Abstract
Starting out from dipotassium 1,5-oligosiloxanylene diide 2, a 3,7,10-trioxa-octasilabicyclo[3.3.3]undecane was prepared, which represents the third known example of this cage structure type. Reaction of 1,3-dichlorotetramethyldisiloxane with 1,1’-bis[bis(trimethylsilyl)potassiosilyl]ferrocene gave a ferrocenophane with a disiloxane containg bridge. The compound can be further derivatized by conversion into a 1,5-oligosilanyl diide. Reacting 1,5-oligosiloxanylene diide 2 with SnCl2 or GeCl2·dioxane in the presence of PMe3 gave cyclic disilylated tetrylene PMe3 adducts. Release of the base-free stannylene led to a dimerization process which gave a bicyclic distannene as the final product. Abstraction of the PMe3 from the cyclic disilylated germylene PMe3 adduct with B(C6F5)3 caused oxidative addition of the germylene into a para-C-F bond of Me3P·B(C6F5)3.
1. Introduction
Polysiloxanes (silicones) are an important class of compounds with an almost limitless number of applications. However, despite the huge popularity of these compounds, the vast majority of siloxanes exist as complex mixtures and the synthesis of structurally defined examples has proved to be quite challenging. The reasons for this are strongly associated with the reaction conditions of these acid or base-catalyzed production processes. Siloxanes undergo redistribution reactions of Si-O bonds under these conditions leading to mixtures of different poly- and oligomeric materials together with cyclic compounds.
The recent development of novel synthetic methods, employing neutral conditions has brought the study of oligosiloxanes with exactly defined structures into the focus of several research groups [1,2,3,4,5,6,7,8]. While most of these new methods exploit the activation of hydrosilanes, it has been recognized that the oxidation of oligo- or polysilanes represents another interesting method to obtain oligo- or polysiloxanes retaining the substitution pattern of the starting material [9].
Our own interest in this direction arises from our work on the synthesis of structurally diverse oligosilanes [10]. Utilizing oligosilanyl units as ligands for lanthanides, we recognized the need for additional Lewis basic coordination sites in the backbone of the ligand [11]. We modified the initially used 1,4-tetrasilanylene unit [12,13,14] by introducing an oxygen atom between the two central silicon atoms. The resulting building block is represented by the easily available disiloxane 1. Its treatment with two equivalents of tBuOK leads to the respective oligosilanylene diide 2 (Scheme 1) [11], which can be used as a versatile starting material for the preparation of larger molecular architectures containing oligosiloxane substructures [15].
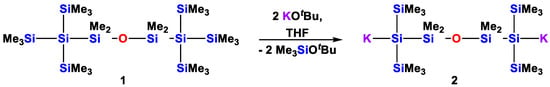
Scheme 1.
Conversion of 1,3-bis[tris(trimethylsilyl)silyl]tetramethyldisiloxane (1) into the 1,5-oligosilanylene diide 2.
Previously, we reported on the use of compound 2 for the synthesis of a number of small cyclic and acyclic oligosiloxanes as well as its application as a ligand for a number of silylated transition metal complexes [15]. In the current account, we wish to extend these studies further and in particular regarding the use of compound 2 as a ligand for germylene and stannylene units.
2. Results and Discussion
Compound 2 was used to incorporate the bis(isotetrasilanyl)siloxane unit into a number of larger oligosiloxanes [15]. Among these, the tricyclic compound 3 (Scheme 2) is noteworthy as it is a rare example of a tricyclic oligosiloxane with a 3,7,10-trioxa-octasilabicyclo[3.3.3]undecane framework [15].
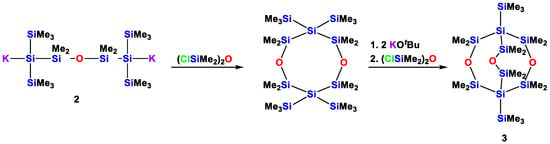
Scheme 2.
Preparation of a 1,5-dioxacyclooctasilane (2), its conversion to the respective 3,7-disilanide, which is further transformed to 1,5-bis(trimethylsilyl)-3,7,10-trioxa-octasilabicyclo[3.3.3]undecane (3).
The only other two known instances of this compound class were reported by Lickiss [16] and more recently by Krempner [17] (Chart 1). Lickiss’ compound is very similar to compound 3, with the only difference being that the trimethylsilyl groups attached to the bridgehead atoms are replaced by dimethylchlorosilyl groups. The compound was formed by careful hydrolysis of (ClMe2Si)4Si. Although a crystal structure analysis was performed on the compound, the poor quality of the structural solution does not allow an in depth structural discussion [16]. Krempner’s compound is of lower symmetry and was prepared by the reaction of a more complex lithium oligosilanyltriolate [18] with a trichloroisotetrasilane [17]. Furthermore, for this compound, single crystal XRD analysis data are available [17].
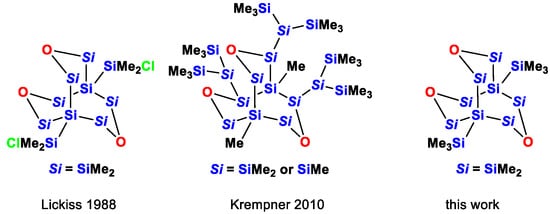
Chart 1.
Known examples of compounds containing the 3,7,10-trioxa-octasilabicyclo[3.3.3]undecane framework.
It took us some time to find the correct space group (tetragonal, P41) for compound 3 due to its merohedral twinning. The molecular structure features a regular symmetric 3,7,10-trioxa-octasilabicyclo[3.3.3]undecane with trimethylsilyl groups attached to the bridgehead silicon atoms (Figure 1). The Me3Si-Si and Si-SiMe2O bond distances of 2.35 and 2.34 Å indicate a non strained cage geometry. All Si-O distances are close to 1.64 Å, and the Si-O-Si angles range between 147.9 and 151.7 deg, which is quite typical for this class of compounds. A distance of 4.695 Å was detected between the two bridgehead atoms of compound 3. This corresponds nicely to a distance of 4.706 Å found for the Lickiss compound. For Krempner’s compound, this distance amounts only to 4.473 Å [17]. The reason for this is likely that the bulky 2-trisilanyl groups in the 2-, 8-, and 9-positions cause a twisting of the 3,7,10-trioxa-octasilabicyclo[3.3.3]undecane cage, which shortens its lengths. For compound 3, 29Si NMR resonances at 10.9, −15.3, and −136.2 ppm for the SiMe3, SiMe2, and bridgehead Si atoms were found [15], which correlate quite nicely with the 5.5 and −136.4 ppm reported for the analogous groups in Lickiss’ compound [16].
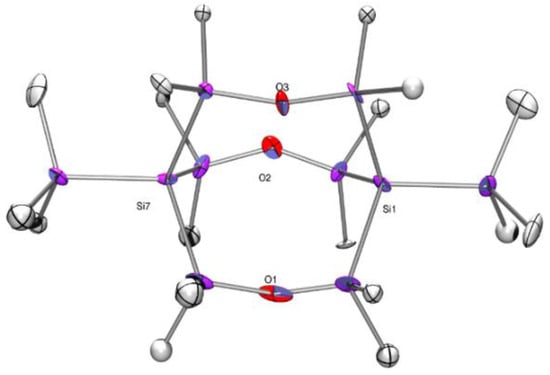
Figure 1.
