‘Acridines’ as New Horizons in Antifungal Treatment


Abstract
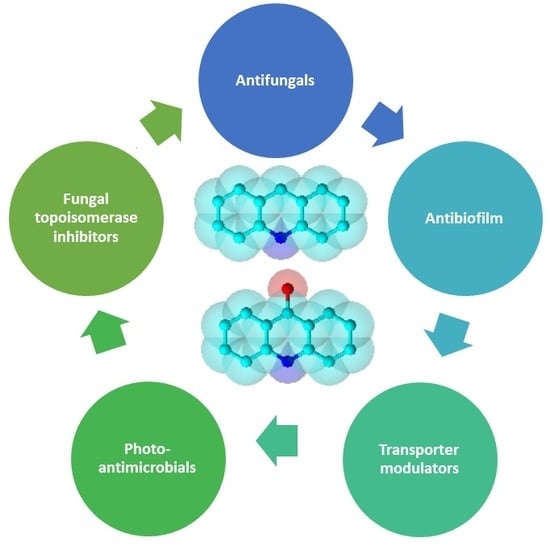
1. Introduction
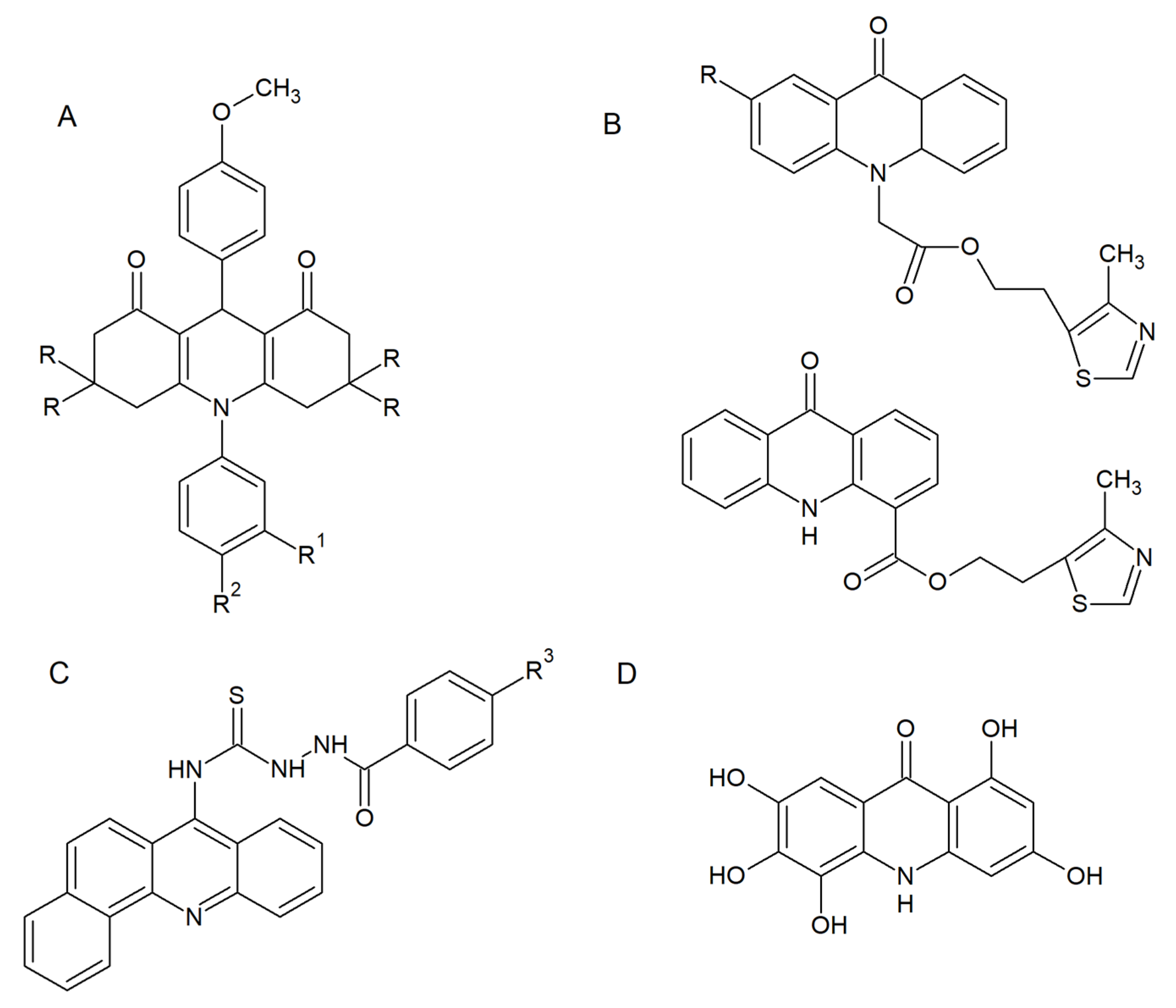
2. Acridine Derivatives as Antifungals
2.1. Antifungal and Antibiofilm Activity
2.2. Acridine/Acridone Derivatives as Efficient Sensitizers
2.3. Fungal Topoisomerases as Drug Targets for Acridine/Acridone Derivatives
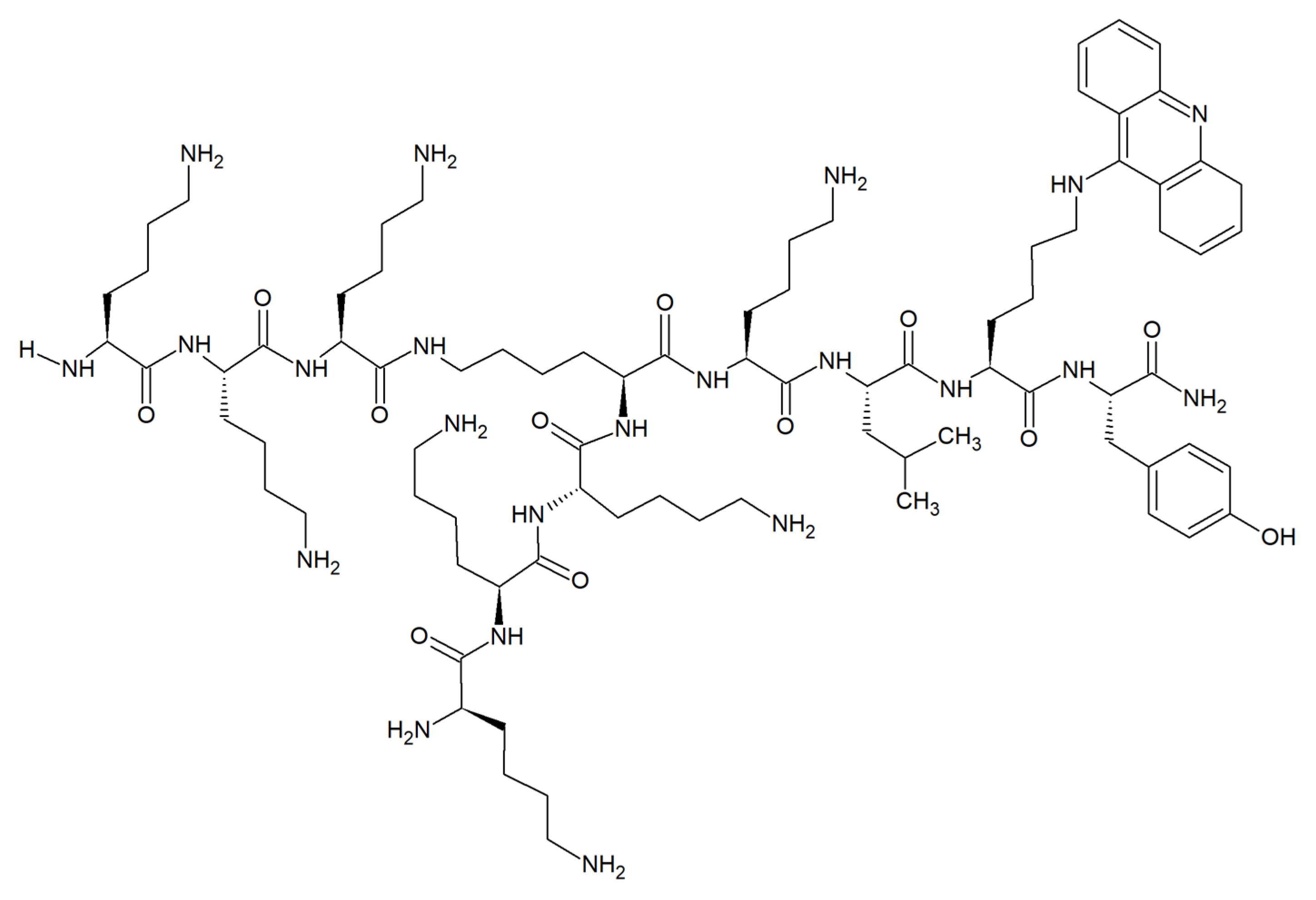
2.4. Antifungal Acridine-Peptide Conjugates
2.5. Acridine Interactions with Efflux/Influx Pumps
3. Conclusion and Future Perspective
Funding
Conflicts of Interest
References
- Wainwright, M. Acridine-a neglected antibacterial chromophore. J. Antimicrob. Chemother. 2001, 47, 1–13. [Google Scholar] [CrossRef]
- Cardenas, M.E.; Cruz, M.C.; Del Poeta, M.; Chung, N.; Perfect, J.R.; Heitman, J. Antifungal Activities of Antineoplastic Agents: Saccharomyces cerevisiae as a Model System to Study Drug Action. Clin. Microbiol. Rev. 1999, 12, 583–611. [Google Scholar] [CrossRef] [PubMed]
- Ebara, S.; Naito, H.; Nakazawa, K.; Ishii, F.; Nakamura, M. FTR1335 is a novel synthetic inhibitor of Candida albicans N-myristoyltransferase with fungicidal activity. Biol. Pharm. Bull. 2005, 28, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Petraitiene, R.; Petraitis, V.; Kelaher, A.M.; Sarafandi, A.A.; Mickiene, D.; Groll, A.H.; Sein, T.; Bacher, J.; Walsh, T.J. Efficacy, plasma pharmacokinetics, and safety of icofungipen, an inhibitor of Candida isoleucyl-tRNA synthetase, in treatment of experimental disseminated candidiasis in persistently neutropenic rabbits. Antimicrob. Agents Chemother. 2005, 49, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, I.; Vetter, N.D.; Palmer, D.R.J.; Milewska, M.J.; Wojciechowski, M.; Milewski, S. Homoisocitrate dehydrogenase from Candida albicans: Properties, inhibition, and targeting by an antifungal pro-drug. FEMS Yeast Res. 2013, 13, 143–155. [Google Scholar] [CrossRef]
- Milewska, M.J.; Prokop, M.; Gabriel, I.; Wojciechowski, M.; Milewski, S. Antifungal activity of homoaconitate and homoisocitrate analogs. Molecules 2012, 17, 14022–14036. [Google Scholar] [CrossRef]
- Liu, J.; Bolstad, D.B.; Smith, A.E.; Priestley, N.D.; Wright, D.L.; Anderson, A.C. Structure-guided development of efficacious antifungal agents targeting Candida glabrata dihydrofolate reductase. Chem. Biol. 2008, 15, 990–996. [Google Scholar] [CrossRef]
