Evodiamine Mitigates Cellular Growth and Promotes Apoptosis by Targeting the c-Met Pathway in Prostate Cancer Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
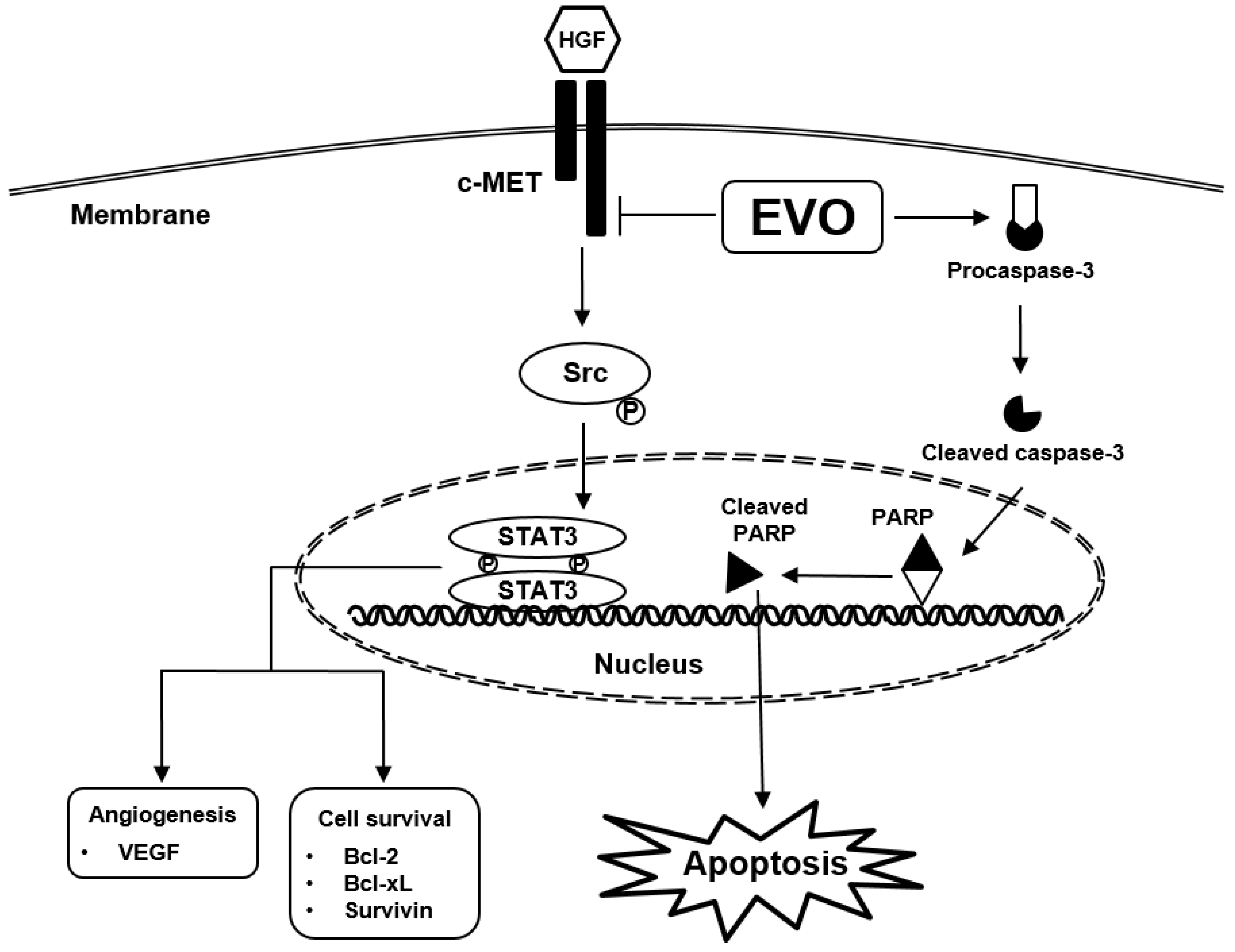
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines and Culture Conditions
2.3. MTT Assay
2.4. Real-Time Cell Proliferation Analysis (RTCA)
2.5. Cell Morphology
2.6. Western blot Analysis
2.7. Immunocytochemistry for STAT3 Localization
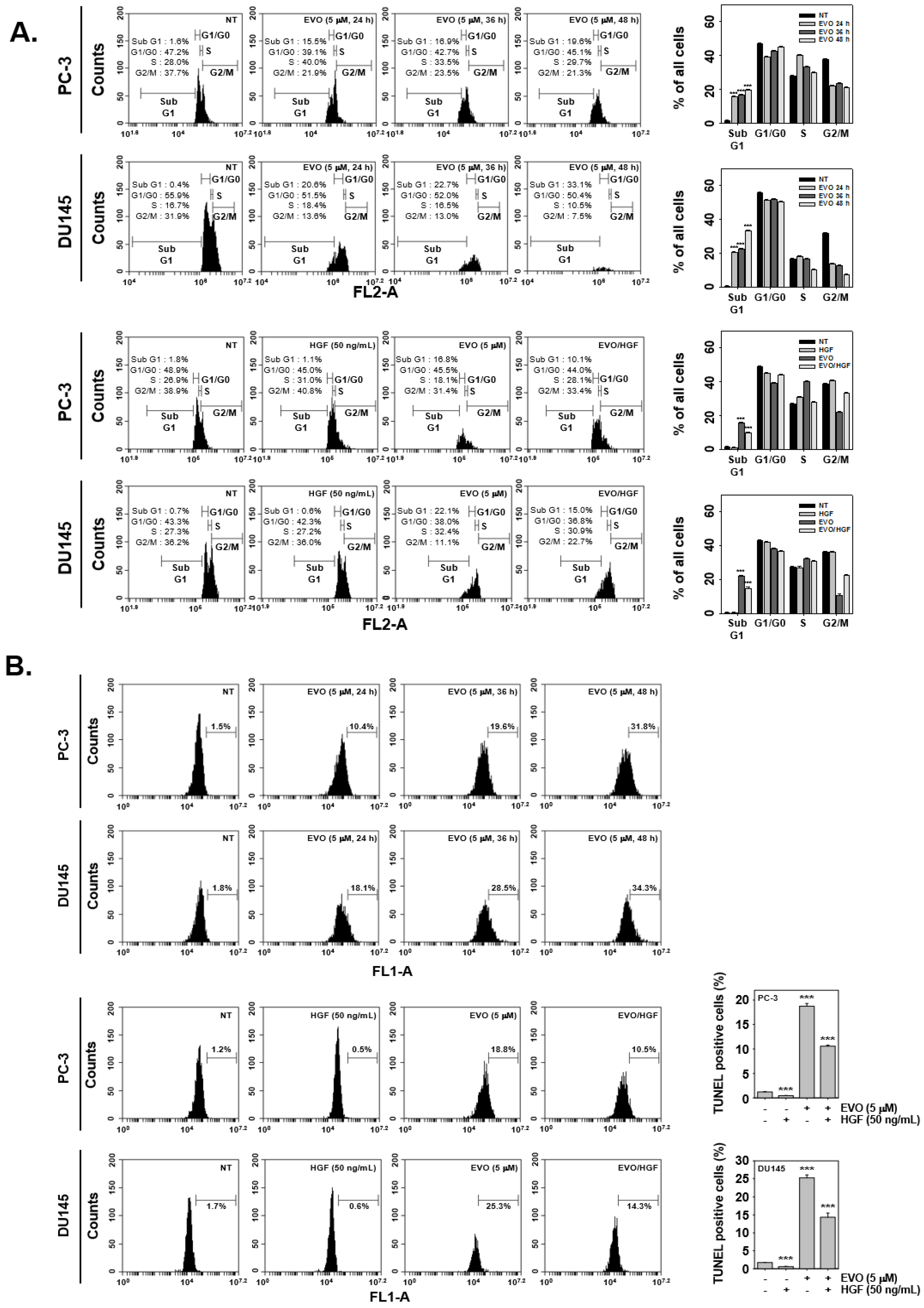
2.8. Cell Cycle Analysis
2.9. TUNEL Assay
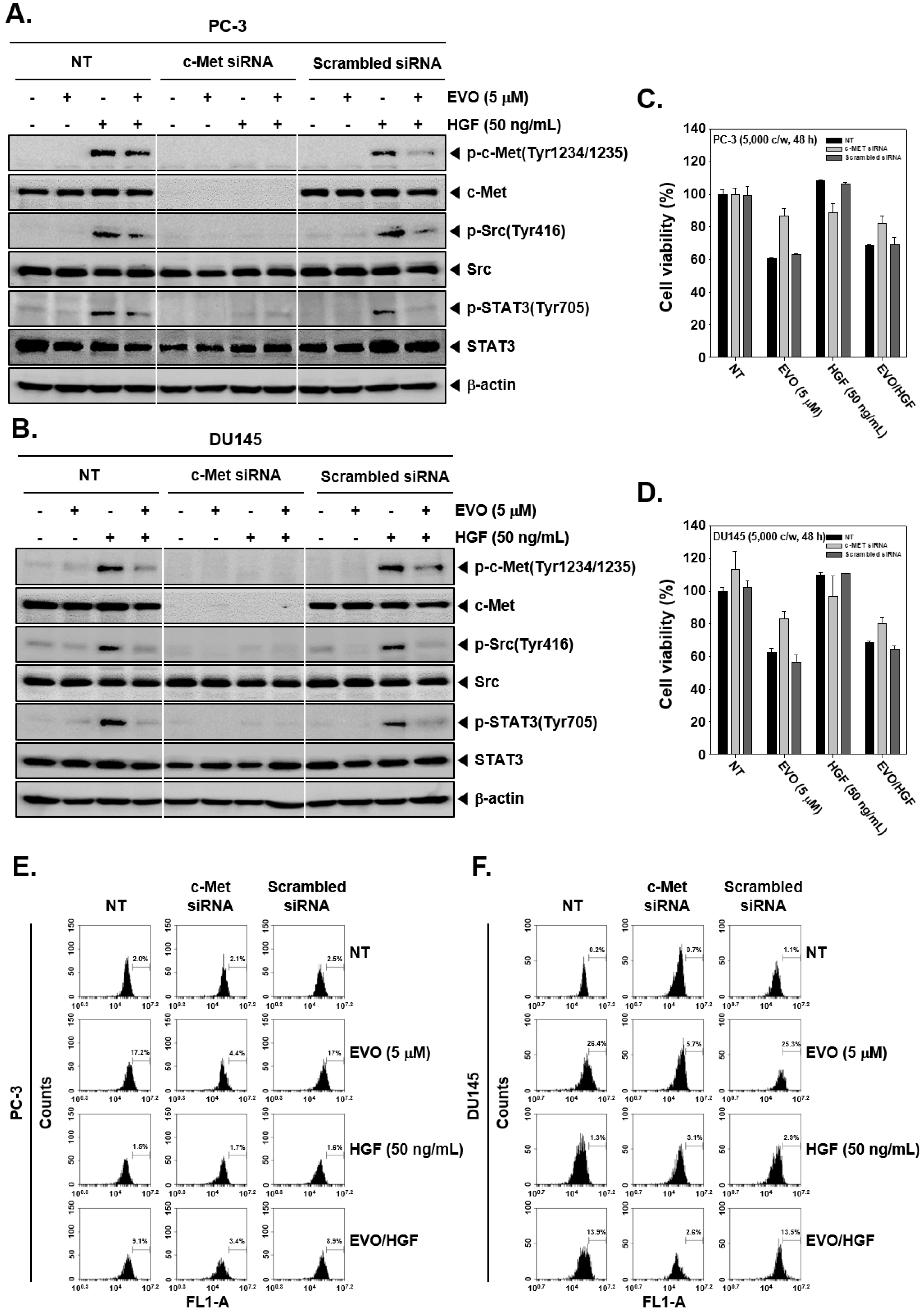
2.10. Transfection of siRNA
2.11. Statistical Analysis
3. Results
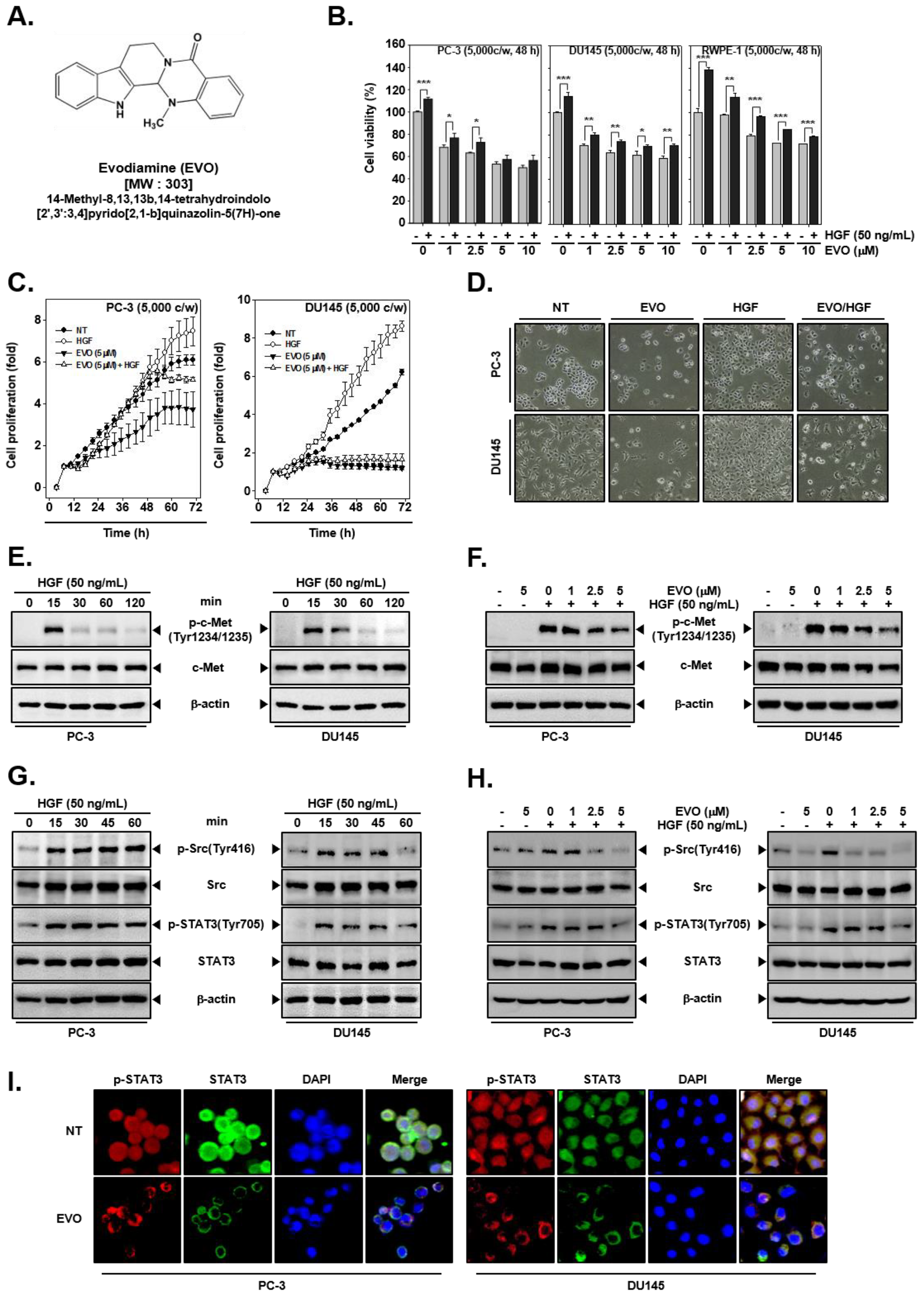
3.1. EVO Suppressed Cellular Growth and Cell Proliferation.
3.2. EVO Attenauted c-Met/Src/STAT3 Phosphorylation in PC-3 and DU145 Cells.
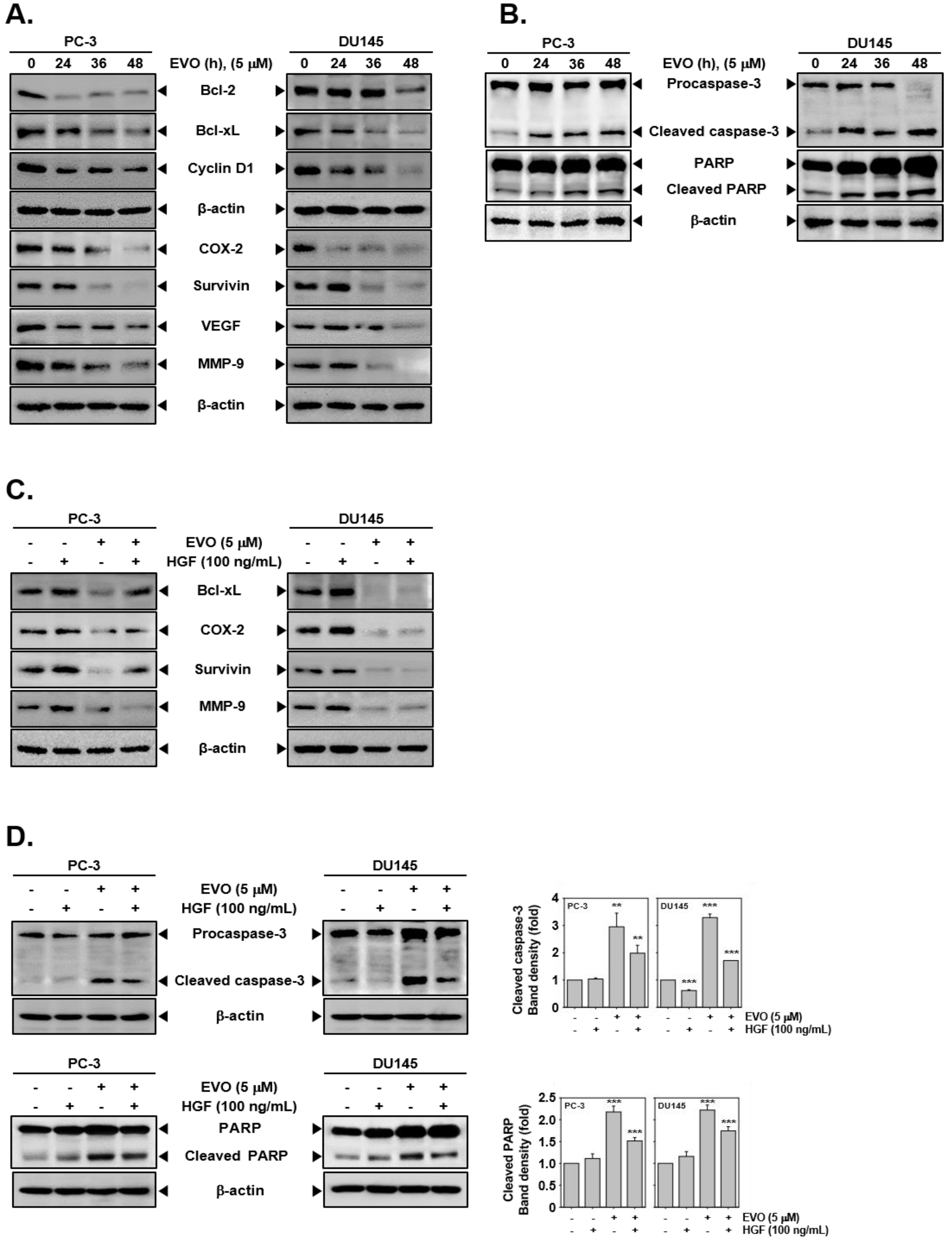
3.3. EVO Caused DNA Damage and Induced Apoptotic Cell Death.
3.4. EVO Downregulated the Expression of Carcinogenic Proteins.
3.5. c-Met Knockdown Blocked Src/STAT3 Signaling in Prostate Cancer Cells.
3.6. c-MET Knockdown Increased Cell Viability and Decreased EVO-Induced Cell Death.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HGF: | hepatocyte growth factor |
| PARP: | Poly(ADP-ribose) polymerase |
| Bcl-2: | B-cell lymphoma 2 |
| VEGF: | Vascular endothelial growth factor |
| COX-2: | Cyclooxygenase 2 |
| MMP-9: | Matrix metallopeptidases 9 |
| GAPDH: | Glyceraldehyde 3-phosphate dehydrogenase |
| TUNEL: | TdT-mediated dUTP nick-end labeling |
| SDS: | Sodium dodecyl sulfate |
| PBS: | Phosphate buffered saline |
| TBS: | Tris buffered saline |
| ECL: | Enhanced chemiluminescence |
| PI: | Propidium iodide |
| FBS: | Fetal bovine serum |
| MTT: | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| siRNA: | Small interfering RNA |
References
- Caires-Dos-Santos, L.; da Silva, S.V.; Smuczek, B.; de Siqueira, A.S.; Cruz, K.S.P.; Barbuto, J.A.M.; Augusto, T.M.; Freitas, V.M.; Carvalho, H.F.; Jaeger, R.G. Laminin-derived peptide C16 regulates Tks expression and reactive oxygen species generation in human prostate cancer cells. J. Cell. Physiol. 2020, 235, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.P.; Gurung, S.K.; Inam, A.; Nigam, L.; Bist, A.; Mohapatra, D.; Senapati, S.; Subbarao, N.; Azam, A.; Mondal, N. CID-6033590 inhibits p38MAPK pathway and induces S-phase cell cycle arrest and apoptosis in DU145 and PC-3 cells. Toxicol. Vitro 2019, 60, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Sikka, S.; Chen, L.; Sethi, G.; Kumar, A.P. Targeting PPARgamma Signaling Cascade for the Prevention and Treatment of Prostate Cancer. PPAR Res. 2012, 2012, 968040. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sikka, S.; Siveen, K.S.; Lee, J.H.; Um, J.Y.; Kumar, A.P.; Chinnathambi, A.; Alharbi, S.A.; Basappa, V.K.; Rangappa, K.S.; et al. Cardamonin represses proliferation, invasion, and causes apoptosis through the modulation of signal transducer and activator of transcription 3 pathway in prostate cancer. Apoptosis 2017, 22, 158–168. [Google Scholar] [CrossRef]
- Zhang, J.; Ahn, K.S.; Kim, C.; Shanmugam, M.K.; Siveen, K.S.; Arfuso, F.; Samym, R.P.; Deivasigamanim, A.; Lim, L.H.; Wang, L. Nimbolide-Induced Oxidative Stress Abrogates STAT3 Signaling Cascade and Inhibits Tumor Growth in Transgenic Adenocarcinoma of Mouse Prostate Model. Antioxid. Redox Signal. 2016, 24, 575–589. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, C.; Baek, S.H.; Ko, J.H.; Lee, S.G.; Yang, W.M.; Um, J.Y.; Sethi, G.; Ahn, K.S. Capsazepine inhibits JAK/STAT3 signaling, tumor growth, and cell survival in prostate cancer. Oncotarget 2017, 8, 17700–17711. [Google Scholar] [CrossRef]
- Jung, K.W.; Won, Y.J.; Kong, H.J.; Lee, E.S.; Community of Population-Based Regional Cancer R. Cancer Statistics in Korea: Incidence, Mortality, Survival, and Prevalence in 2015. Cancer Res. Treat. 2018, 50, 303–316. [Google Scholar] [CrossRef]
- Lim, S.L.; Park, S.Y.; Kang, S.; Park, D.; Kim, S.H.; Um, J.Y.; Jang, H.J.; Lee, J.H.; Jeong, C.H.; Jang, J.H.; et al. Morusin induces cell death through inactivating STAT3 signaling in prostate cancer cells. Am. J. Cancer Res. 2015, 5, 289–299. [Google Scholar]
- Jung, K.W.; Won, Y.J.; Kong, H.J.; Lee, E.S. Prediction of Cancer Incidence and Mortality in Korea, 2019. Cancer Res. Treat. 2019, 51, 431–437. [Google Scholar] [CrossRef]
- Park, S.K.; Sakoda, L.C.; Kang, D.; Chokkalingam, A.P.; Lee, E.; Shin, H.R.; Ahn, Y.O.; Shin, M.H.; Lee, C.W.; Lee, D.H.; et al. Rising prostate cancer rates in South Korea. Prostate 2006, 66, 1285–1291. [Google Scholar] [CrossRef]
- Wu, J.C.; Wang, C.T.; Hung, H.C.; Wu, W.J.; Wu, D.C.; Chang, M.C.; Sung, P.J.; Chou, Y.W.; Wen, Z.H.; Tai, M.H. Heteronemin Is a Novel c-Met/STAT3 Inhibitor Against Advanced Prostate Cancer Cells. Prostate 2016, 76, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, M.D.; Cotter, S.E.; Gargollo, P.C.; Zietman, A.L.; Dahl, D.M.; Smith, M.R. Management of complications of prostate cancer treatment. CA Cancer J. Clin. 2008, 58, 196–213. [Google Scholar] [CrossRef] [PubMed]