Molecular structure of compound 3 (thermal ellipsoid plot drawn at the 30% probability level). All hydrogen atoms are omitted for clarity (bond lengths in Å, angles in deg). Si(1)-Si(4) 2.3404(18), Si(1)-Si(3) 2.3430(18), Si(1)-Si(2) 2.3486(18), Si(1)-Si(5) 2.3497(18), Si(4)-O(3) 1.641(4), Si(6)-O(3) 1.647(4), Si(8)-O(1) 1.648(4), Si(4)-Si(1)-Si(3) 108.90(7), Si(4)-Si(1)-Si(2) 110.01(7), Si(3)-Si(1)-Si(2) 110.54(7), Si(4)-Si(1)-Si(5) 110.13(7), Si(3)-Si(1)-Si(5) 107.46(7), Si(2)-Si(1)-Si(5) 109.76(7), Si(10)-O(2)-Si(3) 147.9(2), Si(4)-O(3)-Si(6) 148.2(2).
In the course of our previous studies of oligosilanyl dianions, we investigated numerous molecular bridges to connect two tris(trimethylsilyl)silyl groups [19,20,21,22,23,24,25]. In one example, we used a 1,1′-ferrocenylene unit as a spacer. Starting out from tris(trimethylsilyl)silylcyclopentadiene, it is fairly easy to prepare 1,1′-bis[tris(trimethylsilyl)silyl]ferrocene [19]. Reaction of the latter with two equivalents of potassium tert-butoxide gives the dianion 4, which could be used to prepare a number of ansa-oligosilanylene bridged ferrocenes [19]. In a similar way, we could react compound 4 with bis(chlorodimethylsilyl)siloxane to obtain compound 5 (Scheme 3).
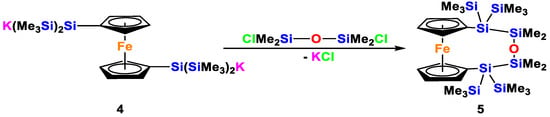
Scheme 3.
Preparation of the oligosiloxane bridged ferrocene 5 from dianion 4.
With the exception of the SiMe2O unit, 1H, 13C, and 29Si NMR spectra of compound 5 are similar to that of a related 1,4-tetrasilanylene bridged compound. A 29Si NMR chemical shift of 7.0 ppm for the SiMe2 group reflects the presence of the comparatively electronegative Cp unit. As there is free rotation around the Cp-Fe bonds, extended lengths of the ansa-bridge does not cause steric strain but only increased the separation of the ipso-Cp carbon atoms. This can be clearly observed in the solid-state structure of 5 (Figure 2). If we compare the Si-C-C-Si torsional angles of compounds where the ansa-bridge consists of either a 1,2-disilanylene [19] or a 1,4-tetrasilanylene [19] unit with that of compound 5, we observe an increase in the value from ca. 20 deg to 55 deg and 70 deg for compound 5. The Si-O-Si angle is 156.12 deg, which is a typical value for this unit in acyclic or larger ring systems [15].
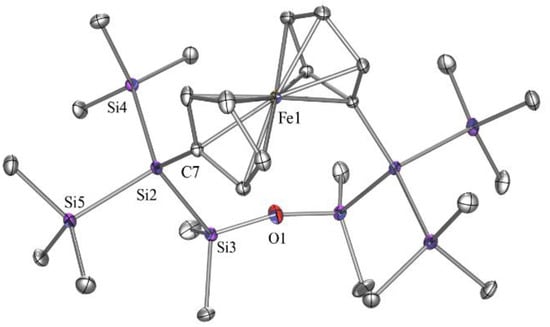
Figure 2.
Molecular structure of compound 5 (thermal ellipsoid plot drawn at the 30% probability level). All hydrogen atoms are omitted for clarity (bond lengths in Å, angles in deg). Fe(1)-C(7) 2.080(3), O(1)-Si(3) 1.6250(11), Si(2)-C(7) 1.880(3), Si(2)-Si(3) 2.3474(12), Si(2)-Si(4) 2.3507(12), Si(2)-Si(5) 2.3555(12), C(7)-Si(2)-Si(3) 108.30(10), C(7)-Si(2)-Si(4) 114.63(10),Si(3)-Si(2)-Si(4) 111.57(4), Si(2)-C(7)-Fe(1) 131.76(15).
In an attempt to generate an oligosilanyl dianion from 5, we reacted it with two equivalents of KOtBu. Despite a seemingly very fast reaction, we were not able to detect the supposedly formed dianionic compound 6 NMR spectroscopically. In addition to formation of Me3SiOtBu, the spectra (1H, 13C and 29Si) only showed the formation of rather broad peaks in the trimethylsilyl region. Nevertheless, within minutes of reaction, a deep red precipitate formed, which reacts upon addition of trimethylchlorosilane to give compound 5, and when treated with phenyldimethylchlorosilane gives a mixture of two diasteromers 5a in an approximate 1:1 ratio (Scheme 4). We observed such a behavior before [19,20], which is caused by the configurational lability of silanides.
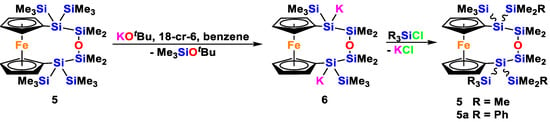
Scheme 4.
Reaction of oligosiloxane bridged ferrocene 5 with two equivalents of tBuOK to form the expected dianionic compound 6, which was derivatized either with Me3SiCl or PhMe2SiCl.
For the last part of this study, we decided to react disilanide 2 with GeCl2·dioxane and SnCl2 in the presence of PMe3 to test the formation of germylene and stannylene phosphane adducts (Scheme 5 and Scheme 6). Related reactions of two equivalents of potassium tris(trimethylsilyl)silanide with either ECl2·NHC (E = Ge, Sn; NHC = 1,3-diisopropyl-4,5-dimethyl-imidazol-2-ylidene (MeIiPr)) [26] or ECl2·PR3 (E = Ge, Sn; PR3 = PMe3, PEt3) [27,28,29] and of 1,3- and 1,4-oligosilanyldiides with ECl2·PR3 (E = Ge, Sn; PR3 = PMe3, PEt3) [30,31,32,33] gave Lewis base adducts of either acyclic or four- and five-membered cyclic disilylated germylenes and stannylenes.
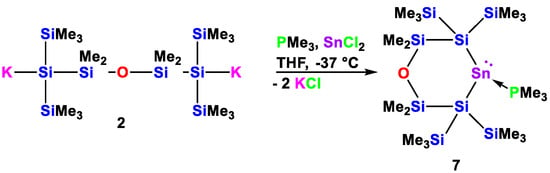
Scheme 5.
Reaction of 1,5-oligosilanylene diide 2 with SnCl2·PMe3 to the cyclic silylated stannylene PMe3 adduct 7.
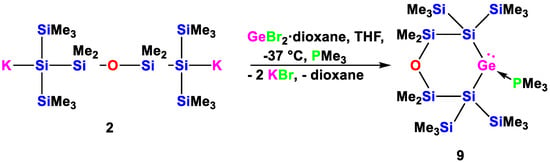
Scheme 6.
Reaction of 1,5-oligosilanylene diide 2 with GeCl2 in the presence of PMe3 to the cyclic silylated germylene PMe3 adduct 9.
Not unexpectedly, a clean formation of stannylene PMe3 adduct 7 was observed in the reaction of compound 2 with SnCl2·PMe3 (Scheme 5) at low temperature.