- Farmakiotis, D.; Kontoyiannis, D.P. Epidemiology of antifungal resistance in human pathogenic yeasts: Current viewpoint and practical recommendations for management. Int. J. Antimicrob. Agents 2017, 50, 318–324. [Google Scholar] [CrossRef]
- Prasher, P.; Sharma, M. Medicinal chemistry of acridine and its analogues. Med. Chem. Commun. 2018, 9, 1589–1618. [Google Scholar] [CrossRef]
- Avers, C.J.; Pfeffer, C.R.; Rancourt, M.W. Acriflavine induction of different kinds of “petite” mitochondrial populations in Saccharomyces cerevisiae. J. Bacteriol. 1965, 90, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Steinert, M.; Assel, S.V. The loss of kinetoplastic DNA in two species of Trypanosomatidae treated with acriflavine. J. Cell Biol. 1967, 34, 489–503. [Google Scholar] [CrossRef]
- Keyhani, E.; Khavari-Nejad, S.; Keyhani, J.; Attar, F. Acriflavine-mediated apoptosis and necrosis in yeast Candida utilis. Ann. N. Y. Acad. Sci. 2009, 1171, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Kaya, M.; Yildirir, Y.; Celik, G.Y. Synthesis, Characterization, and In Vitro Antimicrobial and Antifungal Activity of Novel Acridines. Pharm. Chem. J. 2015, 48, 722–726. [Google Scholar] [CrossRef]
- Markovich, Y.D.; Kudryavtseva, T.N.; Bogatyrev, K.V.; Sysoev, P.I.; Klimova, L.G.; Nazarov, G.V. Synthesis of 2-(4-methyl-1,3-thiazol-5-yl) ethyl esters of acridone carboxylic acids and evaluation of their antibacterial activity. Russ. Chem. Bull. 2014, 63, 1153–1158. [Google Scholar] [CrossRef]
- Chen, R.; Huo, L.; Jaiswal, Y.; Huang, J.; Zhong, Z.; Zhong, J.; Williams, L.; Xia, X.; Liang, Y.; Yan, Z. Design, Synthesis, Antimicrobial, and Anticancer Activities of Acridine Thiosemicarbazides Derivatives. Molecules 2019, 24, 2065. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, D.B.C.; Silva, L.B.; da Silva, B.V.; Borges, T.C.; Marques, B.C.; Dos Santos, M.B.; de Oliveira, L.F.; Bolzani, V.S.; Rodrigues, A.R.A.; Regasini, L.O.; et al. A new acridone with antifungal properties against Candida spp. and dermatophytes, and antibiofilm activity against C. albicans. J. Appl. Microbiol. 2019, 127, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Kulkarny, V.V.; Chavez-Dozal, A.; Rane, H.S.; Jahng, M.; Bernardo, S.M.; Parra, K.J.; Lee, S.A. Quinacrine inhibits Candida albicans growth and filamentation at neutral pH. Antimicrob. Agents. Chemother. 2014, 58. [Google Scholar] [CrossRef]