- Maluf, F.C.; Smaletz, O.; Herchenhorn, D. Castration-resistant prostate cancer: Systemic therapy in 2012. Clinics (Sao Paulo) 2012, 67, 389–394. [Google Scholar] [CrossRef]
- Kan, S.F.; Yu, C.H.; Pu, H.F.; Hsu, J.M.; Chen, M.J.; Wang, P.S. Anti-proliferative effects of evodiamine on human prostate cancer cell lines DU145 and PC3. J. Cell. Biochem. 2007, 101, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Mendes, D.E.; Berkman, C.E. From AR to c-Met: Androgen deprivation leads to a signaling pathway switch in prostate cancer cells. Int. J. Oncol. 2013, 43, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Siemann, D.W. BMS-777607, a small-molecule met kinase inhibitor, suppresses hepatocyte growth factor-stimulated prostate cancer metastatic phenotype in vitro. Mol. Cancer Ther. 2010, 9, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Naughton, M.; Picus, J.; Zhu, X.; Catalona, W.J.; Vollmer, R.T.; Humphrey, P.A. Scatter factor-hepatocyte growth factor elevation in the serum of patients with prostate cancer. J. Urol. 2001, 165, 1325–1328. [Google Scholar] [CrossRef]
- Knudsen, B.S.; Edlund, M. Prostate cancer and the met hepatocyte growth factor receptor. Adv. Cancer Res. 2004, 91, 31–67. [Google Scholar]
- Humphrey, P.A.; Halabi, S.; Picus, J.; Sanford, B.; Vogelzang, N.J.; Small, E.J.; Kantoff, P.W. Prognostic significance of plasma scatter factor/hepatocyte growth factor levels in patients with metastatic hormone- refractory prostate cancer: Results from cancer and leukemia group B 150005/9480. Clin. Genitourin. Cancer 2006, 4, 269–274. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Warrier, S.; Kumar, A.P.; Sethi, G.; Arfuso, F. Potential Role of Natural Compounds as Anti-Angiogenic Agents in Cancer. Curr. Vasc. Pharmacol. 2017, 15, 503–519. [Google Scholar] [CrossRef]
- Hsieh, Y.S.; Yang, S.F.; Sethi, G.; Hu, D.N. Natural bioactives in cancer treatment and prevention. Biomed. Res. Int. 2015, 2015, 182835. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.F.; Weng, C.J.; Sethi, G.; Hu, D.N. Natural bioactives and phytochemicals serve in cancer treatment and prevention. Evid. Based Complement Altern. Med. 2013, 2013, 698190. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Targeting TNF-related apoptosis-inducing ligand (TRAIL) receptor by natural products as a potential therapeutic approach for cancer therapy. Exp. Biol. Med. (Maywood) 2015, 240, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Karamanou, K.; Franchi, M.; Vynios, D.; Brezillon, S. Epithelial-to-mesenchymal transition and invadopodia markers in breast cancer: Lumican a key regulator. