
Thermal Behavior of Green Cellulose-Filled Thermoplastic Elastomer Polymer Blends



Abstract

1. Introduction
- hemicellulose—degrades at around 300 °C and it may be easily hydrolyzed due to its random amorphous structure [25],
- lignin—composed of three different kinds of benzene-propane units which are dense crosslinked, exhibits a very high molecular weight and an increased thermal stability, very hard to decompose; nevertheless, lignin is not a major wood component [26],
- extractives—compounds of a lower molecular weight which decompose at relatively low temperature; may promote ignitability of wood at lower temperatures (consequence of high volatility) and accelerate the degradation process (the degradation of one component may cause an earlier decomposition of other wood compounds) [27].
2. Materials and Methods
2.1. Preparation of the Specimens
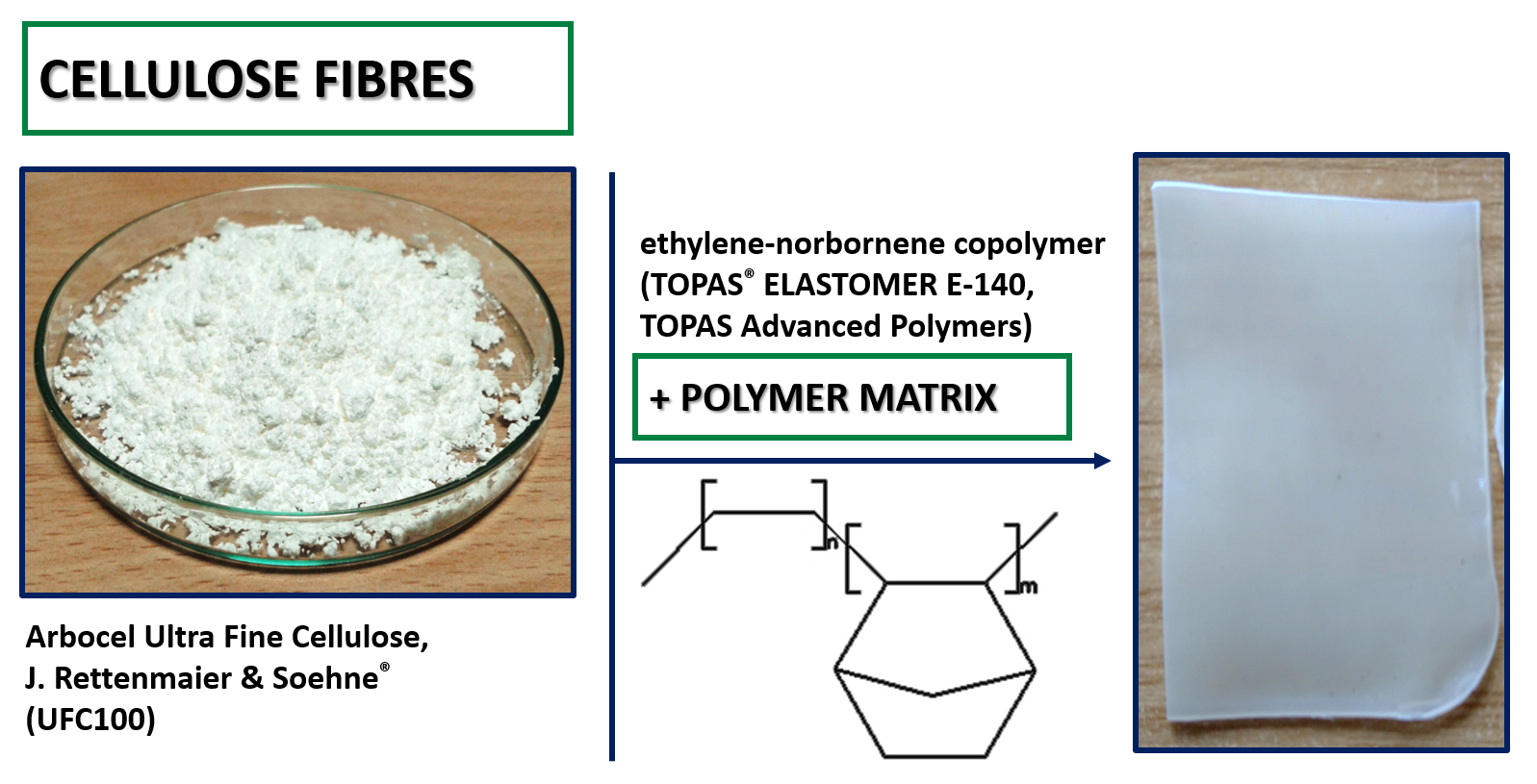
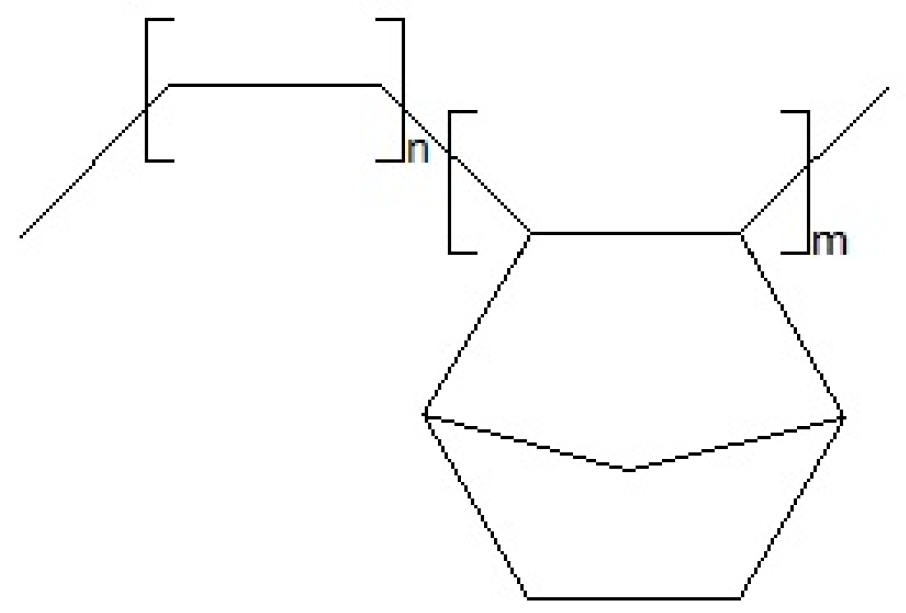
2.1.1. Materials
2.1.2. Cellulose Fibers Hybrid Chemical Modification
2.1.3. Polymer Blend Samples Preparation
2.2. Polymer Blend Samples Characterization
2.2.1. Thermogravimetric Analysis (TGA)
2.2.2. Differential Scanning Calorimetry (DSC)
2.2.3. Surface Free Energy (SFE) Determination
3. Results and Discussion
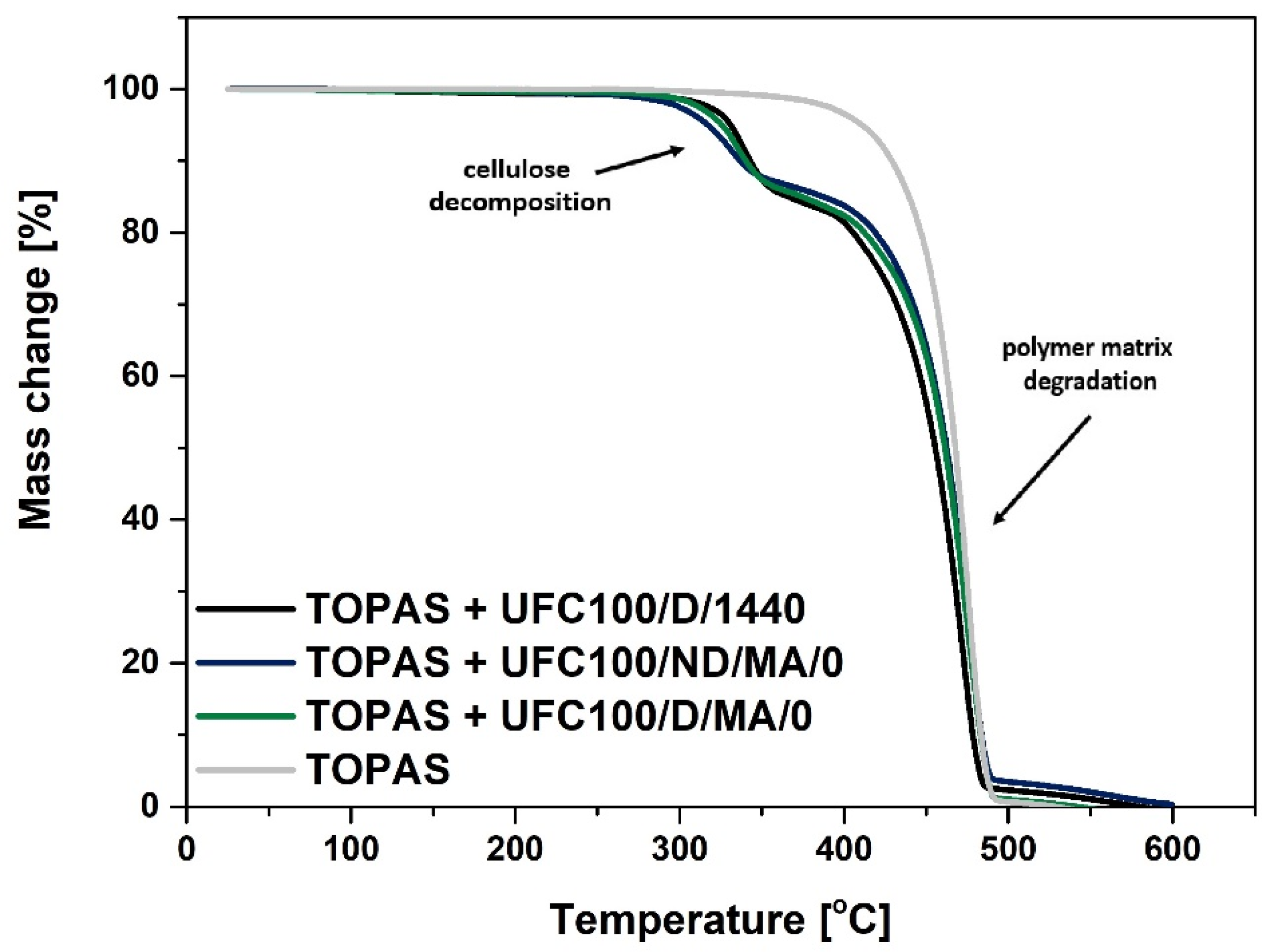
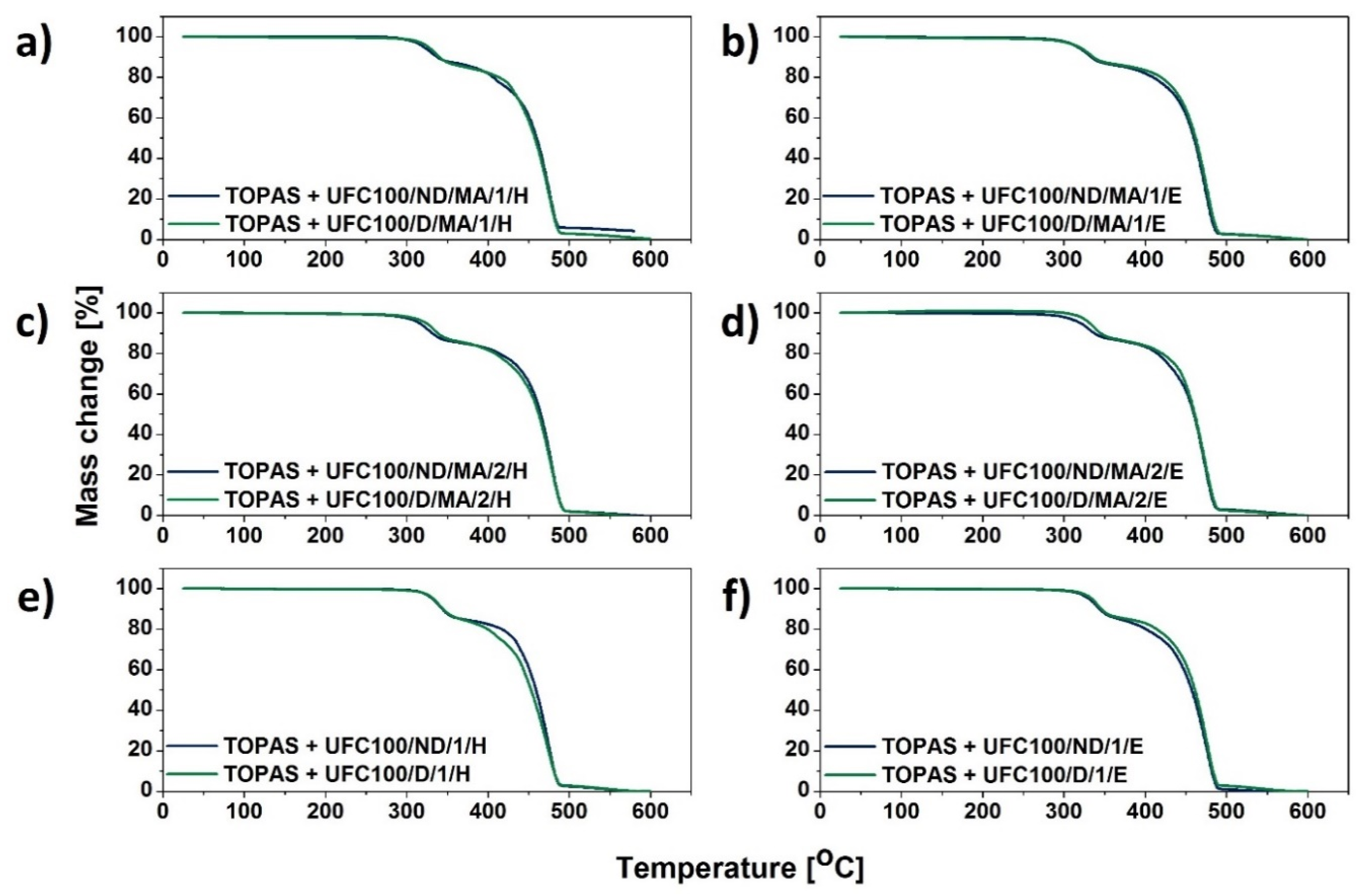
3.1. TGA Characterization
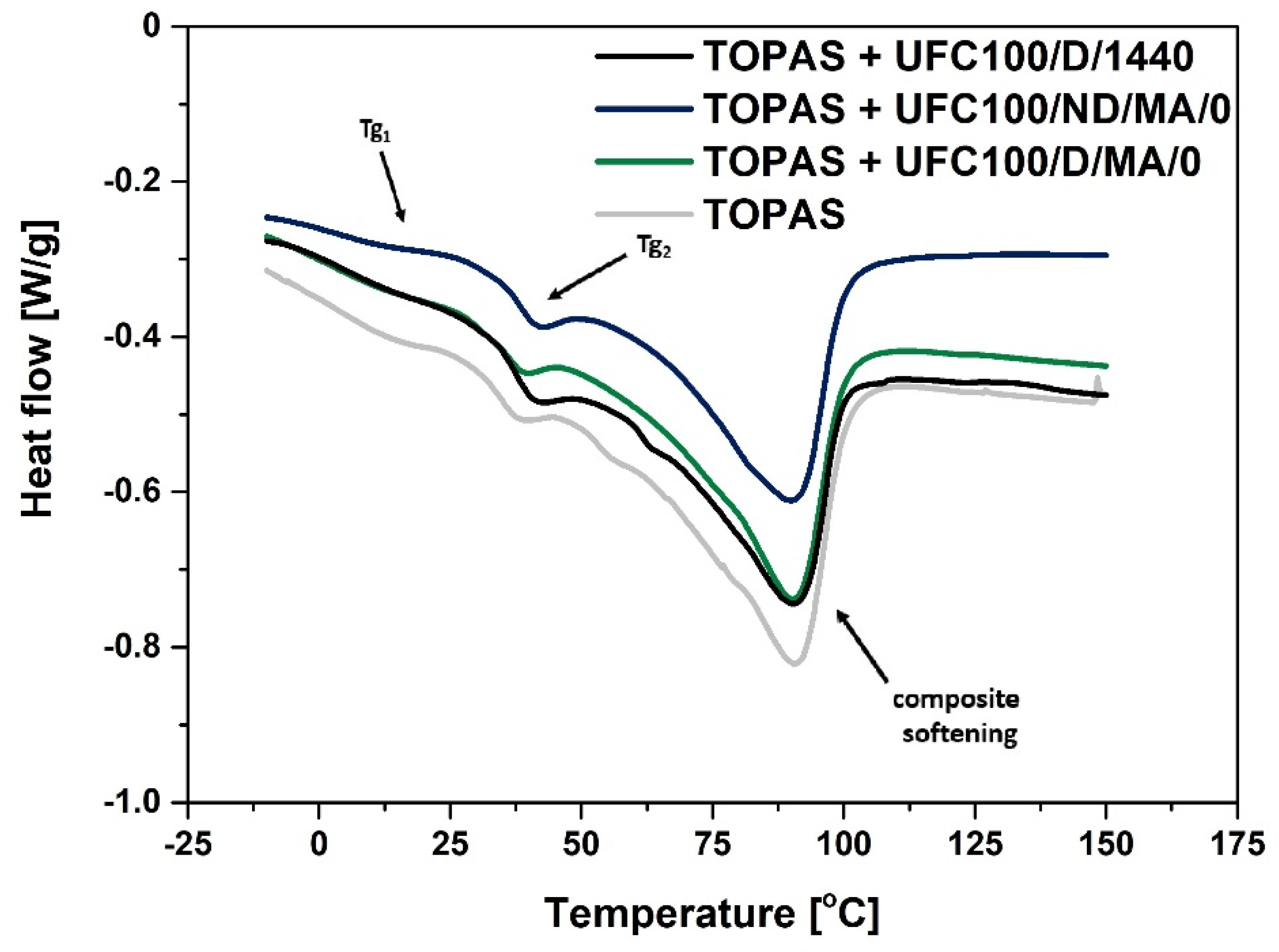
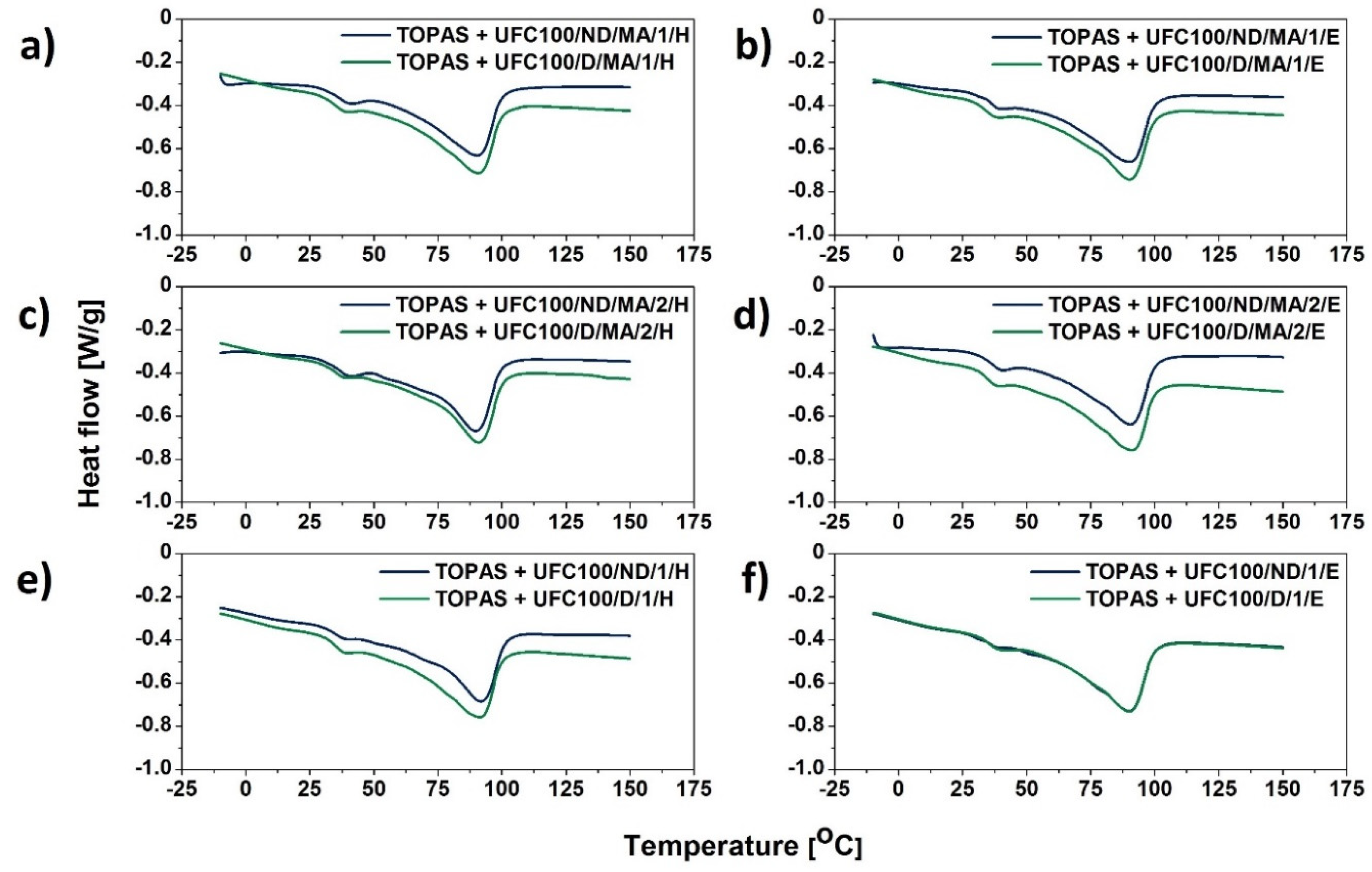
3.2. DSC Analysis
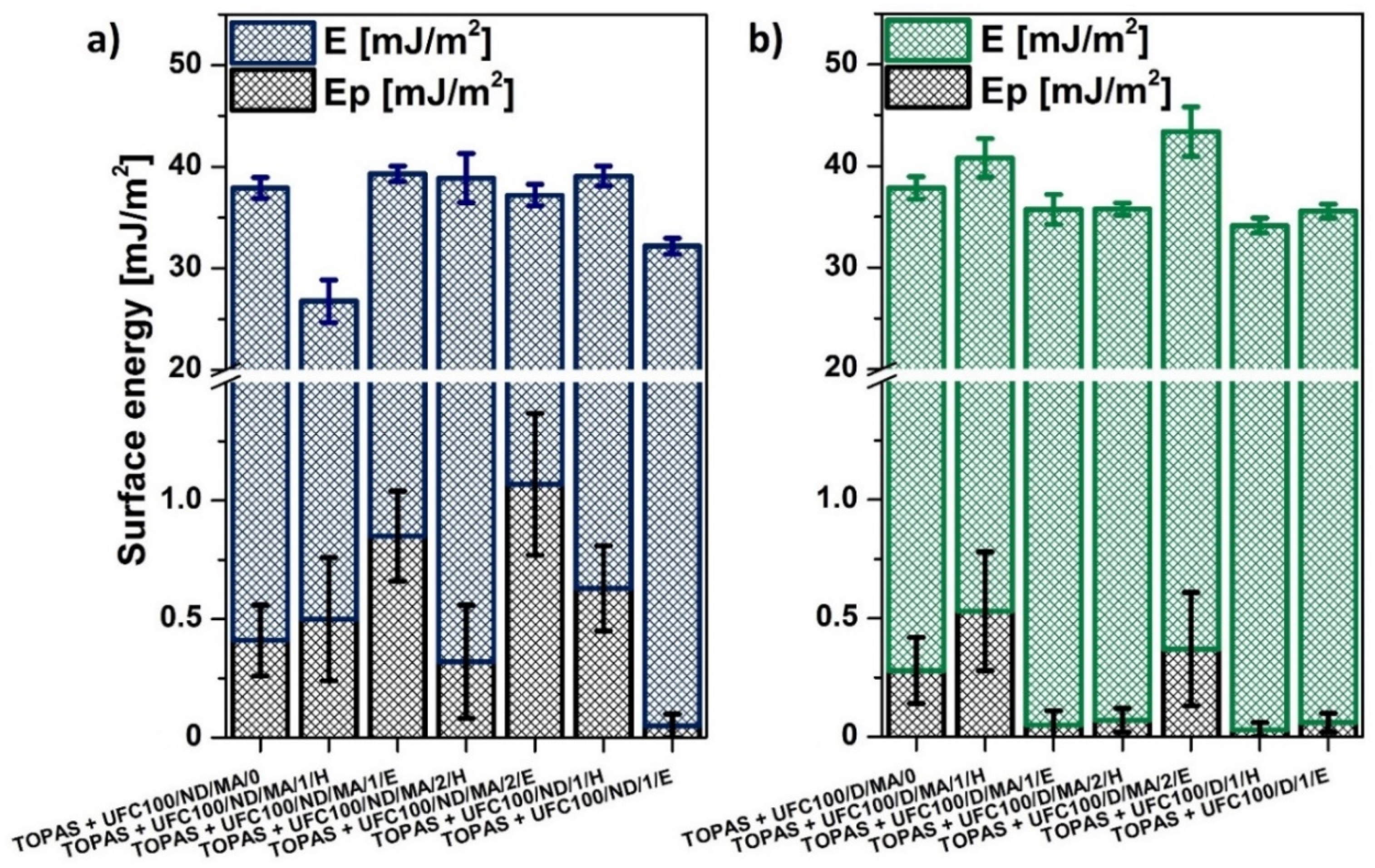
3.3. SFE Determination
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Liu, W.J.; Jiang, H.; Yu, H.Q. Development of Biochar-Based Functional Materials: Toward a Sustainable Platform Carbon Material. Chem. Rev. 2015, 115, 12251–12285. [Google Scholar] [CrossRef]
- Cho, M.J.; Park, B.D. Tensile and thermal properties of nanocellulose-reinforced poly(vinyl alcohol) nanocomposites. J. Ind. Eng. Chem. 2011, 17, 36–40. [Google Scholar] [CrossRef]
- Mandal, A.; Chakrabarty, D. Studies on the mechanical, thermal, morphological and barrier properties of nanocomposites based on poly(vinyl alcohol) and nanocellulose from sugarcane bagasse. J. Ind. Eng. Chem. 2014, 20, 462–473. [Google Scholar] [CrossRef]
- Funazukuri, T.; Ozawa, S. Effects of pretreatment with ionic liquids on cellulose hydrolysis under hydrothermal conditions. Molecules 2019, 24, 3572. [Google Scholar] [CrossRef] [PubMed]