1H, 13C, and 29Si NMR spectra of compound 7 show two signal sets for the methyl and trimethylsilyl groups on the different sides of the ring, indicating configurational stability of the tin atom with a stereochemically active lone pair. The same was observed before at the stannacyclopentasilane analog with PEt3 serving as base [30]. The 119Sn NMR resonance of compound 7 was found at −167.9 ppm, which is somewhat downfield from the −224.4 ppm observed for the related stannacyclopentasilane analog [30]. For the two examples of bis[tris(trimethylsilyl)silyl]stannylene phosphane adducts with PEt3 [28] and PMe3 [33], the respective 119Sn NMR resonances were detected at −113.3 and −89.9 ppm. When these values are correlated with the Si-Sn-Si angles observed in the solid state, it becomes evident that the chemical shift goes downfield with increasing Si-Sn-Si angle, which correlates with a diminished singlet–triplet gap. Although the 31P NMR resonance of compound 7 (−62.5 ppm) is very close to that of free PMe3, coupling to 117Sn and 119Sn can be observed very well.
The single crystal XRD analysis of compound 7 (Figure 3) exhibits Sn-Si distances of 2.6465(8) and 2.6662(8) Å, a Sn-P distance of 2.6059(9) Å, and a Si-Sn-Si angle of 103.35(2) deg. While the distances in the stannacyclopentasilane analog are very similar, the Si-Sn-Si angle of that compound is smaller (98.17(9) deg). For the acyclic compounds [(Me3Si)3Si]2Sn·PR3 (R = Et. Me) [28,33], the Sn-Si distances range between 2.686(3) and 2.717(1) Å and the Si-Sn-Si angles increase to 114.21(4) (PEt3) and 117.53(6) (PMe3) deg.
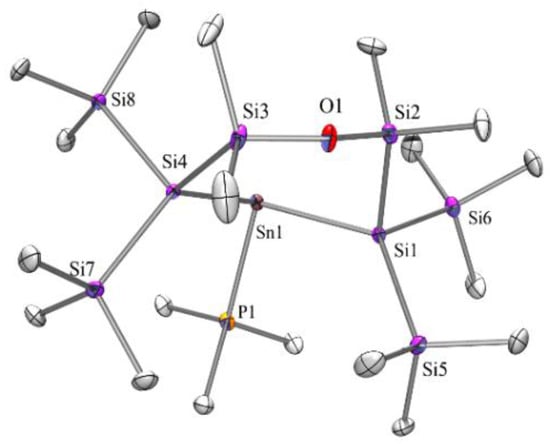
Figure 3.
Molecular structure of compound 7 (thermal ellipsoid plot drawn at the 30% probability level). All hydrogen atoms are omitted for clarity (bond lengths in Å, angles in deg). C(1)-P(1) 1.814(3), C(4)-Si(2) 1.866(3), O(1)-Si(3) 1.6378(19), O(1)-Si(2) 1.642(2), P(1)-Sn(1) 2.6059(8), Si(1)-Si(2) 2.3397(11), Si(1)-Sn(1) 2.6464(8), Si(4)-Sn(1) 2.6662(8), Si(3)-O(1)-Si(2) 145.51(13), Si(2)-Si(1)-Sn(1) 96.56(3), Si(3)-Si(4)-Sn(1) 109.20(3), P(1)-Sn(1)-Si(1) 99.58(2), P(1)-Sn(1)-Si(4) 104.33(2), Si(1)-Sn(1)-Si(4) 103.35(2).
When the reaction of compound 2 with SnCl2 was repeated in the absence of a strongly donating agent (PR3, NHC), the formation of a bicyclic distannene (8) was observed. In addition, this reaction course follows what we previously observed for the reactions of dipotassio α,α,ω,ω-tetrakis(trimethylsilyl)oligosilanyl-α,ω-diiides with SnCl2 [30,33]. For this reason we suppose that compound 8 forms by dimerization of an initially formed six-membered cyclic disilylated stannylene to an exocyclic distannene, which finally rearranges via two 1,2-silyl shifts to compound 8 (Scheme 7). For the bicyclic distannene with two annulated six-membered rings, support for this reaction mechanism was obtained by a computational study [30].
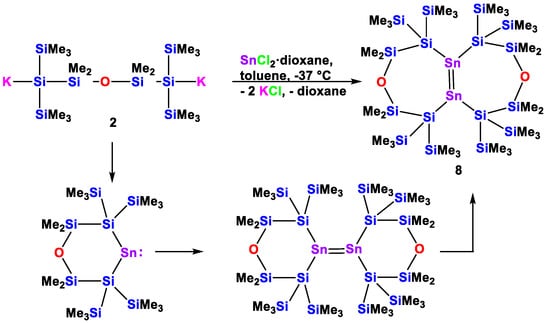
Scheme 7.
Reaction of 1,5-oligosilanylene diide 2 with SnCl2 in the absence of a donating Lewis base to the bicyclic tetrasilylated distannene 8.
Compound 8 adds to a growing number of known tetrasilylated distannenes. Starting with Klinkhammer’s bis[tris(trimethylsilyl)silyl]stannylene, which exists as a distannene in the solid state [34], followed by Sekiguchi’s tetrakis(di-tert-butylmethylsilyl)distannene [35], our group added two further examples of bicyclic distannenes [30,33] (Chart 2).
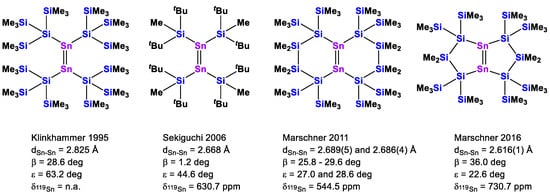
Chart 2.
Known tetrasilylated distannenes with key structural values of Sn-Sn bond distance, bending angle β, twisting angle ε, and 119Sn NMR chemical shift value.
The 119Sn NMR chemical shift of compound 8 (636.9 ppm) is very close to Sekiguchi’s compound (tBu2MeSi)2Sn = Sn(SiMetBu2)2 (see Chart 2). In the crystal structure of compound 8 (Figure 4), the bending angle is 31.1 deg, but its twisting angle is rather large. However, while (tBu2MeSi)2Sn = Sn(SiMetBu2)2 is a rather symmetric molecule with respect to the twisting of the stannylene units, the situation in compound 8 is somewhat more complicated. The two rings of the bicyclic molecules exhibit different conformations, with one ring showing a small Si-Sn-Sn-Si torsional angle of 17.0 deg, while for the other ring, the angle measured was 81.6 deg. The Sn=Sn double bond distance of 2.7409(9) Å, as observed for compound 8, is the longest of the three examples of bicyclic disilylated stannanes (Chart 2) [30,33]. It seems that increasing ring size and likely the associated increased ring strain cause a diminished interaction of the two stannylene units. This does not, however, correlate with the respective 119Sn NMR chemical shifts.
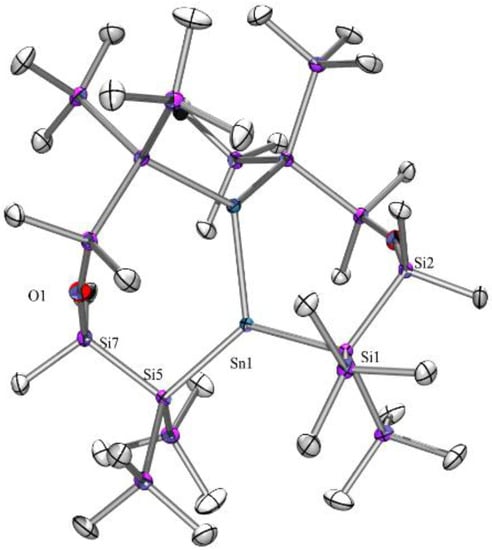
Figure 4.