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Stefanescu, R.; Stanciu, G.D.; Luca, A.; Caba, I.C.; Tamba, B.I.; Mihai, C.T. Contributions of Mass Spectrometry to the Identification of Low Molecular Weight Molecules Able to Reduce the Toxicity of Amyloid-beta Peptide to Cell Cultures and Transgenic Mouse Models of Alzheimer’s Disease. Molecules 2019, 24, 1167. [Google Scholar] [CrossRef]
- Prasannan, R.; Kalesh, K.A.; Shanmugam, M.K.; Nachiyappan, A.; Ramachandran, L.; Nguyen, A.H.; Kumar, A.P.; Lakshmanan, M.; Ahn, K.S.; Sethi, G. Key cell signaling pathways modulated by zerumbone: Role in the prevention and treatment of cancer. Biochem. Pharmacol. 2012, 84, 1268–1276. [Google Scholar] [CrossRef]
- Ahn, K.S.; Sethi, G.; Chaturvedi, M.M.; Aggarwal, B.B. Simvastatin, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, suppresses osteoclastogenesis induced by receptor activator of nuclear factor-kappaB ligand through modulation of NF-kappaB pathway. Int. J. Cancer 2008, 123, 1733–1740. [Google Scholar] [CrossRef]
- Manna, S.K.; Aggarwal, R.S.; Sethi, G.; Aggarwal, B.B.; Ramesh, G.T. Morin (3,5,7,2′,4′-Pentahydroxyflavone) abolishes nuclear factor-kappaB activation induced by various carcinogens and inflammatory stimuli, leading to suppression of nuclear factor-kappaB-regulated gene expression and up-regulation of apoptosis. Clin. Cancer Res. 2007, 13, 2290–2297. [Google Scholar] [CrossRef]
- Chua, A.W.; Hay, H.S.; Rajendran, P.; Shanmugam, M.K.; Li, F.; Bist, P.; Koay, E.S.; Lim, L.H.; Kumar, A.P.; Sethi, G. Butein downregulates chemokine receptor CXCR4 expression and function through suppression of NF-kappaB activation in breast and pancreatic tumor cells. Biochem. Pharmacol. 2010, 80, 1553–1562. [Google Scholar] [CrossRef]
- Nair, A.S.; Shishodia, S.; Ahn, K.S.; Kunnumakkara, A.B.; Sethi, G.; Aggarwal, B.B. Deguelin, an Akt inhibitor, suppresses IkappaBalpha kinase activation leading to suppression of NF-kappaB-regulated gene expression, potentiation of apoptosis, and inhibition of cellular invasion. J. Immunol. 2006, 177, 5612–5622. [Google Scholar] [CrossRef]
