
Synthesis of Tetrabenazine and Its Derivatives, Pursuing Efficiency and Selectivity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Discussion
2.1. Racemic Synthesis of TBZ
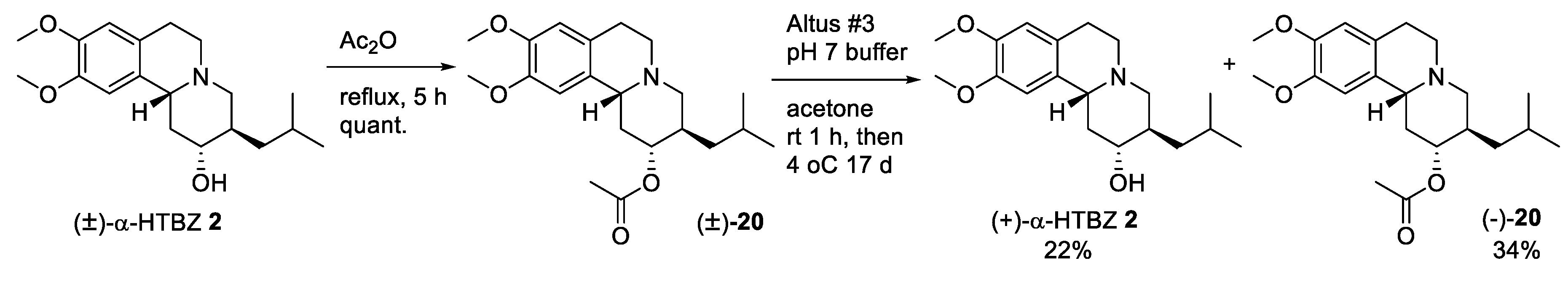