Molecular structure of compound 8 (thermal ellipsoid plot drawn at the 30% probability level). All hydrogen atoms are omitted for clarity (bond lengths in Å, angles in deg). Sn(1)-Si(1) 2.6131(8), Sn(1)-Si(5) 2.6179(8), Sn(1)-Sn(1A) 2.7409(9), Si(1)-Si(2) 2.3557(10), Si(7)-O(1) 1.6302(8), Si(7)-C(14) 1.866(3), Si(1)-Sn(1)-Si(5) 122.90(3), Si(1)-Sn(1)-Sn(1A) 105.336(17), Si(5)-Sn(1)-Sn(1A) 122.683(18), Si(7)-O(1)-Si(7A) 168.10(19).
Reacting oligosilanyldiide 2 with GeCl2·dioxane in the presence of PMe3 gave the germylene PMe3 adduct (Scheme 6) in a clean reaction. Again, this is in agreement with similar reactions to acyclic [29,36] and cyclic [32,37] disilylated germylenes we have reported before. The obtained compound 9 features the expected six-membered ring with a germylene being coordinated by a PMe3 molecule as base.
1H, 13C, and 29Si NMR spectra of compound 9 again show two signal sets for the methyl and trimethylsilyl groups on the different sides of the ring, thus indicating configurational stability of the germylene unit. However, the trimethylsilyl groups show clear signs of signal broadening, suggesting that at rt conditions, coalescence has already started. For the related germacyclopentasilane analog with PEt3 serving as base [37], the signals at rt are much broader, indicating a higher degree of inversion. However, it is not clear whether the increased configurational stability of compound 9 is based on the different ligand and different Si-Ge-Si angle or the stronger interaction between the germylene and PMe3 compared to PEt3. The Si-Ge-Si angle of compound 9 in the solid state (Figure 5) of 107.88(4) deg is larger than the respective angle of compound 7 (103.35(2) deg), which can be explained by the shorter Si-Ge bonds. The same angle in the five-membered ring examples [37,38] is also smaller (101.3(3) and 102.70(1) deg). It is known that the geometry of the tetrylene has a decisive influence on the singlet–triplet gap [39], so it can be assumed that the respective free germylene of compound 9 exhibits a higher reactivity than the examples with smaller Si-Ge-Si angles.
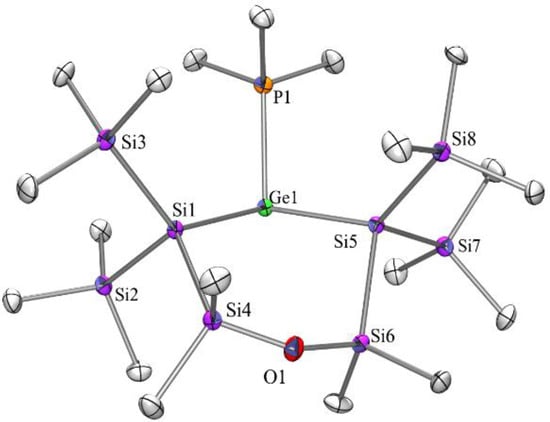
Figure 5.
Molecular structure of compound 9 (thermal ellipsoid plot drawn at the 30% probability level). All hydrogen atoms are omitted for clarity (bond lengths in Å, angles in deg). Ge(1)-P(1) 2.3570(13), Ge(1)-Si(5) 2.4463(13), Ge(1)-Si(1) 2.4664(13), O(1)-Si(4) 1.625(3), O(1)-Si(6) 1.642(3), P(1)-C(1) 1.818(5), Si(1)-Si(4) 2.3656(17), Si(2)-C(9) 1.879(5), Si(5)-Si(6) 2.3537(17), P(1)-Ge(1)-Si(5) 100.20(5), P(1)-Ge(1)-Si(1) 101.87(4), Si(5)-Ge(1)-Si(1) 107.88(5), Si(4)-O(1)-Si(6) 148.4(2), Si(4)-Si(1)-Ge(1) 110.79(6).
In previous studies, we found that the addition of strong donor molecules like phosphanes or N-heterocyclic carbenes (NHCs) is essential to achieve a clean germylene formation in the reaction of silanides with GeCl2. In order to get access to the free germylene from its phosphane adduct, treatment with a fairly strong Lewis acid proved to be a good strategy. The Lewis acid would abstract the donor and release the germylene. Massey’s borane B(C6F5)3 [40] was found to be a particularly good reagent in this respect. When compound 9 was treated with B(C6F5)3, we expected PMe3 abstraction, followed by the formation of the free germylene, which could dimerize in a similar way as shown above for the analogous stannnylene. Alternatively, we also observed several instances where the germylene undergoes a 1,2-silyl shift to the germanium atom, thus forming a silagermene, which might undergo a 2 + 2 cycloaddition reaction in a subsequent step. However, in the current case, our expectations were not met and another reaction pathway was observed. While the PMe3 abstraction seemingly occurred as expected, the free germylene reacted by insertion into a p-C-F bond of (C6F5)3B·PMe3 (Scheme 8). This is quite unexpected as we have never before observed activation of C-F bonds in related reactions. Nevertheless, the reaction is not completely without precedence. Roesky and co-workers observed C-F activation of heteroleptic chloro- and amino-silylenes (LSiX, X = Cl, NR2, L = PhC(NtBu)2) stabilized by a benzamidinato ligand with tert-butyl substituents on the nitrogen atoms [41,42]. Related reactivity was even observed with germylenes and stannylenes [43]. One particular aspect of this chemistry is that there is some completion between oxidative addition of C-F bonds and fluoride metathesis to products, where the divalent character of the tetrylene is retained and only the substituent is exchanged for fluoride. This chemistry has also been studied theoretically [44].
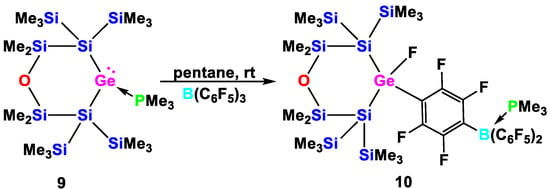
Scheme 8.
Reaction of germylene adduct 9 with B(C6F5)3 to abstract the PMe3 to release the free germylene, which in a subsequent step inserts into a para-C-F bond of B(C6F5)3·PMe3 to give compound 10.
The fact that this oxidative addition occurs with the free germylene of compound 9 but was not observed with the previously studied cyclic disilylated germylenes suggests a higher reactivity, which is likely caused by the larger Si-Ge-Si bond angle which favors a smaller singlet–triplet gap [39].
NMR spectra of compound 10 feature a combination of the cyclooligosilyloxygermanium part similar to in compound 9 with signals similar to that of Me3P·B(C6F5)3 [45] and B(p-C6F4H)3 [46]. The 1H, 13C, and 29Si spectra mainly represent the cyclooligosilyloxygermanium fragment. With two types of methyl and trimethylsilyl resonances indicating ring-side differentiation. The borate adduct part of compound 10 is mainly represented in the 19F and 31P NMR spectra. The resonance for the Ge-F unit in the 19F NMR spectrum was detected at −209.2 ppm. While no similar germanium fluoride is known, the chemical shift of the related silane [(Me3Si)2MeSi]2PhSiF at −193.5 ppm [47] supports the assignment.
In the solid-state structure of compound 10 (Figure 6), the rather short Si-Ge distances of 2.385(1) and 2.397(1) Å reflect the fact that bonds to tetravalent germanium are shorter. For the same bonds in compound 9 (2.446(2) and 2.466(1) Å), significantly longer distances were observed.