- Manu, K.A.; Shanmugam, M.K.; Ramachandran, L.; Li, F.; Fong, C.W.; Kumar, A.P.; Tan, P.; Sethi, G. First evidence that gamma-tocotrienol inhibits the growth of human gastric cancer and chemosensitizes it to capecitabine in a xenograft mouse model through the modulation of NF-kappaB pathway. Clin. Cancer Res. 2012, 18, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Nguyen, A.H.; Lee, J.H.; Li, F.; Singh, S.S.; Kumar, A.P.; Low, G.; Jha, S.; Tergaonkar, V.; Ahn, K.S.; et al. Negative regulation of signal transducer and activator of transcription-3 signalling cascade by lupeol inhibits growth and induces apoptosis in hepatocellular carcinoma cells. Br. J. Cancer 2014, 111, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Ren, L.; Wen, L.; Wang, Y.; Qi, J. Effect of evodiamine on the proliferation and apoptosis of A549 human lung cancer cells. Mol. Med. Rep. 2016, 14, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.J.; Wu, N.; Liu, Y.; Shu, K.J.; Zou, X.; Zhang, R.X.; Pi, C.J.; He, B.C.; Ke, Z.Y.; Chen, L.; et al. Evodiamine inhibits the proliferation of human osteosarcoma cells by blocking PI3K/Akt signaling. Oncol. Rep. 2015, 34, 1388–1396. [Google Scholar] [CrossRef]
- Feng, H.; Guo, B.; Kong, X.; Wu, B. [Evodiamine enhances the radiosensitivity of esophageal squamous cell cancer Eca-109 cells]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2016, 32, 940–944. [Google Scholar]
- Shi, L.; Yang, F.; Luo, F.; Liu, Y.; Zhang, F.; Zou, M.; Liu, Q. Evodiamine exerts anti-tumor effects against hepatocellular carcinoma through inhibiting beta-catenin-mediated angiogenesis. Tumour Biol. 2016, 37, 12791–12803. [Google Scholar] [CrossRef]
- Hu, C.Y.; Wu, H.T.; Su, Y.C.; Lin, C.H.; Chang, C.J.; Wu, C.L. Evodiamine Exerts an Anti-Hepatocellular Carcinoma Activity through a WWOX-Dependent Pathway. Molecules 2017, 22, 1175. [Google Scholar] [CrossRef]
- Su, T.; Yang, X.; Deng, J.H.; Huang, Q.J.; Huang, S.C.; Zhang, Y.M.; Zheng, H.M.; Wang, Y.; Lu, L.L.; Liu, Z.Q. Evodiamine, a Novel NOTCH3 Methylation Stimulator, Significantly Suppresses Lung Carcinogenesis in Vitro and in Vivo. Front. Pharmacol. 2018, 9, 434. [Google Scholar] [CrossRef]
- Hong, J.Y.; Park, S.H.; Min, H.Y.; Park, H.J.; Lee, S.K. Anti-proliferative effects of evodiamine in human lung cancer cells. J. Cancer Prev. 2014, 19, 7–13. [Google Scholar] [CrossRef]
- Du, J.; Sun, Y.; Lu, Y.Y.; Lau, E.; Zhao, M.; Zhou, Q.M.; Su, S.B. Berberine and Evodiamine Act Synergistically Against Human Breast Cancer MCF-7 Cells by Inducing Cell Cycle Arrest and Apoptosis. Anticancer Res. 2017, 37, 6141–6151. [Google Scholar]