- Elkhaoulani, A.; Arrakhiz, F.Z.; Benmoussa, K.; Bouhfid, R.; Qaiss, A. Mechanical and thermal properties of polymer composite based on natural fibers: Moroccan hemp fibers/polypropylene. Mater. Des. 2013, 49, 203–208. [Google Scholar] [CrossRef]
- Thakur, V.K.; Thakur, M.K. Processing and characterization of natural cellulose fibers/thermoset polymer composites. Carbohydr. Polym. 2014, 109, 102–117. [Google Scholar] [CrossRef]
- Csikós, Á.; Faludi, G.; Domján, A.; Renner, K.; Móczó, J.; Pukánszky, B. Modification of interfacial adhesion with a functionalized polymer in PLA/wood composites. Eur. Polym. J. 2015, 68, 592–600. [Google Scholar] [CrossRef]
- Naseem, A.; Tabasum, S.; Zia, K.M.; Zuber, M.; Ali, M.; Noreen, A. Lignin-derivatives based polymers, blends and composites: A review. Int. J. Biol. Macromol. 2016, 93, 296–313. [Google Scholar] [CrossRef]
- Lin, K.H.; Enomae, T.; Chang, F.C. Cellulose Nanocrystal Isolation from Hardwood Pulp using Various Hydrolysis Conditions. Molecules 2019, 24, 3724. [Google Scholar] [CrossRef]
- Castro-Muñoz, R.; González-Valdez, J. New trends in biopolymer-based membranes for pervaporation. Molecules 2019, 24, 3584. [Google Scholar] [CrossRef]
- Kang, W.Y.; Li, C.F.; Liu, Y.X. Antioxidant phenolic compounds and flavonoids of Mitragyna rotundifolia (Roxb.) Kuntze in vitro. Med. Chem. Res. 2010, 19, 1222–1232. [Google Scholar] [CrossRef]
- Wang, N.; Xin, H.; Zhang, Q.; Jiang, Y.; Wang, X.; Shou, D.; Qin, L. Carbon nanotube-polymer composite for effervescent pipette tip solid phase microextraction of alkaloids and flavonoids from Epimedii herba in biological samples. Talanta 2017, 162, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Peltzer, M.; Jimenez, A. Natural antioxidants for polypropylene stabilization. Recent Adv. Res. Biodegrad. Polym. Sustain. Compos. 2009, 1, 13–21. [Google Scholar]
- Jaafar, J.; Ismail, A.F.; Matsuura, T.; Nagai, K. Performance of SPEEK based polymer-nanoclay inorganic membrane for DMFC. J. Memb. Sci. 2011, 382, 202–211. [Google Scholar] [CrossRef]
- Liu, S.P. Flame retardant and mechanical properties of polyethylene/magnesium hydroxide/montmorillonite nanocomposites. J. Ind. Eng. Chem. 2014, 20, 2401–2408. [Google Scholar] [CrossRef]
- Olsson, E.; Johansson, C.; Larsson, J.; Järnström, L. Montmorillonite for starch-based barrier dispersion coating - Part 2: Pilot trials and PE-lamination. Appl. Clay Sci. 2014, 97, 167–173. [Google Scholar] [CrossRef]