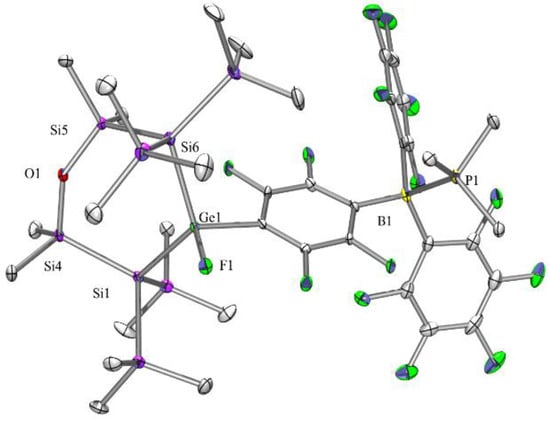
Figure 6.
Molecular structure of compound 10 (thermal ellipsoid plot drawn at the 30% probability level). All hydrogen atoms are omitted for clarity (bond lengths in Å, angles in deg). Ge(1)-F(1) 1.769(2), Ge(1)-C(17) 1.969(4), Ge(1)-Si(6) 2.3849(13), Ge(1)-Si(1) 2.3966(12), O(1)-Si(5) 1.654(3), O(1)-Si(4) 1.654(3), P(1)-C(35) 1.805(5), P(1)-B(1) 2.055(5), Si(1)-Si(4) 2.3775(17), Si(2)-C(1) 1.860(5), Si(5)-Si(6) 2.3447(17), B(1)-C(29) 1.632(7), F(10)-C(28) 1.347(6), F(1)-Ge(1)-Si(6) 105.08(9), F(1)-Ge(1)-Si(1) 104.92(9), Si(6)-Ge(1)-Si(1) 119.41(4), Si(5)-O(1)-Si(4) 139.1(2), Si(4)-Si(1)-Ge(1) 107.01(6), Si(5)-Si(6)-Ge(1) 101.08(5).
3. Experimental Section
3.1. General Remarks
All reactions involving air-sensitive compounds were carried out under an atmosphere of dry nitrogen or argon using either Schlenk techniques or a glove box. Solvents were dried using a column-based solvent purification system [48]. 1,3-Bis[tris(trimethylsilyl)silyl]-1,1,3,3-tetramethyldisiloxane (1) [11], 2 [11], 3 [15], and 4 [19] were prepared according to previously published procedures. All other chemicals were obtained from different suppliers and used without further purification.
1H (300 MHz), 13C (75.4 MHz), 19F ((282.2 MHz), 29Si (59.3 MHz), 31P (124.4 MHz), and 119Sn (111.8 MHz) NMR spectra were recorded on a Varian INOVA 300 spectrometer and are referenced to tetramethylsilane (TMS) for 1H, 13C, and 29Si, to CFCl3 for 19F, to 85% H3PO4 for 31P and to Me4Sn for 119Sn. If not otherwise noted, the solvent used was C6D6, and samples were measured at rt. In case of reaction samples, a D2O capillary was used to provide an external lock frequency signal. To compensate for the low isotopic abundance of 29Si, the INEPT pulse sequence [49,50] was used for the amplification of the signal for some of the spectra.
Elementary analyses were carried out using a Heraeus VARIO ELEMENTAR instrument. For a number of compounds, no good elemental analysis values could be obtained, which is a typical problem for these compounds, caused primarily by silicon carbide formation during the combustion process. Multinuclear NMR spectra (1H, 13C, 29Si) of these compounds are presented in the supplementary materials (Figures S1–S22) as proof of purity.
3.2. X-ray Structure Determination
For X-ray structure analyses, the crystals were mounted onto the tip of glass fibers, and data collection was performed with a BRUKER-AXS SMART APEX CCD diffractometer using graphite-monochromated Mo Kα radiation (0.71073 Å). The data were reduced to F2o and corrected for absorption effects with SAINT [51] and SADABS [52,53], respectively. The structures were solved by direct methods and refined using the full-matrix least-squares method (SHELXL97) [54]. If not otherwise noted, all non-hydrogen atoms were refined with anisotropic displacement parameters, and all hydrogen atoms were located in calculated positions to correspond to standard bond lengths and angles. Crystallographic data (excluding structure factors) for the structures of compounds 3, 5, 7, 8, 9, and 10 reported in this paper have been deposited with the Cambridge Crystallographic Data Center as supplementary publication (Table S1) No. CCDC-1982093 (3), 1982094 (5), 1982096 (7), 1982098 (8), 1982097 (9), and 1982095 (10). Data can be obtained free of charge at: http://www.ccdc.cam.ac.uk/products/csd/request/. Figures of solid-state molecular structures were generated using Ortep-3 as implemented in WINGX [55] and rendered using POV-Ray 3.6 [56].
3.3. Synthesis
3.3.1. 1,1′-ansa-[2,2,4,4-Tetramethyl-1,1,5,5-tetrakis(trimethylsilyl)-3-oxa-1,2,4,5-tetrasila-1,5-diyl]ferrocene (5)
1,1′-Bis[tris(trimethylsilyl)silyl]ferrocene (588 mg, 0.87 mmol) and KOtBu (200 mg, 1.78 mmol) were dissolved in THF (5 mL) and after 20 min a bright red color was observed. After 22 h, the solution was added dropwise within 2 min to a solution of 1,3-dichloro-1,1,3,3-tetramethyldisiloxane (182 mg, 0.90 mmol) in toluene (5 mL), and the reaction mixture was stirred for 6 h. Volatiles were removed under reduced pressure, pentane was added (3 × 3 mL), centrifuged, and filtrated. Orange crystals of compound 5 (560 mg, 97%) were obtained from pentane at −62 °C. NMR (δ in ppm): 1H: 4.23 (m, 4H), 4.11 (m, 4H), 0.51 (s, 12H), 0.26 (s, 36H).13C: 75.9, 70.7, 66.7, 7.2, 1.3. 29Si: 7.0 (s, SiMe2), −13.9 (SiMe3), −84.0 (Siq).
3.3.2. 1,1’-ansa-[1,5-Dipotassio-2,2,4,4-tetramethyl-1,5-bis(trimethylsilyl)-3-oxa-1,2,4,5-tetrasila-1,5-diyl]ferrocene (6)
A solution of compound 5 (100 mg, 0.15 mmol) in benzene (1 mL) was added dropwise to a solution of KOtBu (35 mg, 0.31 mmol) and 18-cr-6 (86 mg, 0.33 mmol) in benzene (1 mL). Immediately, the color changed from orange to bright red. After 1 min, a luminous bright orange precipitate formed. After 5 h at rt, the supernatant was decanted and the precipitate was dissolved in THF and subjected to NMR-analysis, which showed only a single signal at −11.1 ppm along with trace amounts of silyl ether in the 29Si NMR spectrum, hence not giving clear evidence for the formation of compound 6. Therefore, the solution was treated with chlorodimethylphenylsilane (89 mg, 0.52 mmol) in THF (1 mL). After 2.5 h at rt, the reaction mixture was subjected to NMR-analysis showing a diastereotopic mixture of compound 5a. NMR (δ in ppm): 29Si: 7.5/7.4 (s, SiMe2), −13.4/−13.5 (SiMe3), −17.7/−18.1 (SiPhMe2), −83.2/−83.4 (Siq).
