latz, A. Classification of interfacial wetting behavior in binary liquid mixtures. Phys. Rev. B 1989, 40, 9204–9237. [Google Scholar] [CrossRef]
- Costas, M.E.; Varea, C.; Robledo, A. Global phase diagram for the wetting transition at interfaces in fluid mixtures. Phys. Rew. Lett. 1983, 51, 2394–2397. [Google Scholar] [CrossRef]
- Evans, M.J.B. Measurement of Surface and Interfacial Tension. In Measurement of the Thermodynamic Properties of Multiple Phases; Weir, R.D.D., de Loos, T.W.W., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 7. [Google Scholar]
- Mejía, A.; Cartes, M.; Segura, H.; Müller, E.A. Use of equations of state and coarse grained simulations to complement experiments: Describing the interfacial properties of carbon dioxide + decane and carbon dioxide + eicosane mixtures. J. Chem. Eng. Data 2014, 59, 2928–2941. [Google Scholar] [CrossRef]
- Mori, Y.H.; Tsul, N.; Klyomlya, M. Surface and interfacial tensions and their combined properties in seven binary, immiscible liquid-liquid-vapor systems. J. Chem. Eng. Data 1984, 29, 407–412. [Google Scholar] [CrossRef]
- Van der Waals, J.D. Thermodynamische Theorie der Kapillarität unter voraussetzung Stetiger dichteänderung. Zeit. Phys. Chem. 1893, 13, 657–725. [Google Scholar]
- Carey, B.S. The Gradient Theory of Fluid Interfaces. Ph.D. Thesis, University of Minnesota, Minneapolis, MN, USA, 1979. [Google Scholar]
- Telo da Gama, M.M.; Evans, R. The structure and surface tension of the liquid-vapour interface near the upper critical end point of a binary mixture of Lennard-Jones fluids: I. The two phase region. Mol. Phys. 1983, 48, 229–250. [Google Scholar] [CrossRef]
- Tarazona, P.; Telo da Gama, M.M.; Evans, R. Wetting transitions at fluid-fluid interfaces: I. The order of the transition. Mol. Phys. 1983, 49, 283–300. [Google Scholar] [CrossRef]
- Mejía, A.; Segura, H. Interfacial behavior in systems Type IV. Int. J. Thermophys. 2004, 25, 1395–1445. [Google Scholar] [CrossRef]
- Mejía, A.; Segura, H. On the interfacial behavior about the shield region. Int. J. Thermophys. 2005, 26, 13–29. [Google Scholar] [CrossRef]
- Mejía, A.; Pàmies, J.C.; Duque, D.; Segura, H.; Vega, L.F. Phase and interface behavior in type I and type V Lennard-Jones mixtures: Theory and Simulations. J. Chem. Phys. 2005, 123, 034505–034515. [Google Scholar] [CrossRef]
- Mejía, A.; Vega, L.F. Perfect wetting along a three-phase line: Theory and molecular dynamics simulations. J. Chem. Phys. 2006, 124, 2445051–2445057. [Google Scholar] [CrossRef]
- Cornelisse, P.M.W.; Peters, C.J.; de Swaan Arons, J. Interfacial phase transitions at solid-fluid and liquid-vapor interfaces. Int. J. Thermophys. 1998, 19, 1501–1509. [Google Scholar] [CrossRef]
- Hulshof, H. Ueber die Oberflächenspannung. Ann. Phys. 1901, 309, 165–186. [Google Scholar] [CrossRef]
- Irving, J.H.; Kirkwood, J.G. The statistical mechanical theory of transport processes. IV. The equations of hydrodynamics. J. Chem. Phys. 1950, 18, 817–829. [Google Scholar] [CrossRef]
- Garrido, J.; Quinteros-Lama, H.; Piñeiro, M.M.; Mejía, A.; Segura, H. On the phase and interface behavior along the three-phase line of ternary Lennard-Jones mixtures. A collaborative approach based on square gradient theory and molecular dynamics simulations. J. Chem. Phys. 2014. [Google Scholar] [CrossRef]