- Vieyra, H.; Aguilar-Méndez, M.A.; San Martín-Martínez, E. Study of biodegradation evolution during composting of polyethylene-starch blends using scanning electron microscopy. J. Appl. Polym. Sci. 2013, 127, 845–853. [Google Scholar] [CrossRef]
- Chang, P.R.; Jian, R.; Yu, J.; Ma, X. Starch-based composites reinforced with novel chitin nanoparticles. Carbohydr. Polym. 2010, 80, 420–425. [Google Scholar] [CrossRef]
- Faruk, O.; Bledzki, A.K.; Fink, H.P.; Sain, M. Progress report on natural fiber reinforced composites. Macromol. Mater. Eng. 2014, 299, 9–26. [Google Scholar] [CrossRef]
- Poletto, M.; Zattera, A.J.; Forte, M.M.C.; Santana, R.M.C. Thermal decomposition of wood: Influence of wood components and cellulose crystallite size. Bioresour. Technol. 2012, 109, 148–153. [Google Scholar] [CrossRef]
- Poletto, M.; Ornaghi Júnior, H.L.; Zattera, A.J. Native cellulose: Structure, characterization and thermal properties. Materials 2014, 7, 6105–6119. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Seney, R.; Bayliss, P.C.; Kitchens, C.L. Carbon nanotube and cellulose nanocrystal hybrid films. Molecules 2019, 24, 2662. [Google Scholar] [CrossRef] [PubMed]
- Abushammala, H.; Mao, J. A Review of the Surface Modification of Cellulose and Nanocellulose Using Aliphatic and Aromatic Mono- and Di-Isocyanates. Molecules 2019, 24, 2782. [Google Scholar] [CrossRef] [PubMed]
- Freire, C.S.R.; Silvestre, A.J.D.; Neto, C.P.; Gandini, A.; Martin, L.; Mondragon, I. Composites based on acylated cellulose fibers and low-density polyethylene: Effect of the fiber content, degree of substitution and fatty acid chain length on final properties. Compos. Sci. Technol. 2008, 68, 3358–3364. [Google Scholar] [CrossRef]
- Zhou, X.; Li, W.; Mabon, R.; Broadbelt, L.J. A critical review on hemicellulose pyrolysis. Energy Technol. 2017, 5, 52–79. [Google Scholar] [CrossRef]
- Brebu, M.; Vasile, C. Thermal degradation of lignin - A review. Cellul. Chem. Technol. 2010, 44, 353–363. [Google Scholar]
- Pelaez-Samaniego, M.R.; Yadama, V.; Lowell, E.; Espinoza-Herrera, R. A review of wood thermal pretreatments to improve wood composite properties. Wood Sci. Technol. 2013, 47, 1285–1319. [Google Scholar] [CrossRef]
- Lionetto, F.; Del Sole, R.; Cannoletta, D.; Vasapollo, G.; Maffezzoli, A. Monitoring wood degradation during weathering by cellulose crystallinity. Materials 2012, 5, 1910–1922. [Google Scholar] [CrossRef]