- Persinoti, G.F.; de Aguiar Peres, N.T.; Jacob, T.R.; Rossi, A.; Vêncio, R.Z.; Martinez-Rossi, N.M. RNA-sequencing analysis of Trichophyton rubrum transcriptome in response to sublethal doses of acriflavine. BMC Genomics 2014, 15. [Google Scholar] [CrossRef]
- Kabir, M.A.; Hussain, M.A.; Ahmad, Z. Candida albicans: A Model Organism for Studying Fungal Pathogens. ISRN Microbiol. 2012, 2012. [Google Scholar] [CrossRef]
- Ramage, G.; Rajendran, R.; Sherry, L.; Williams, C. Fungal Biofilm Resistance. Int. J. Microbiol. 2012, 2012. [Google Scholar] [CrossRef]
- Gauwerky, K.; Borelli, C.; Korting, H.C. Targeting virulence: A new paradigm for antifungals. Drug Discov. Today 2009, 14, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, M. ‘Safe’ photoantimicrobials for skin and soft-tissue infections. Int. J. Antimicrob. Agents 2010, 36, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Tavares, A.; Carvalho, C.M.; Faustino, M.A.; Neves, M.G.; Tome, J.P.; Tome, A.C.; Cavaleiro, J.A.; Cunha, A.; Gomes, N.C.; Alves, E.; et al. Antimicrobial photodynamic therapy: Study of bacterial recovery viability and potential development of resistance after treatment. Mar. Drugs 2010, 8, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, F.; Martinelli, M.; Cocchi, A.; Arbia, D.; Fantetti, L.; Roncucci, G. In vitro resistance selection studies of RLP068/Cl, a new Zn(II) phthalocyanine suitable for antimicrobial photodynamic therapy. Antimicrob. Agents Chemother. 2010, 54, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.F.; McCarron, P.A.; Tunney, M.M. Antifungal photodynamic therapy. Microbiol. Res. 2008, 163, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Felip-León, C.; Martínez-Arroyo, O.; Díaz-Oltra, S.; Miravet, J.F.; Apostolova, N.; Galindo, F. Synthesis, spectroscopic studies and biological evaluation of acridine derivatives: The role of aggregation on the photodynamic efficiency. Bioorg. Med. Chem. Lett. 2018, 28, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Taraszkiewicz, A.; Grinholc, M.; Bielawski, K.P.; Kawiak, A.; Nakonieczna, J. Imidazoacridinone derivatives as efficient sensitizers in photoantimicrobial chemotherapy. Appl. Environ. Microbiol. 2013, 79, 3692–3702. [Google Scholar] [CrossRef]