- Müller, E.A.; Mejía, A. Resolving discrepancies in the measurements of the interfacial tension for the CO2 + H2O mixture by computer simulation. J. Phys. Chem. Lett. 2014, 5, 1267–1271. [Google Scholar] [CrossRef] [PubMed]
- Daubert, T.E.; Danner, R.P. Physical and Thermodynamic Properties of Pure Chemicals. Data Compilation; Taylor & Francis: Bristol, PA, USA, 1989. [Google Scholar]
- Kamilov, I.K.; Stepanov, G.V.; Abdulagatov, I.M.; Rasulov, A.R.; Milikhina, E.I. Liquid-liquid-vapor, liquid-liquid, and liquid-vapor phase transitions in aqueous n-hexane mixtures from isochoric heat capacity measurements. J. Chem. Eng. Data 2001, 46, 1556–1567. [Google Scholar] [CrossRef]
- Bertrand, E.; Dobbs, H.; Broseta, D.; Indekeu, J.; Bonn, D.; Meunier, J. First-order and critical wetting of alkanes on water. Phys. Rev. Lett. 2000, 85, 1282–1285. [Google Scholar] [CrossRef] [PubMed]
- Mejía, A.; Herdes-Moreno, C.; Müller, E.A. Force fields for coarse-grained molecular simulations from a corresponding states correlation. Ind. Eng. Chem. Res. 2014, 53, 4131–4141. [Google Scholar] [CrossRef]
- Lobanova, O.; Avendaño, C.; Lafitte, T.; Müller, E.A.; Jackson, G. SAFT-γ force field for the simulation of molecular fluids: 4. A single-site coarse-grained model of water applicable over a wide temperature range. Mol. Phys. 2015, 113, 1228–1249. [Google Scholar] [CrossRef]
- Lafitte, T.; Apostolakou, A.; Avendaño, C.; Galindo, A.; Adjiman, C.S.; Müller, E.A.; Jackson, G. Accurate statistical associating fluid theory for chain molecules formed from Mie segments. J. Chem Phys. 2013. [Google Scholar] [CrossRef]
- Garrido, J.M.; Piñeiro, M.M.; Blas, F.J.; Müller, E.A.; Mejía, A. Interfacial tensions of industrial fluids from a molecular-based square gradient theory. AIChE J. 2016, 62, 1781–1794. [Google Scholar] [CrossRef]
- Mie, G. Zur kinetischen Theorie der einatomigen Körper. Ann. Phys. 1903, 316, 657–697. [Google Scholar] [CrossRef]
- Lobanova, O. Development of Coarse-Grained Force Fields from a Molecular Based Equation of State for Thermodynamic and Structural Properties of Complex Fluids. Ph.D. Thesis, Imperial College London, London, UK, 2014. [Google Scholar]
- Rowlinson, J.S.; Swinton, F. Liquids and Liquids Mixtures; Butterworths: London, UK, 1982. [Google Scholar]
- Müller, E.A.; Ervik, Å.; Mejía, A. Best Practices for Computing Interfacial Properties from Molecular Dynamics Simulations. Available online: https://www.livecomsjournal.org (accessed on 24 March 2020).
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon: Oxford, UK, 2017. [Google Scholar]
- Holcomb, C.D.; Clancy, P.; Zollweg, J.A. A critical study of the simulation of the liquid-vapour interface of a Lennard-Jones fluid. Mol. Phys. 1993, 78, 437–459. [Google Scholar] [CrossRef]
- Müller, E.A.; Jackson, G. Force-field parameters from the SAFT-γ equation of state for use in coarse-grained molecular simulations. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 405–427. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Martinez-Veracoechea, F.; Müller, E.A. Temperature-quench molecular dynamics simulations for fluid phase equilibria. Mol. Simulat. 2005, 31, 33–43. [Google Scholar] [CrossRef]
- Duque, D.; Vega, L.F. Some issues on the calculation of interfacial properties by molecular simulation. J. Chem. Phys. 2004, 121, 8611–8617. [Google Scholar] [CrossRef] [PubMed]