- Bledzki, A.K.; Faruk, O.; Huque, M. Physico-mechanical studies of wood fiber reinforced composites. Polym. Plast. Technol. Eng. 2002, 41, 435–451. [Google Scholar] [CrossRef]
- Ciolacu, D.; Ciolacu, F.; Popa, V.I. Amorphous cellulose—Structure and characterization. Cellul. Chem. Technol. 2011, 45, 13–21. [Google Scholar]
- Basen, M.; Rhaesa, A.M.; Kataeva, I.; Prybol, C.J.; Scott, I.M.; Poole, F.L.; Adams, M.W.W. Degradation of high loads of crystalline cellulose and of unpretreated plant biomass by the thermophilic bacterium Caldicellulosiruptor bescii. Bioresour. Technol. 2014, 152, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.E.M.; Dos Santos, V.; Mármol, G.; Frías, M.; Fiorelli, J. Potential of the hornification treatment on eucalyptus and pine fibers for fiber-cement applications. Cellulose 2017, 24, 2275–2286. [Google Scholar] [CrossRef]
- Minor, J.L. Hornification -Its origin and meaning. Progrress Pap. Recycl. 1994, 3, 93–95. [Google Scholar]
- Fernandes Diniz, J.M.B.; Gil, M.H.; Castro, J.A.A.M. Hornification - Its origin and interpretation in wood pulps. Wood Sci. Technol. 2004, 37, 489–494. [Google Scholar] [CrossRef]
- Zimmermann, R.; Nitschke, C.; Potthast, A.; Werner, C.; Freudenberg, U.; Scherr, G.; Müller, Y.; Eichhorn, K.-J.; Tot, I.; Rosenau, T. The impact of esterification reactions on physical properties of cellulose thin films. Soft Matter 2010, 6, 3680. [Google Scholar]
- Park, S.; Venditti, R.A.; Jameel, H.; Pawlak, J.J. Changes in pore size distribution during the drying of cellulose fibers as measured by differential scanning calorimetry. Carbohydr. Polym. 2006, 66, 97–103. [Google Scholar] [CrossRef]
- Vuoti, S.; Laatikainen, E.; Heikkinen, H.; Johansson, L.S.; Saharinen, E.; Retulainen, E. Chemical modification of cellulosic fibers for better convertibility in packaging applications. Carbohydr. Polym. 2013, 96, 549–559. [Google Scholar] [CrossRef]
- Cichosz, S.; Masek, A. Cellulose Fibers Hydrophobization via a Hybrid Chemical Modification. Polymers 2019, 11, 1174. [Google Scholar] [CrossRef]
- Broido, A. A simple, sensitive graphical method of treating thermogravimetric analysis data. J. Polym. Sci. Part A-2 Polym. Phys. 1969, 7, 1761–1773. [Google Scholar] [CrossRef]
- Encinas, N.; Pantoja, M.; Abenojar, J.; Martínez, M.A. Control of wettability of polymers by surface roughness modification. J. Adhes. Sci. Technol. 2010, 24, 1869–1883. [Google Scholar] [CrossRef]