- Taraszkiewicz, A.; Szewczyk, G.; Sarna, T.; Bielawski, K.P.; Nakonieczna, J. Photodynamic Inactivation of Candida albicans with Imidazoacridinones: Influence of Irradiance, Photosensitizer Uptake and Reactive Oxygen Species Generation. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Bailly, C. Contemporary challenges in the design of topoisomerase II inhibitors for cancer chemotherapy. Chem. Rev. 2012, 112. [Google Scholar] [CrossRef]
- Capizzi, R.L.; Roman, L.A.; Tjulandin, S.; Smirnova, I.; Manikhas, A.; Paterson, J.S.; Major, A.; Lundberg, A.S.; Fumoleau, P. Phase II trial of C1311, a novel inhibitor of topoisomerase II in advanced breast cancer. J. Clin. Oncol. 2008, 26, 1055. [Google Scholar] [CrossRef]
- Baltazar, L.M.; Ray, A.; Santos, D.A.; Cisalpino, P.S.; Friedman, A.J.; Nosanchuk, J.D. Antimicrobial photodynamic therapy: An effective alternative approach to control fungal infections. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A. Acridine derivatives as chemotherapeutic agents. Curr. Med. Chem. 2002, 9, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Belmont, P.; Bosson, J.; Godet, T.; Tiano, M. Acridine and acridone derivatives, anticancer properties and synthetic methods: Where are we now? Anti-Cancer Agents Med. Chem. 2007, 7, 139–169. [Google Scholar] [CrossRef]
- Wu, C.C.; Li, Y.C.; Wang, Y.R.; Li, T.K.; Chan, N.L. On the structural basis and design guidelines for type II topoisomerase-targeting anticancer drugs. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef]
- Lemke, K.; Wojciechowski, M.; Laine, W.; Bailly, C.; Colson, P.; Baginski, M.; Larsen, A.K.; Skladanowski, A. Induction of unique structural changes in guanine-rich DNA regions by the triazoloacridone C-1305, a topoisomerase II inhibitor with antitumor activities. Nucleic Acids Res. 2005, 33, 6034–6047. [Google Scholar] [CrossRef]
- Wesierska-Gadek, J.; Schloffer, D.; Gueorguieva, M.; Uhl, M.; Skladanowski, A. Increased susceptibility of poly (ADP-ribose) polymerase-1 knockout cells to antitumor triazoloacridone C-1305 is associated with permanent G2 cell cycle arrest. Cancer Res. 2004, 64, 4487–4497. [Google Scholar] [CrossRef]
- Cholody, M.W.; Martelli, S.; Paradziej-Lukowicz, J.; Konopa, J. 5-[(Aminoalkyl)amino]imidazo[4,5,1-de]acridin-6-ones as a novel class of antineoplastic agents. Synthesis and biological activity. J. Med. Chem. 1990, 33, 49–52. [Google Scholar] [CrossRef]
- Mazerska, Z.; Sowinski, P.; Konopa, J. Molecular mechanism of the enzymatic oxidation investigated for imidazoacridinone antitumor drug, C-1311. Biochem. Pharmacol. 2003, 66, 1727–1736. [Google Scholar] [CrossRef]