- Aasen, A.; Hammer, M.; Müller, E.A.; Wilhelmsen, Ø. Equation of state and force fields for Feynman–Hibbs-corrected Mie fluids. II. Application to mixtures of helium, neon, hydrogen, and deuterium. J. Chem. Phys 2020. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluid | msi | λr,ii | εii/kB/K | σii/Å |
|---|---|---|---|---|
| n-hexane (n-C6H14) | 2 | 19.57 | 376.35 | 4.508 |
| water (H2O) | 1 | 8.00 | −4.806 × 10−4 T2 + 0.6107 T + 165.9 | −6.455 × 10−9 T3 + 9.1 x 10−6 T2 − 4.291 x 10−3 T + 3.543 |
| T/K | γVL1/mN m−1 | γL1L2/mN m−1 | γVL2/mN m−1 |
|---|---|---|---|
| 290 | 68.001 | 53.001 | 20.241 |
| 320 | 64.201 | 50.901 | 16.643 |
| 350 | 59.062 | 46.473 | 12.362 |
| 380 | 54.683 | 43.202 | 9.202 |
| 410 | 45.743 | 40.612 | 5.134 |
| 440 | 40.751 | 35.965 | 2.833 |
| 470 | 32.052 | 30.303 | 1.002 |
| 486.3 c | 27.93 | 27.93 | 0.00 |
| Organic (n-hexane rich) phase | ||||
| T/K | x1 | ρ1/g cm−3 | ρ2/g cm−3 | ρ/g cm−3 |
| 290 | 0.0471 | 0.00711 | 0.6612 | 0.6683 |
| 320 | 0.0811 | 0.01211 | 0.6263 | 0.6383 |
| 350 | 0.1162 | 0.01633 | 0.5871 | 0.6032 |
| 380 | 0.1691 | 0.02332 | 0.5434 | 0.5662 |
| 410 | 0.2323 | 0.03122 | 0.4891 | 0.5203 |
| 440 | 0.3381 | 0.04421 | 0.4122 | 0.4563 |
| 470 | 0.4554 | 0.05812 | 0.3311 | 0.3891 |
| 479.10 b | 0.501 | 0.134 | 0.134 | 0.268 |
| Aqueous (water rich) phase | ||||
| T/K | x1 | ρ1/g cm−3 | ρ2/g cm−3 | ρ/g cm−3 |
| 290 | 1.000 | 1.0031 | 0.000 | 1.0031 |
| 320 | 1.000 | 0.9942 | 0.000 | 0.9942 |
| 350 | 1.000 | 0.9792 | 0.000 | 0.9792 |
| 380 | 1.000 | 0.9581 | 0.000 | 0.9581 |
| 410 | 1.000 | 0.9343 | 0.000 | 0.9343 |
| 440 | 1.000 | 0.9051 | 0.000 | 0.9051 |
| 470 | 1.000 | 0.8752 | 0.000 | 0.8752 |
| 479.1b | 1.000 | 0.864 | 0.000 | 0.864 |
| vapor phase | ||||
| T/K | x1 | ρ1/g cm−3 | ρ2/g cm−3 | ρ/g cm−3 |
| 290 | 0.9641 | 0.0041 | 0.0011 | 0.0051 |
| 320 | 0.9251 | 0.0061 | 0.0031 | 0.0091 |
| 350 | 0.9012 | 0.0102 | 0.0063 | 0.0162 |
| 380 | 0.8491 | 0.0152 | 0.0132 | 0.0282 |
| 410 | 0.8073 | 0.0241 | 0.0272 | 0.0511 |
| 440 | 0.7551 | 0.0343 | 0.0532 | 0.1192 |
| 470 | 0.5713 | 0.1274 | 0.0956 | 0.2231 |
| 479.1 b | 0.501 | 0.134 | 0.134 | 0.268 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso, G.; Chaparro, G.; Cartes, M.; Müller, E.A.; Mejía, A. Probing the Interfacial Behavior of Type IIIa Binary Mixtures Along the Three-Phase Line Employing Molecular Thermodynamics. Molecules 2020, 25, 1499. https://doi.org/10.3390/molecules25071499
Alonso G, Chaparro G, Cartes M, Müller EA, Mejía A. Probing the Interfacial Behavior of Type IIIa Binary Mixtures Along the Three-Phase Line Employing Molecular Thermodynamics. Molecules. 2020; 25(7):1499. https://doi.org/10.3390/molecules25071499
Chicago/Turabian StyleAlonso, Gerard, Gustavo Chaparro, Marcela Cartes, Erich A. Müller, and Andrés Mejía. 2020. "Probing the Interfacial Behavior of Type IIIa Binary Mixtures Along the Three-Phase Line Employing Molecular Thermodynamics" Molecules 25, no. 7: 1499. https://doi.org/10.3390/molecules25071499
APA StyleAlonso, G., Chaparro, G., Cartes, M., Müller, E. A., & Mejía, A. (2020). Probing the Interfacial Behavior of Type IIIa Binary Mixtures Along the Three-Phase Line Employing Molecular Thermodynamics. Molecules, 25(7), 1499. https://doi.org/10.3390/molecules25071499