- Perisić, M.; Radojević, V.; Uskoković, P.S.; Stojanović, D.; Jokić, B.; Aleksić, R. Wood-thermoplastic composites based on industrial waste and virgin high-density polyethylene (HDPE). Mater. Manuf. Process. 2009, 24, 1207–1213. [Google Scholar] [CrossRef]
- Müller-Hagedorn, M.; Bockhorn, H.; Krebs, L.; Müller, U. A comparative kinetic study on the pyrolysis of three different wood species. J. Anal. Appl. Pyrolysis 2003, 68, 231–249. [Google Scholar] [CrossRef]
- Singh, S.; Mohanty, A.K. Wood fiber reinforced bacterial bioplastic composites: Fabrication and performance evaluation. Compos. Sci. Technol. 2007, 67, 1753–1763. [Google Scholar] [CrossRef]
- Ashori, A.; Nourbakhsh, A. Composites: Part B Performance properties of microcrystalline cellulose as a reinforcing agent in wood plastic composites. Compos. Part B 2010, 41, 578–581. [Google Scholar] [CrossRef]
- Fernandes, E.G.; Pietrini, M.; Chiellini, E. Bio-Based Polymeric Composites Comprising Wood Flour as Filler. Biomacromolecules 2004, 7, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Sailaja, R.R.N.; Seetharamu, S. Mechanical and Thermal Properties of LDPE-Cellulose Acetate Phthalate Blends—effect of Maleic AnhydrideGrafted LDPE Compatibilizer. J. Appl. Polym. Sci. 2008, 112, 650–659. [Google Scholar]
- Georgopoulos, S.T.; Tarantili, P.A.; Avgerinos, E.; Andreopoulos, A.G.; Koukios, E.G. Thermoplastic polymers reinforced with fibrous agricultural residues. Polym. Degrad. Stab. 2005, 90, 303–312. [Google Scholar] [CrossRef]
- Roland, A.I.; Schmidt-Naake, G. Thermal degradation of polystyrene produced by nitroxide-controlled radical polymerization. J. Anal. Appl. Pyrolysis 2001, 58, 143–154. [Google Scholar] [CrossRef]
- Östlund, Å.; Idström, A.; Olsson, C.; Larsson, P.T.; Nordstierna, L. Modification of crystallinity and pore size distribution in coagulated cellulose films. Cellulose 2013, 20, 1657–1667. [Google Scholar] [CrossRef]
- Pasquini, D.; De Morais Teixeira, E.; Da Silva Curvelo, A.A.; Belgacem, M.N.; Dufresne, A. Surface esterification of cellulose fibers: Processing and characterisation of low-density polyethylene/cellulose fibers composites. Compos. Sci. Technol. 2008, 68, 193–201. [Google Scholar] [CrossRef]