3.3.3. 2-Stanna-5-oxa-1,1,3,3-tetrakis(trimethylsilyl)tetramethylcyclohexasilan-2-ylidene·PMe3 (7)
A mixture of SnCl2 (30 mg, 0.16 mmol) and PMe3 (18 mg, 0.24 mmol) was dissolved in THF (2 mL), stirred for 10 min at rt, and cooled to −37 °C. To this, a solution of compound 2 (0.16 mmol) in THF (1 mL) was added dropwise resulting in an red–brown solution. After stirring for 1 h, the solvent was removed in a vacuum and the residue was extracted with pentane (3 × 2 mL). Filtration over celite was followed by concentration of the volume. Crystallization at −37 °C provided yellow crystalline blocks of compound 7 (47 mg, 44%). Mp.: 154–156 °C. NMR (δ in ppm): 1H: 1.10 (d, 9H, 2JH-P = 8.6 Hz, PMe3), 0.57 (bs, 6H, OSiMe2), 0.51 (bs, 6H, OSiMe2), 0.45 (bs, 18H, SiMe3), 0.39 (bs, 18H, SiMe3). 13C: 18.2 (d, 1JC-P = 16.4 Hz, PMe3), 8.7 (OSiMe2), 8.4 (OSiMe2), 5.0 (SiMe3), 3.5 (SiMe3). 29Si: 12.9 (d, 3JSi-P = 9 Hz, OSiMe2), −6.1 (d, 3JSi-P = 14 Hz, SiMe3), −9.9 (bs, SiMe3), −142.4 (d, 3JSi-P = 16 Hz, d, 1JSi117-119Sn = 569/575 Hz, SiSn). 31P: −62.5 (1JP-Sn117/119Sn = 1991/2084 Hz, SnPMe3). 119Sn: −167.9 (1JP-119Sn = 2084 Hz, SnPMe3).
3.3.4. 1,7-Distanna-2,2,6,6,8,8,12,12-octakis(trimethylsilyl)-4,10-dioxabicyclo[5.5.0]-dodecasil-1,7-ene (8)
Compound 8 was obtained in an analogous way as described below for compound 9 using SnCl2 (60 mg, 0.32 mmol), tBuOK (74 mg, 0.78 mmol), and 2 (200mg, 0.16 mmol). Green crystals (which turn to orange/red under polarized light) of compound 8 (47 mg, 64%) were formed in a toluene solution at −37 °C. Mp.: 167–170 °C. NMR (δ in ppm): 1H: 0.59 (s, 24H, OSiMe2), 0.44 (s, 72H, SiMe3). 13C: 8.3 (OSiMe2), 4.1 (SiMe3).29Si: 13.75 (OSiMe2), −9.0 (SiMe3), −84.9 (SiSn). 119Sn: 636.9.
3.3.5. 2-Germa-5-oxa-1,1,3,3-tetrakis(trimethylsilyl)tetramethylcyclohexasilan-2-ylidene·PMe3 (9)
A mixture of GeBr2·dioxane (102 mg, 0.32 mmol) and PMe3 (36 mg, 0.48 mmol) was dissolved in THF (3 mL), stirred for 10 min at rt, and cooled to −37 °C. To this, a solution of compound 2 (0.32 mmol) in THF (2 mL) was added dropwise resulting in an orange–brown solution. After stirring for 1 h, the solvent was removed in a vacuum and the residue was extracted with pentane (3 × 2 mL). Filtration over celite was followed by concentration of the volume. Crystallization at −37 °C provided compound 9 (75 mg, 37%) as yellow–orange crystalline blocks. Mp.: 134–136 °C. NMR (δ in ppm): 1H: 1.13 (d, 9H, 2JH-P = 9.9 Hz, PMe3), 0.58 (s, 12H, SiMe2), 0.44 (s, 18H, SiMe3), 0.39 (s, 18H, SiMe3). 13C: 18.6 (1JC-P = 23 Hz, PMe3), 8.1 (OSiMe2), 4.6 (SiMe3), 3.1 (SiMe3). 29Si: 12.5 (d, 3JSi-P = 16 Hz, OSiMe2), −9.2 (bs, SiMe3), −10.7 (bs, SiMe3), −134.3 (d, 2JSi-P = 16 Hz, SiGe). 31P: −21.2.
3.3.6. C-F Insertion Product from the Reaction of 9 with B(C6F5)3 (10)
To a solution of tris(pentafluorophenyl)borane (64 mg, 0.13 mmol) in pentane (4 mL), germylene adduct 9 in pentane (3 mL) was added dropwise. Immediately, the formation of a white precipitate in a clear and colorless solution was observed. After stirring for 30 min, the solvent was removed in a vacuum to yield a mixture of an oily residue and colorless crystalline blocks. The oily residue was removed by washing with cold pentane to leave compound 10 (87 mg, 64%). NMR (δ in ppm): 1H: 0.62 (d, 9H, 2JP-H = 11.0 Hz, PMe3), 0.53 (s, 6H, SiMe2), 0.47 (s, 6H, SiMe2), 0.45 (s, 18H, SiMe3), 0.25 (s, 18H, SiMe3). 13C: 10.7 (bd, 1JC-P = 40 Hz, PMe3), 7.7 (2 signals, SiMe2), 3.2 (SiMe3), 2.5 (SiMe3), signals for the fluorinated borate could not be detected. 19F: −127.5 (td, J = 16 Hz, 28 Hz, C6F4), −129.9 (pd, J = 16 Hz, C6F4), −130.0 (d, J = 26 Hz, o-C6F5), −155.8 (t, J = 23 Hz, p-C6F5), −163.3 (dt, J = 9 Hz, 24 Hz, m-C6F5), −209.2 (t, J = 17 Hz, Ge-F). 29Si: 11.4 (d, 3JSi-F = 2.9 Hz, OSi), −6.5 (d, 3JSi-F = 3.3 Hz, SiMe3), −8.6 (d, 3JSi-F = 5.7 Hz, SiMe3), −109.9 (d, 2JSi-F = 8.7 Hz, GeSi). 31P: −8.6 (bs, PMe3).
4. Conclusions
A number of compounds containing a 1,5-oligosiloxanylene unit were prepared. Repetitive formation of 1,5-oligosiloxanylene diides allowed the synthesis of a 3,7,10-trioxa-octasilabicyclo[3.3.3]undecane, with trimethylsilyl groups at the bridgehead positions. Reaction of 1,3-dichlorotetramethyldisiloxane with 1,1′-bis[bis(trimethylsilyl)potassiosilyl]ferrocene gave the respective ferrocenophane with a disiloxane containg bridge. The compound can be further derivatized by reaction with two equivalents of KOtBu into the 1,5-oligosilanyl diide. NMR spectroscopic evidence for the presence of the latter was difficult. This was most likely due to fast configurational inversion of the silanide atoms; the dynamic of these processes leads to very broad resonances. Nevertheless, unequivocal proof for the dianion formation was obtained by derivatization with chlorosilanes.
Reacting 1,5-oligosiloxanylene diide 2 with SnCl2 or GeCl2·dioxane in the presence of PMe3 gave cyclic disilylated tetrylene PMe3 adducts. Spectroscopic and structural characteristics of these compounds are very similar to that of related compounds with 1,4-tetrasilanylene backbones.
Release of the base-free stannylene led to a dimerization process, which gave a distannene with two annulated seven membered rings. 119Sn NMR spectroscopic analysis showed the compound to be a distannene with a slight stannylene character also in solution featuring a resonance at 636.9 ppm. Abstraction of the PMe3 from the cyclic disilylated germylene PMe3 adduct with B(C6F5)3 was carried out to obtain a base-free disilylated germylene. Thus far, disilylated germylenes either dimerized to digermanes or underwent 1,2-silyl shift reactions to silagermenes, which either can dimerize in a [2 + 2] cycloaddition reaction or react further to a silylgermylsilylene, which inserts into a remote Si-Si bond. In the current case, however, oxidative addition of the germylene into a para-C-F bond of Me3P·B(C6F5)3 was observed. We assume that the novel reaction pattern is a consequence of a diminished singlet-triplet gap of the germylene, which is caused by the geometrical constraint of the elongated backbone resulting in a larger Si-Ge-Si angle and thus an increased triplet character.