- Yang, F.; Shi, L.; Liang, T.; Ji, L.; Zhang, G.; Shen, Y.; Zhu, F.; Xu, L. Anti-tumor effect of evodiamine by inducing Akt-mediated apoptosis in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2017, 485, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hu, J. Evodiamine Induces Apoptosis, G2/M Cell Cycle Arrest, and Inhibition of Cell Migration and Invasion in Human Osteosarcoma Cells via Raf/MEK/ERK Signalling Pathway. Med. Sci. Monit. 2018, 24, 5874–5880. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Jin, X.; Cao, Z.; Li, W. [Evodiamine induces extrinsic and intrinsic apoptosis of ovarian cancer cells via the mitogen-activated protein kinase/phosphatidylinositol-3-kinase/protein kinase B signaling pathways]. J. Tradit. Chin. Med. 2016, 36, 353–359. [Google Scholar] [PubMed]
- Wang, R.; Deng, D.; Shao, N.; Xu, Y.; Xue, L.; Peng, Y.; Liu, Y.; Zhi, F. Evodiamine activates cellular apoptosis through suppressing PI3K/AKT and activating MAPK in glioma. Onco Targets Ther. 2018, 11, 1183–1192. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, C.; Um, J.Y.; Sethi, G.; Ahn, K.S. Casticin-Induced Inhibition of Cell Growth and Survival Are Mediated through the Dual Modulation of Akt/mTOR Signaling Cascade. Cancers (Basel) 2019, 11, 254. [Google Scholar] [CrossRef]
- Lee, J.H.; Rangappa, S.; Mohan, C.D.; Lin, Z.X.; Rangappa, K.S.; Ahn, K.S.; Basappa; Sethi, G. Brusatol, a Nrf2 Inhibitor Targets STAT3 Signaling Cascade in Head and Neck Squamous Cell Carcinoma. Biomolecules 2019, 9, 550. [Google Scholar] [CrossRef]
- Lee, J.H.; Chinnathambi, A.; Alharbi, S.A.; Shair, O.H.M.; Sethi, G.; Ahn, K.S. Farnesol abrogates epithelial to mesenchymal transition process through regulating Akt/mTOR pathway. Pharmacol. Res. 2019, 150, 104504. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Shanmugam, M.K.; Chinnathambi, A.; Alharbi, S.A.; Shair, O.H.M.; Um, J.Y.; Sethi, G.; Ahn, K.S. Fangchinoline, a Bisbenzylisoquinoline Alkaloid can Modulate Cytokine-Impelled Apoptosis via the Dual Regulation of NF-kappaB and AP-1 Pathways. Molecules 2019, 24, 3127. [Google Scholar] [CrossRef]
- Yang, M.H.; Lee, J.H.; Ko, J.H.; Jung, S.H.; Sethi, G.; Ahn, K.S. Brassinin Represses Invasive Potential of Lung Carcinoma Cells through Deactivation of PI3K/Akt/mTOR Signaling Cascade. Molecules 2019, 24, 1584. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Shanmugam, M.K.; Narula, A.S.; Kim, C.; Lee, J.H.; Namjoshi, O.A.; Blough, B.E.; Sethi, G.; Ahn, K.S. Oxymatrine Attenuates Tumor Growth and Deactivates STAT5 Signaling in a Lung Cancer Xenograft Model. Cancers (Basel) 2019, 11, 49. [Google Scholar] [CrossRef]