- Ishii, D.; Tatsumi, D.; Matsumoto, T. Effect of solvent exchange on the solid structure and dissolution behavior of cellulose. Biomacromolecules 2003, 4, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Vuoti, S.; Talja, R.; Johansson, L.S.; Heikkinen, H.; Tammelin, T. Solvent impact on esterification and film formation ability of nanofibrillated cellulose. Cellulose 2013, 20, 2359–2370. [Google Scholar] [CrossRef]
- Cichosz, S.; Masek, A.; Wolski, K. Innovative cellulose fibers reinforced ethylene-norbornene copolymer composites of an increased degradation potential. Polym. Degrad. Stab. 2019, 159, 174–183. [Google Scholar] [CrossRef]
- Marcovich, N.E.; Villar, M.A. Thermal and mechanical characterization of linear low-density polyethylene/wood flour composites. J. Appl. Polym. Sci. 2003, 90, 2775–2784. [Google Scholar] [CrossRef]
- Lodha, P.; Netravali, A.N. Thermal and mechanical properties of environment-friendly “green” plastics from stearic acid modified-soy protein isolate. Ind. Crops Prod. 2005, 21, 49–64. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Y.; Abidi, N.; Cabrales, L. Wettability and Surface Free Energy of Graphene Films. Langmuir 2009, 25, 11078–11081. [Google Scholar] [CrossRef]
- Cantero, G.; Arbelaiz, A.; Llano-ponte, R. Effects of fiber treatment on wettability and mechanical behaviour of flax/polypropylene composites. Compos. Sci. Technol. 2003, 63, 1247–1254. [Google Scholar] [CrossRef]
- Rudawska, A.; Jakubowska, P.; Kloziński, A. Surface free energy of composite materials with high calcium carbonate filler content. Polimery 2017, 434–440. [Google Scholar] [CrossRef]
- Liu, H.; Jiang, Y.; Tan, W.; Wang, X. Enhancement of the laser transmissionweldability between polyethylene and polyoxymethylene by plasma surface treatment. Materials 2017, 11, 29. [Google Scholar] [CrossRef]
- Arbelaiz, A.; Fernández, B.; Ramos, J.A.; Retegi, A.; Llano-Ponte, R.; Mondragon, I. Mechanical properties of short flax fiber bundle/polypropylene composites: Influence of matrix/fiber modification, fiber content, water uptake and recycling. Compos. Sci. Technol. 2005, 65, 1582–1592. [Google Scholar] [CrossRef]
- Aranberri-Askargorta, I.; Lampke, T.; Bismarck, A. Wetting behavior of flax fibers as reinforcement for polypropylene. J. Coll. Interface Sci. 2003, 263, 580–589. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Acetone | Ethanol (99.9%) | Hexane |
|---|---|---|---|
| boiling point (°C) | 55–57 | 78 | 68 |
| viscosity at 20 °C (mPas) | 0.330 | 1.078 | 0.310 |
| density at 20 °C (g/cm3) | 0.791 | 0.790 | 0.660 |