Supplementary Materials
The supplementary materials are available online. Figure S1–S22, 1H, 13C, 19F, 29Si, 31P, and 119Sn NMR spectra of compounds 5, 5a, 7, 8, 9, and 10, Table S1, Tabulated crystallographic data and ORTEP plots for compounds 3, 5, 7, 8, 9, and 10.
Author Contributions
The manuscript was written through contributions of all authors. R.Z., A.P., J.B., and C.M. conceived the experiments. R.Z., A.P. and to a minor extent J.B. did all the synthetic and characterization work. C.M. and J.B. provided supervision, financial support via projects and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Austrian Science Fund (Fonds zur Förderung der wissenschaftlichen Forschung) (FWF) via projects P-30955 (J.B.) and P-26417 (C.M.). Open Access Funding by the Graz University of Technology is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Liu, Y.; Onodera, K.; Takeda, N.; Ouali, A.; Unno, M. Synthesis and Characterization of Functionalizable Silsesquioxanes with Ladder-type Structures. Organometallics 2019, 38, 4373–4376. [Google Scholar] [CrossRef]
- Dankert, F.; Weigend, F.; von Hänisch, C. Not Non-Coordinating at All: Coordination Compounds of the Cyclodimethylsiloxanes Dn (D = Me2SiO; n = 6, 7) and Group 2 Metal Cations. Inorg. Chem. 2019, 58, 15417–15422. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Shiba, H.; Yoshikawa, M.; Wada, H.; Shimojima, A.; Kuroda, K. Synthesis of Polycyclic and Cage Siloxanes by Hydrolysis and Intramolecular Condensation of Alkoxysilylated Cyclosiloxanes. Chem. Eur. J. 2019, 25, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Oba, Y.; Nakajima, Y.; Shimada, S.; Sato, K. One-Pot Iterative Synthesis of Sequence-Controlled Oligosiloxanes. Angew. Chem. 2018, 130, 4727–4731. [Google Scholar] [CrossRef]
- Brook, M.A. New Control Over Silicone Synthesis Using SiH Chemistry: The Piers Rubinsztajn Reaction. Chem. Eur. J. 2018, 24, 8458–8469. [Google Scholar] [CrossRef]
- Yokouchi, Y.; Ishida, S.; Onodera, T.; Oikawa, H.; Iwamoto, T. Facile synthesis and bridgehead-functionalization of bicyclo[3.3.3]pentasiloxanes. Chem. Commun. 2017, 54, 268–270. [Google Scholar] [CrossRef]
- Thompson, D.B.; Brook, M.A. Rapid Assembly of Complex 3D Siloxane Architectures. J. Am. Chem. Soc. 2008, 130, 32–33. [Google Scholar] [CrossRef]
- Cui, C.; Qin, G. Controlled synthesis cyclosiloxanes by NHC-catalyzed hydrolytic oxidation of dihydrosilanes. Dalton Trans. 2017, 46, 8746–8750. [Google Scholar]
- Unno, M.; Tanaka, R.; Tanaka, S.; Takeuchi, T.; Kyushin, S.; Matsumoto, H. Oligocyclic Ladder Polysiloxanes: Alternative Synthesis by Oxidation. Organometallics 2005, 24, 765–768. [Google Scholar] [CrossRef]
- Marschner, C. Preparation and Reactions of Polysilanyl Anions and Dianions. Organometallics 2006, 25, 2110–2125. [Google Scholar] [CrossRef]
- Zitz, R.; Hlina, J.; Aghazadeh Meshgi, M.; Krenn, H.; Marschner, C.; Szilvási, T.; Baumgartner, J. Using Functionalized Silyl Ligands to Suppress Solvent Coordination to Silyl Lanthanide(II) Complexes. Inorg. Chem. 2017, 56, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Zitz, R.; Hlina, J.; Arp, H.; Kinschel, D.; Marschner, C.; Baumgartner, J. Group 4 Metal and Lanthanide Complexes in the Oxidation State +3 with Tris(trimethylsilyl)silyl Ligands. Inorg. Chem. 2019, 58, 7107–7117. [Google Scholar] [CrossRef]
- Zitz, R.; Arp, H.; Hlina, J.; Walewska, M.; Marschner, C.; Szilvási, T.; Blom, B.; Baumgartner, J. Open-Shell Lanthanide(II+) or -(III+) Complexes Bearing σ-Silyl and Silylene Ligands: Synthesis, Structure, and Bonding Analysis. Inorg. Chem. 2015, 54, 3306–3315. [Google Scholar] [CrossRef] [PubMed]
- Zitz, R.; Hlina, J.; Gatterer, K.; Marschner, C.; Szilvási, T.; Baumgartner, J. Neutral “Cp-Free” Silyl-Lanthanide(II) Complexes: Synthesis, Structure, and Bonding Analysis. Inorg. Chem. 2015, 54, 7065–7072. [Google Scholar] [CrossRef] [PubMed]
- Zitz, R.; Baumgartner, J.; Marschner, C. Chemistry of a 1,5-Oligosilanylene Dianion Containing a Disiloxane Unit. Organometallics 2019, 38, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Al-Juaid, S.S.; Derouiche, Y.; Hitchcock, P.B.; Lickiss, P.D. Preparation and X-ray crystal structure of an unusual bicyclic trisilane-tris(disoloxane). J. Organomet. Chem. 1988, 341, 241–245. [Google Scholar] [CrossRef]
- Jäger-Fiedler, U.; Köckerling, M.; Reinke, H.; Krempner, C. Discrete oxygen containing oligosilane dendrimers-modelling oxygen defects in silicon nanomaterials. Chem. Commun. 2010, 46, 4535–4537. [Google Scholar] [CrossRef]
- Krempner, C.; Jager-Fiedler, U.; Kockerling, M.; Reinke, H. Synthesis and Structures of Titanium and Zirconium Trisiloxides. Organometallics 2009, 28, 382–385. [Google Scholar] [CrossRef]
- Wagner, H.; Baumgartner, J.; Marschner, C. 1,1‘-Oligosilylferrocene Compounds. Organometallics 2007, 26, 1762–1770. [Google Scholar] [CrossRef]
- Fischer, R.; Konopa, T.; Ully, S.; Baumgartner, J.; Marschner, C. Route Si6 revisited. J. Organomet. Chem. 2003, 685, 79–92. [Google Scholar] [CrossRef]
- Mechtler, C.; Marschner, C. Polysilyldianions—Synthesis and reactivity. Tetrahedron Lett. 1999, 40, 7777–7778. [Google Scholar] [CrossRef]
- Kayser, C.; Kickelbick, G.; Marschner, C. Simple Synthesis of Oligosilyl-α,ω-dipotassium Compounds. Angew. Chem. Int. Ed. 2002, 41, 989–992. [Google Scholar] [CrossRef]
- Fischer, R.; Frank, D.; Gaderbauer, W.; Kayser, C.; Mechtler, C.; Baumgartner, J.; Marschner, C. α,ω-Oligosilyl Dianions and Their Application in the Synthesis of Homo- and Heterocyclosilanes. Organometallics 2003, 22, 3723–3731. [Google Scholar] [CrossRef]
- Fischer, R.; Konopa, T.; Baumgartner, J.; Marschner, C. Small Cyclosilanes: Syntheses and Reactions toward Mono- and Dianions. Organometallics 2004, 23, 1899–1907. [Google Scholar] [CrossRef]