- Hu, C.; Gao, X.; Han, Y.; Guo, Q.; Zhang, K.; Liu, M.; Wang, Y.; Wang, J. Evodiamine sensitizes BGC-823 gastric cancer cells to radiotherapy in vitro and in vivo. Mol. Med. Rep. 2016, 14, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Chien, C.C.; Wu, M.S.; Chen, Y.C. Evodiamine from Evodia rutaecarpa induces apoptosis via activation of JNK and PERK in human ovarian cancer cells. Phytomedicine 2016, 23, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Sachita, K.; Kim, Y.; Yu, H.J.; Cho, S.D.; Lee, J.S. In Vitro Assessment of the Anticancer Potential of Evodiamine in Human Oral Cancer Cell Lines. Phytother. Res. 2015, 29, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.M.; Guh, J.H.; Huang, Y.T.; Chueh, S.C.; Chiang, P.C.; Teng, C.M. Induction of mitotic arrest and apoptosis in human prostate cancer pc-3 cells by evodiamine. J. Urol. 2005, 173, 256–261. [Google Scholar] [CrossRef]
- Nishida, S.; Hirohashi, Y.; Torigoe, T.; Inoue, R.; Kitamura, H.; Tanaka, T.; Takahashi, A.; Asanuma, H.; Masumori, N.; Tsukamoto, T.; et al. Prostate cancer stem-like cells/cancer-initiating cells have an autocrine system of hepatocyte growth factor. Cancer Sci. 2013, 104, 431–436. [Google Scholar] [CrossRef]
- van Leenders, G.J.; Sookhlall, R.; Teubel, W.J.; de Ridder, C.M.; Reneman, S.; Sacchetti, A.; Vissers, K.J.; van Weerden, W.; Jenster, G. Activation of c-MET induces a stem-like phenotype in human prostate cancer. PLoS ONE 2011, 6, e26753. [Google Scholar] [CrossRef]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.Q.; Sethi, G.; Goh, B.C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef]
- Sethi, G.; Chatterjee, S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Wong, K.F.; Kumar, A.P.; Senapati, P.; Behera, A.K.; Hui, K.M.; et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer 2014, 13, 66. [Google Scholar] [CrossRef]
- Rajendran, P.; Li, F.; Shanmugam, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Ahn, K.S.; Kumar, A.P.; Sethi, G. Honokiol inhibits signal transducer and activator of transcription-3 signaling, proliferation, and survival of hepatocellular carcinoma cells via the protein tyrosine phosphatase SHP-1. J. Cell. Physiol. 2012, 227, 2184–2195. [Google Scholar] [CrossRef]
- Tan, S.M.; Li, F.; Rajendran, P.; Kumar, A.P.; Hui, K.M.; Sethi, G. Identification of beta-escin as a novel inhibitor of signal transducer and activator of transcription 3/Janus-activated kinase 2 signaling pathway that suppresses proliferation and induces apoptosis in human hepatocellular carcinoma cells. J. Pharmacol. Exp. Ther. 2010, 334, 285–293. [Google Scholar] [CrossRef]