| Sample | Dried Before Modification (D) | Not Dried Before Modification (ND) | Solvent Exchange | MA Treated | |||
|---|---|---|---|---|---|---|---|
| Before MA Treatment | After MA Treatment | ||||||
| H | E | H | E | ||||
| UFC100/ND/MA/0 | ------ | ✔ | ------ | ------ | ------ | ------ | ✔ |
| UFC100/ND/MA/1/H | ------ | ✔ | ✔ | ------ | ------ | ------ | ✔ |
| UFC100/ND/MA/1/E | ------ | ✔ | ------ | ✔ | ------ | ------ | ✔ |
| UFC100/ND/MA/2/H | ------ | ✔ | ------ | ------ | ✔ | ------ | ✔ |
| UFC100/ND/MA/2/E | ------ | ✔ | ------ | ------ | ------ | ✔ | ✔ |
| UFC100/ND/1/H | ------ | ✔ | ✔ | ------ | ------ | ------ | ------ |
| UFC100/ND/1/E | ------ | ✔ | ------ | ✔ | ------ | ------ | ------ |
| UFC100/D/MA/0 | ✔ | ------ | ------ | ------ | ------ | ------ | ✔ |
| UFC100/D/MA/1/H | ✔ | ------ | ✔ | ------ | ------ | ------ | ✔ |
| UFC100/D/MA/1/E | ✔ | ------ | ------ | ✔ | ------ | ------ | ✔ |
| UFC100/D/MA/2/H | ✔ | ------ | ------ | ------ | ✔ | ------ | ✔ |
| UFC100/D/MA/2/E | ✔ | ------ | ------ | ------ | ------ | ✔ | ✔ |
| UFC100/D/1/H | ✔ | ------ | ✔ | ------ | ------ | ------ | ------ |
| UFC100/D/1/E | ✔ | ------ | ------ | ✔ | ------ | ------ | ------ |
| Sample | T05% (°C) | T10% (°C) | T15% (°C) | T20% (°C) | T50% (°C) | T80% (°C) | T90% (°C) |
|---|---|---|---|---|---|---|---|
| TOPAS | 411 | 430 | 440 | 447 | 468 | 479 | 483 |
| TOPAS + UFC100/D/1440 | 331 | 343 | 366 | 405 | 456 | 473 | 478 |
| TOPAS + UFC100/ND/MA/0 | 317 | 337 | 387 | 419 | 462 | 478 | 484 |
| TOPAS + UFC100/ND/MA/1/H | 321 | 338 | 381 | 406 | 460 | 478 | 483 |
| TOPAS + UFC100/ND/MA/1/E | 317 | 333 | 375 | 410 | 460 | 477 | 482 |
| TOPAS + UFC100/ND/MA/2/H | 315 | 332 | 369 | 414 | 465 | 481 | 486 |
| TOPAS + UFC100/ND/MA/2/E | 320 | 338 | 385 | 415 | 460 | 476 | 481 |
| TOPAS + UFC100/ND/1/H | 332 | 344 | 367 | 417 | 459 | 476 | 481 |
| TOPAS + UFC100/ND/1/E | 330 | 344 | 368 | 401 | 457 | 476 | 482 |
| TOPAS + UFC100/D/MA/0 | 326 | 340 | 374 | 412 | 462 | 478 | 484 |
| TOPAS + UFC100/D/MA/1/H | 327 | 340 | 372 | 413 | 458 | 477 | 482 |
| TOPAS + UFC100/D/MA/1/E | 318 | 336 | 384 | 420 | 462 | 478 | 484 |
| TOPAS + UFC100/D/MA/2/H | 324 | 339 | 375 | 409 | 462 | 480 | 486 |
| TOPAS + UFC100/D/MA/2/E | 332 | 345 | 389 | 421 | 461 | 477 | 483 |
| TOPAS + UFC100/D/1/H | 333 | 345 | 366 | 400 | 453 | 474 | 481 |
| TOPAS + UFC100/D/1/E | 335 | 346 | 377 | 414 | 461 | 477 | 483 |
| Sample | ||