- Khan, T.; Sankhe, K.; Suvarna, V.; Sherje, A.; Patel, K.; Dravyakar, B. DNA gyrase inhibitors: Progress and synthesis of potent compounds as antibacterial agents. Biomed. Pharmacother. 2018, 103, 923–938. [Google Scholar] [CrossRef]
- Goto, T.; Wang, J.C. Cloning of yeast TOP1, the gene encoding DNA topoisomerase I and DNA topoisomerase II. Proc. Natl. Acad. Sci. USA 1985, 82, 7178–7182. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Roles of DNA topoisomerases in chromosomal replication and segregation. Adv. Pharmacol. 1994, 29A, 103–134. [Google Scholar] [PubMed]
- Holm, C.; Goto, T.; Wang, J.C.; Botstein, D. DNA topoisomerase II is required at the time of mitosis in yeast. Cell 1985, 41, 553–563. [Google Scholar] [CrossRef]
- Del Poeta, M.; Toffaletti, D.L.; Rude, T.H.; Dykstra, C.C.; Heitman, J.; Perfect, J.R. Topoisomerase I is essential in Cryptococcus neoformans: Role in pathobiology and as an antifungal target. Genetics 1999, 152, 167–178. [Google Scholar]
- Jiang, W.; Gerhold, D.; Kmiec, E.B.; Hauser, M.; Becker, J.M.; Koltin, Y. The topoisomerase I gene from Candida albicans. Microbiology 1997, 143, 377–386. [Google Scholar] [CrossRef][Green Version]
- Kwok, S.C.; Schelenz, S.; Wang, X.; Steverding, D. In vitro effect of DNA topoisomerase inhibitors on Candida albicans. Med. Mycol. 2010, 48, 155–160. [Google Scholar] [CrossRef]
- Steverding, D.; Evans, P.; Msika, L.; Riley, B.; Wallington, J.; Schelenz, S. In vitro antifungal activity of DNA topoisomerase inhibitors. Med. Mycol. 2012, 50. [Google Scholar] [CrossRef]
- Shen, L.L.; Baranowski, J.; Fostel, J.; Montgomery, D.A.; Lartey, P.A. DNA topoisomerases from pathogenic fungi: Targets for the discovery of antifungal drugs. Antimicrob. Agents Chemother. 1992, 36, 2778–2784. [Google Scholar] [CrossRef] [PubMed]
- Fostel, J.; Montgomery, D.; Lartey, P. Comparison of responses of DNA topoisomerase I from Candida albicans and human cells to four new agents which stimulate topoisomerase-dependent DNA nicking. FEMS Microbiol. Lett. 1996, 138, 105–111. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kukowska, M. Amino acid or peptide conjugates of acridine/acridone and quinoline/quinolone-containing drugs. A critical examination of their clinical effectiveness within a twenty-year time frame in antitumor chemotherapy and treatment of infectious diseases. Eur. J. Pharm. Sci. 2017, 109, 587–615. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.B.; Vuyisich, M.; Gooch, B.D.; Beal, P.A. Preferred RNA binding sites for a threading intercalator revealed by in vitro evolution. Chem. Biol. 2003, 10, 663–672. [Google Scholar] [CrossRef]