- Arora, L.; Kumar, A.P.; Arfuso, F.; Chng, W.J.; Sethi, G. The Role of Signal Transducer and Activator of Transcription 3 (STAT3) and Its Targeted Inhibition in Hematological Malignancies. Cancers (Basel) 2018, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Bharathkumar, H.; Bulusu, K.C.; Pandey, V.; Rangappa, S.; Fuchs, J.E.; Shanmugam, M.K.; Dai, X.; Li, F.; Deivasigamani, A.; et al. Development of a novel azaspirane that targets the Janus kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 34296–34307. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Cho, S.K.; Kapoor, S.; Kumar, A.; Vali, S.; Abbasi, T.; Kim, S.H.; Sethi, G.; Ahn, K.S. beta-Caryophyllene oxide inhibits constitutive and inducible STAT3 signaling pathway through induction of the SHP-1 protein tyrosine phosphatase. Mol. Carcinog. 2014, 53, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.F.; Huang, W.J.; Lin, L.C.; Wang, P.S. Inhibitory effects of evodiamine on the growth of human prostate cancer cell line LNCaP. Int. J. Cancer 2004, 110, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Raffo, A.J.; Perlman, H.; Chen, M.W.; Day, M.L.; Streitman, J.S.; Buttyan, R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995, 55, 4438–4445. [Google Scholar] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, S.T.; Um, J.-Y.; Chinnathambi, A.; Alharbi, S.A.; Narula, A.S.; Namjoshi, O.A.; Blough, B.E.; Ahn, K.S. Evodiamine Mitigates Cellular Growth and Promotes Apoptosis by Targeting the c-Met Pathway in Prostate Cancer Cells. Molecules 2020, 25, 1320. https://doi.org/10.3390/molecules25061320
Hwang ST, Um J-Y, Chinnathambi A, Alharbi SA, Narula AS, Namjoshi OA, Blough BE, Ahn KS. Evodiamine Mitigates Cellular Growth and Promotes Apoptosis by Targeting the c-Met Pathway in Prostate Cancer Cells. Molecules. 2020; 25(6):1320. https://doi.org/10.3390/molecules25061320
Chicago/Turabian StyleHwang, Sun Tae, Jae-Young Um, Arunachalam Chinnathambi, Sulaiman Ali Alharbi, Acharan S. Narula, Ojas A. Namjoshi, Bruce E. Blough, and Kwang Seok Ahn. 2020. "Evodiamine Mitigates Cellular Growth and Promotes Apoptosis by Targeting the c-Met Pathway in Prostate Cancer Cells" Molecules 25, no. 6: 1320. https://doi.org/10.3390/molecules25061320
APA StyleHwang, S. T., Um, J.-Y., Chinnathambi, A., Alharbi, S. A., Narula, A. S., Namjoshi, O. A., Blough, B. E., & Ahn, K. S. (2020). Evodiamine Mitigates Cellular Growth and Promotes Apoptosis by Targeting the c-Met Pathway in Prostate Cancer Cells. Molecules, 25(6), 1320. https://doi.org/10.3390/molecules25061320