|---|---|---|
| TOPAS | --------- | 225 ± 3 |
| TOPAS + UFC100/D/1440 | 189 ± 2 | 232 ± 2 |
| TOPAS + UFC100/ND/MA/0 | 167 ± 2 | 232 ± 3 |
| TOPAS + UFC100/ND/MA/1/H | 186 ± 3 | 218 ± 4 |
| TOPAS + UFC100/ND/MA/1/E | 174 ± 2 | 228 ± 3 |
| TOPAS + UFC100/ND/MA/2/H | 176 ± 2 | 224 ± 3 |
| TOPAS + UFC100/ND/MA/2/E | 172 ± 2 | 234 ± 3 |
| TOPAS + UFC100/ND/1/H | 230 ± 2 | 243 ± 2 |
| TOPAS + UFC100/ND/1/E | 193 ± 2 | 213 ± 3 |
| TOPAS + UFC100/D/MA/0 | 195 ± 2 | 224 ± 3 |
| TOPAS + UFC100/D/MA/1/H | 195 ± 2 | 233 ± 2 |
| TOPAS + UFC100/D/MA/1/E | 184 ± 3 | 235 ± 3 |
| TOPAS + UFC100/D/MA/2/H | 183 ± 2 | 213 ± 3 |
| TOPAS + UFC100/D/MA/2/E | 194 ± 1 | 234 ± 3 |
| TOPAS + UFC100/D/1/H | 194 ± 2 | 217 ± 3 |
| TOPAS + UFC100/D/1/E | 194 ± 2 | 235 ± 3 |
| Sample | Tg1 (°C) | Tg2 (°C) | Tpeak (°C) | ΔH (J/g) |
|---|---|---|---|---|
| TOPAS | 9 | 36 | 90 | 54 |
| TOPAS + UFC100/D/1440 | 4 | 38 | 90 | 40 |
| TOPAS + UFC100/ND/MA/0 | 4 | 39 | 90 | 41 |
| TOPAS + UFC100/ND/MA/1/H | 4 | 39 | 90 | 42 |
| TOPAS + UFC100/ND/MA/1/E | 4 | 37 | 90 | 43 |
| TOPAS + UFC100/ND/MA/2/H | 4 | 38 | 90 | 41 |
| TOPAS + UFC100/ND/MA/2/E | 4 | 37 | 91 | 42 |
| TOPAS + UFC100/ND/1/H | 13 | 36 | 92 | 43 |
| TOPAS + UFC100/ND/1/E | 11 | 36 | 90 | 47 |
| TOPAS + UFC100/D/MA/0 | 10 | 37 | 90 | 45 |
| TOPAS + UFC100/D/MA/1/H | 11 | 36 | 91 | 45 |
| TOPAS + UFC100/D/MA/1/E | 12 | 35 | 90 | 44 |
| TOPAS + UFC100/D/MA/2/H | 12 | 37 | 91 | 43 |
| TOPAS + UFC100/D/MA/2/E | 13 | 35 | 91 | 45 |
| TOPAS + UFC100/D/1/H | 11 | 35 | 90 | 44 |
| TOPAS + UFC100/D/1/E | 12 | 38 | 90 | 44 |
| E [mJ/m2] | Ep [mJ/m2] |
|---|---|
| 40 ± 1 | 0.10 ± 0.06 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cichosz, S.; Masek, A. Thermal Behavior of Green Cellulose-Filled Thermoplastic Elastomer Polymer Blends. Molecules 2020, 25, 1279. https://doi.org/10.3390/molecules25061279
Cichosz S, Masek A. Thermal Behavior of Green Cellulose-Filled Thermoplastic Elastomer Polymer Blends. Molecules. 2020; 25(6):1279. https://doi.org/10.3390/molecules25061279
Chicago/Turabian StyleCichosz, Stefan, and Anna Masek. 2020. "Thermal Behavior of Green Cellulose-Filled Thermoplastic Elastomer Polymer Blends" Molecules 25, no. 6: 1279. https://doi.org/10.3390/molecules25061279
APA StyleCichosz, S., & Masek, A. (2020). Thermal Behavior of Green Cellulose-Filled Thermoplastic Elastomer Polymer Blends. Molecules, 25(6), 1279. https://doi.org/10.3390/molecules25061279