- Qi, X.; Xia, T.; Roberts, R.W. Acridine-N peptide conjugates display enhanced affinity and specificity for boxB RNA targets. Biochemistry 2010, 49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, X.; Sofang, J.; Zheng, X.; Chen, J.; Ma, P.; Zhang, B.; Wang, R. Conjugation with acridines turn nuclear localization sequence into highly active antimicrobial peptide. Engineering 2015, 1, 500–505. [Google Scholar] [CrossRef]
- Wynn, J.E.; Zhang, W.; Falkinham, J.O., III; Santos, W.L. Branched Peptides: Acridine and Boronic Acid Derivatives as Antimicrobial Agents. ACS Med. Chem. Lett. 2017, 8, 820–823. [Google Scholar] [CrossRef]
- Wakiec, R.; Gabriel, I.; Prasad, R.; Becker, J.M.; Payne, J.W.; Milewski, S. Enhanced susceptibility to antifungal oligopeptides in yeast strains overexpressing ABC multidrug efflux pumps. Antimicrob. Agents Chemother. 2008, 52, 4057–4063. [Google Scholar] [CrossRef]
- Prasad, R.; Kapoor, K. Multidrug resistance in yeast Candida. Int. Rev. Cytol. 2005, 242, 215–248. [Google Scholar]
- Singh, P.; Kaur, J.; Yadav, B.; Komath, S.S. Design, synthesis and evaluations of acridone derivatives using Candida albicans-search for MDR modulators led to the identification of an anti-candidiasis agent. Bioorg. Med. Chem. 2009, 17, 3973–3979. [Google Scholar] [CrossRef]
- Singh, P.; Kaur, J.; Yadav, B.; Komath, S.S. Targeting efflux pumps-In vitro investigations with acridone derivatives and identification of a lead molecule for MDR modulation. Bioorg. Med. Chem. 2010, 18, 4212–4223. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ||||
|---|---|---|---|---|
| Antifungal Spectrum of Activity | Mode of Action | MIC range | Ref. | |
| Acriflavine | S. cerevisiae | Differences in mitochondrial ultrastructure. Considerable variation in mitochondrial enzyme activities. | - | [10] |
| C. utilis | Alteration in cell respiratory control ratio and a decrease in cytochrome content. Chromatin condensation and cytoplasmic lysis. | - | [12] | |
| T. rubrum | Alteration in transmembrane transport and oxidation-reduction reactions. Down-regulation of genes putatively involved in the pathogenicity. | - | [18] | |