- Mechtler, C.; Zirngast, M.; Gaderbauer, W.; Wallner, A.; Baumgartner, J.; Marschner, C. Synthesis of oligosilyldi- and trianions. J. Organomet. Chem. 2006, 691, 150–158. [Google Scholar] [CrossRef]
- Katir, N.; Matioszek, D.; Ladeira, S.; Escudié, J.; Castel, A. Stable N-Heterocyclic Carbene Complexes of Hypermetallyl Germanium(II) and Tin(II) Compounds. Angew. Chem. Int. Ed. 2011, 50, 5352–5355. [Google Scholar] [CrossRef]
- Klinkhammer, K. Dihypersilylstannylene and dihypersilylplumbylene—two Lewis-amphoteric carbene homologues. Polyhedron 2002, 21, 587–598. [Google Scholar] [CrossRef]
- Arp, H.; Marschner, C.; Baumgartner, J.; Zark, P.; Müller, T. Coordination Chemistry of Disilylated Stannylenes with Group 10 d10 Transition Metals: Silastannene vs Stannylene Complexation. J. Am. Chem. Soc. 2013, 135, 7949–7959. [Google Scholar] [CrossRef]
- Walewska, M.; Baumgartner, J.; Marschner, C. Synthesis of vinyl germylenes. Chem. Commun. 2015, 51, 276–278. [Google Scholar] [CrossRef]
- Arp, H.; Baumgartner, J.; Marschner, C.; Müller, T. A Cyclic Disilylated Stannylene: Synthesis, Dimerization, and Adduct Formation. J. Am. Chem. Soc. 2011, 133, 5632–5635. [Google Scholar] [CrossRef]
- Hlina, J.; Baumgartner, J.; Marschner, C.; Zark, P.; Müller, T. Coordination Chemistry of Disilylated Germylenes with Group 4 Metallocenes. Organometallics 2013, 32, 3300–3308. [Google Scholar] [CrossRef] [PubMed]
- Hlina, J.; Baumgartner, J.; Marschner, C.; Albers, L.; Müller, T.; Jouikov, V. Formation and Properties of a Bicyclic Silylated Digermene. Chem. Eur. J. 2014, 20, 9357–9366. [Google Scholar] [CrossRef] [PubMed]
- Walewska, M.; Hlina, J.; Gaderbauer, W.; Wagner, H.; Baumgartner, J.; Marschner, C. NHC Adducts of Disilylated Germylenes and Stannylenes and Their Coordination Chemistry with Group 11 Metals. Z. Anorg. Allg. Chem. 2016, 642, 1304–1313. [Google Scholar] [CrossRef]
- Klinkhammer, K.W.; Schwarz, W. Bis(hypersilyl)tin and Bis(hypersilyl)lead, Two Electron-Rich Carbene Homologs. Angew. Chem. Int. Ed. Engl. 1995, 34, 1334–1336. [Google Scholar] [CrossRef]
- Fukawa, T.; Lee, V.Ya.; Nakamoto, M.; Sekiguchi, A. Tetrakis(di-tert-butylmethylsilyl)distannene and Its Anion Radical. J. Am. Chem. Soc. 2004, 126, 11758–11759. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh Meshgi, M.; Zitz, R.; Walewska, M.; Baumgartner, J.; Marschner, C. Tuning the Si–N Interaction in Metalated Oligosilanylsilatranes. Organometallics 2017, 36, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Hlina, J.; Baumgartner, J.; Marschner, C.; Albers, L.; Müller, T. Cyclic Disilylated and Digermylated Germylenes. Organometallics 2013, 32, 3404–3410. [Google Scholar] [CrossRef]
- Walewska, M.; Hlina, J.; Baumgartner, J.; Müller, T.; Marschner, C. Basic Reactivity Pattern of a Cyclic Disilylated Germylene. Organometallics 2016, 35, 2728–2737. [Google Scholar] [CrossRef]
- Gaspar, P.P.; Xiao, M.; Pae, D.H.; Berger, D.J.; Haile, T.; Chen, T.; Lei, D.; Winchester, W.R.; Jiang, P. The quest for triplet ground state silylenes. J. Organomet. Chem. 2002, 646, 68–79. [Google Scholar] [CrossRef]
- Lawson, J.R.; Melen, R.L. Tris(pentafluorophenyl)borane and Beyond: Modern Advances in Borylation Chemistry. Inorg. Chem. 2017, 56, 8627–8643. [Google Scholar] [CrossRef]
- Swamy, V.S.V.S.N.; Parvin, N.; Raj, K.V.; Vanka, K.; Sen, S.S. C(sp3)–F, C(sp2)–F and C(sp3)–H bond activation at silicon(II) centers. Chem. Commun. 2017, 53, 9850–9853. [Google Scholar] [CrossRef] [PubMed]
- Jana, A.; Samuel, P.P.; Tavčar, G.; Roesky, H.W.; Schulzke, C. Selective Aromatic C-F and C-H Bond Activation with Silylenes of Different Coordinate Silicon. J. Am. Chem. Soc. 2010, 132, 10164–10170. [Google Scholar] [CrossRef] [PubMed]
- Samuel, P.P.; Singh, A.P.; Sarish, S.P.; Matussek, J.; Objartel, I.; Roesky, H.W.; Stalke, D. Oxidative Addition Versus Substitution Reactions of Group 14 Dialkylamino Metalylenes with Pentafluoropyridine. Inorg. Chem. 2013, 52, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; De, S.; Koley, D. DFT Study on C–F Bond Activation by Group 14 Dialkylamino Metalylenes: A Competition between Oxidative Additions versus Substitution Reactions. Inorg. Chem. 2017, 56, 10633–10643. [Google Scholar] [CrossRef] [PubMed]
- Chase, P.A.; Parvez, M.; Piers, W.E. Trimethylphosphine-tris(pentafluorophenyl)borane. Acta Cryst. E 2006, 62, o5181–o5183. [Google Scholar] [CrossRef]
- Ullrich, M.; Lough, A.J.; Stephan, D.W. Reversible, Metal-Free, Heterolytic Activation of H2 at Room Temperature. J. Am. Chem. Soc. 2009, 131, 52–53. [Google Scholar] [CrossRef]
- Hassler, K.; Dzambaski, A.; Baumgartner, J. Dihaloheptasilanes X2Si[SiMe(SiMe3)2]2 as potential precursors for silylenes, disilenes and cyclotrisilanes. Silicon Chem. 2007, 3, 271–288. [Google Scholar] [CrossRef]
- Pangborn, A.B.; Giardello, M.A.; Grubbs, R.H.; Rosen, R.K.; Timmers, F.J. Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar] [CrossRef]
- Morris, G.A.; Freeman, R. Enhancement of Nuclear Magnetic Resonance Signals by Polarization Transfer. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar] [CrossRef]
- Helmer, B.J.; West, R. Enhancement of 29Si NMR Signals by Proton Polarization Transfer. Organometallics 1982, 1, 877–879. [Google Scholar] [CrossRef]
- Bruker-AXS. SAINTPLUS: Software Reference Manual; Version 6.45; Bruker-AXS: Madison, WI, USA, 2003. [Google Scholar]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Cryst. A 1995, 51, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SADABS; Version 2.10; Bruker AXS Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Available online: http://www.povray.org/download/ (accessed on 9 July 2008).
Sample Availability: Samples of the compounds are not available from the authors. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).