| Quinacrine | C. albicans | Growth and filamentation inhibition. Antibiofilm activity. Vacuolar alkalinization and defects in endocytosis. | - | [17] |
| 1,8-dioxoacridine derivatives | C. albicans C. glabrata | Moderate growth inhibition. | - | [13] |
| 2-(4-methyl-1,3-thiazol-5-yl)ethyl esters of acridone carboxylic acids | C. albicans | Moderate growth inhibition. | - | [14] |
| Acridine thiosemicarbazides derivatives | C. albicans | Strong growth inhibition. | 20–80 μM | [15] |
| M14 | Candida spp. C. parapsilosis C. orthopsilosis C. metapsilosis C. albicans C. glabrata C. neorugosa C. tropicalis C. lusitaniae C. guilliermondii Trichophyton spp. T. rubrum T. mentagrophytes T. tonsurans T. verrucosum T. schoenleinii | Fungicidal activity. The inhibition of C. albicans biofilm formation. C. albicans and T. rubrum hyphal growth inhibition. Low toxicity to human fibroblasts. | 7.81–31.25 μg mL-1 | [16] |
| Imidazoacridinone derivatives | C. albicans C. glabrata | Efficient accumulation in the nucleus and vacuoles. Superoxide anion generation. Not effluxed by Candida ABC transporters. | - | [27,28] |
| Acridine conjugates with branched lysine peptides | C. albicans A. niger | Growth and biofilm formation inhibition. Anticandidal activity not affected by the removal of acridine moiety. No effect on fungal membrane integrity. Moderate growth inhibition related to the presence of acridine moiety for A. niger. Negligible hemolytic effects on red blood cells. No toxicity to mammalian cells. | 1–4 μg mL-1 | [54] |
| Acridones carrying hydroxyl amine substituent at N-10 and COOH at C-4 | C. albicans | Growth inhibition. The cell wall rupturing. Multi drug resistance modulators. | 132–405 μg mL-1 | [57,58] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabriel, I. ‘Acridines’ as New Horizons in Antifungal Treatment. Molecules 2020, 25, 1480. https://doi.org/10.3390/molecules25071480
Gabriel I. ‘Acridines’ as New Horizons in Antifungal Treatment. Molecules. 2020; 25(7):1480. https://doi.org/10.3390/molecules25071480
Chicago/Turabian StyleGabriel, Iwona. 2020. "‘Acridines’ as New Horizons in Antifungal Treatment" Molecules 25, no. 7: 1480. https://doi.org/10.3390/molecules25071480
APA StyleGabriel, I. (2020). ‘Acridines’ as New Horizons in Antifungal Treatment. Molecules, 25(7), 1480. https://doi.org/10.3390/molecules25071480