2.2. Resolution of Racemic TBZ
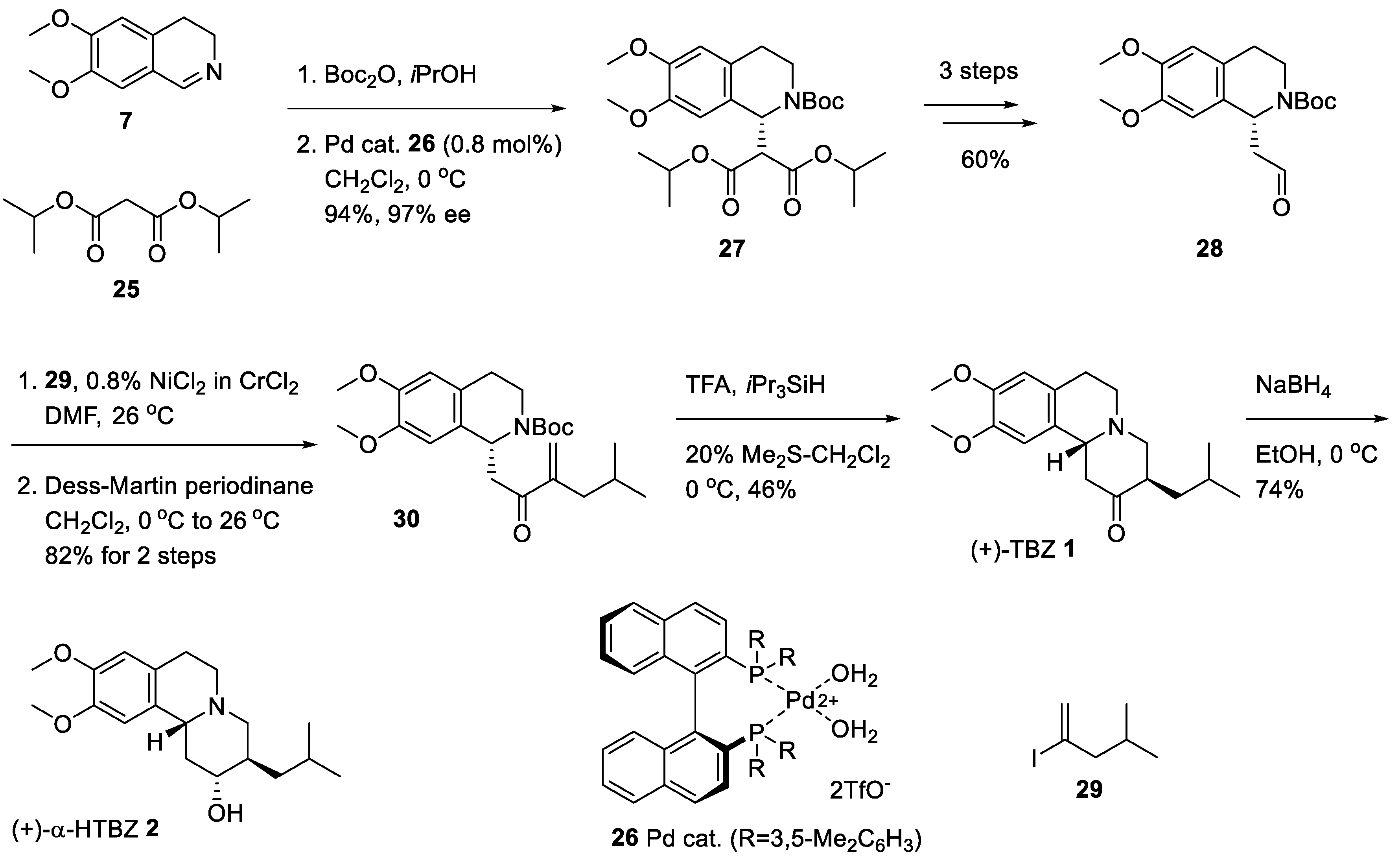
2.3. Asymmetric Synthesis of (+)-TBZ
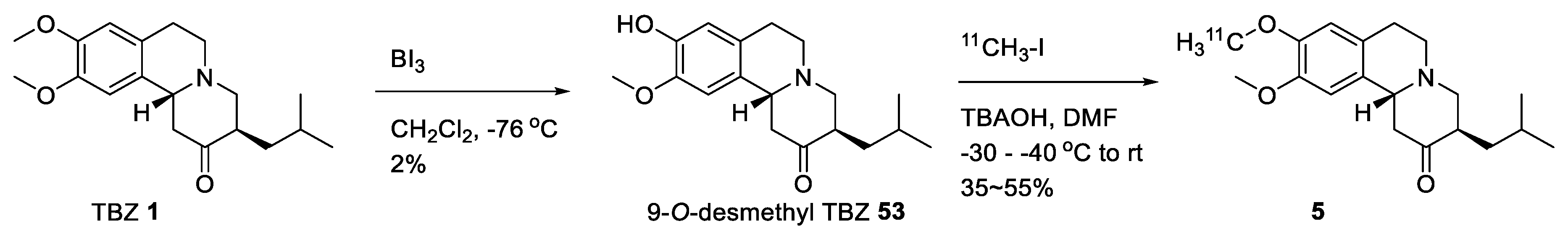
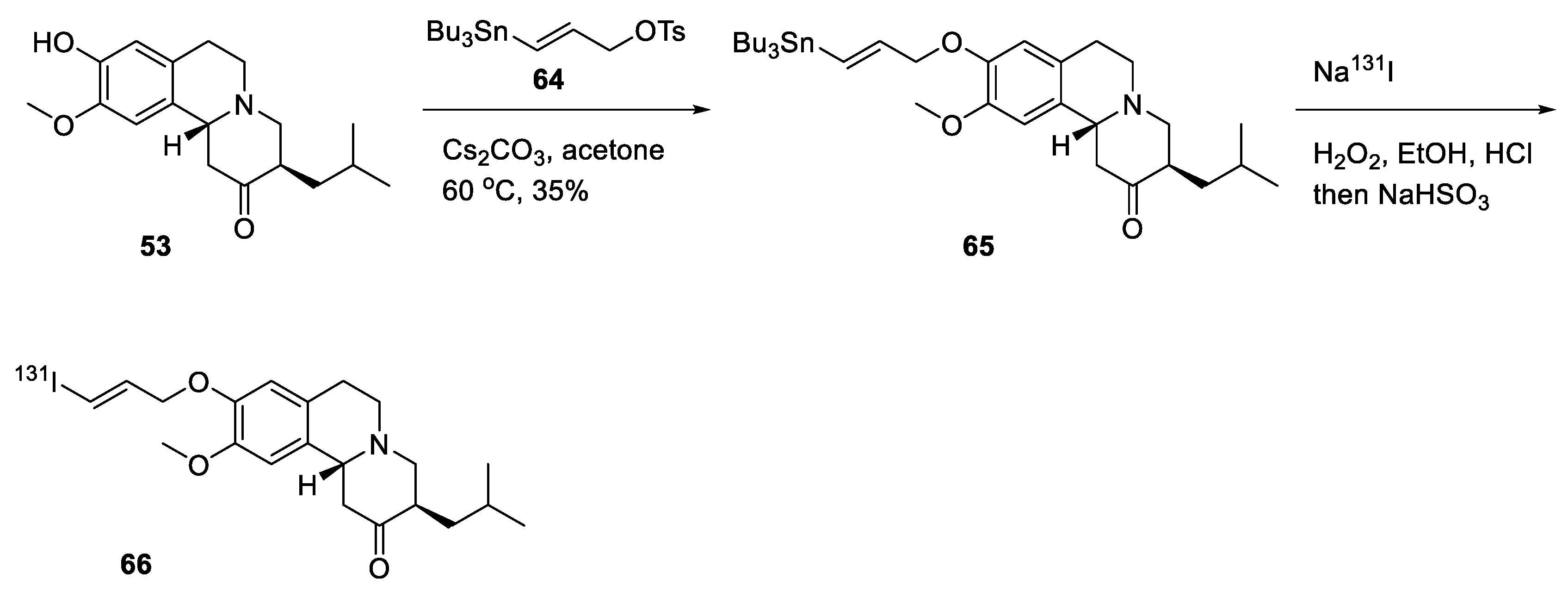
2.4. Preparation of Radioisotopically Labeled TBZ
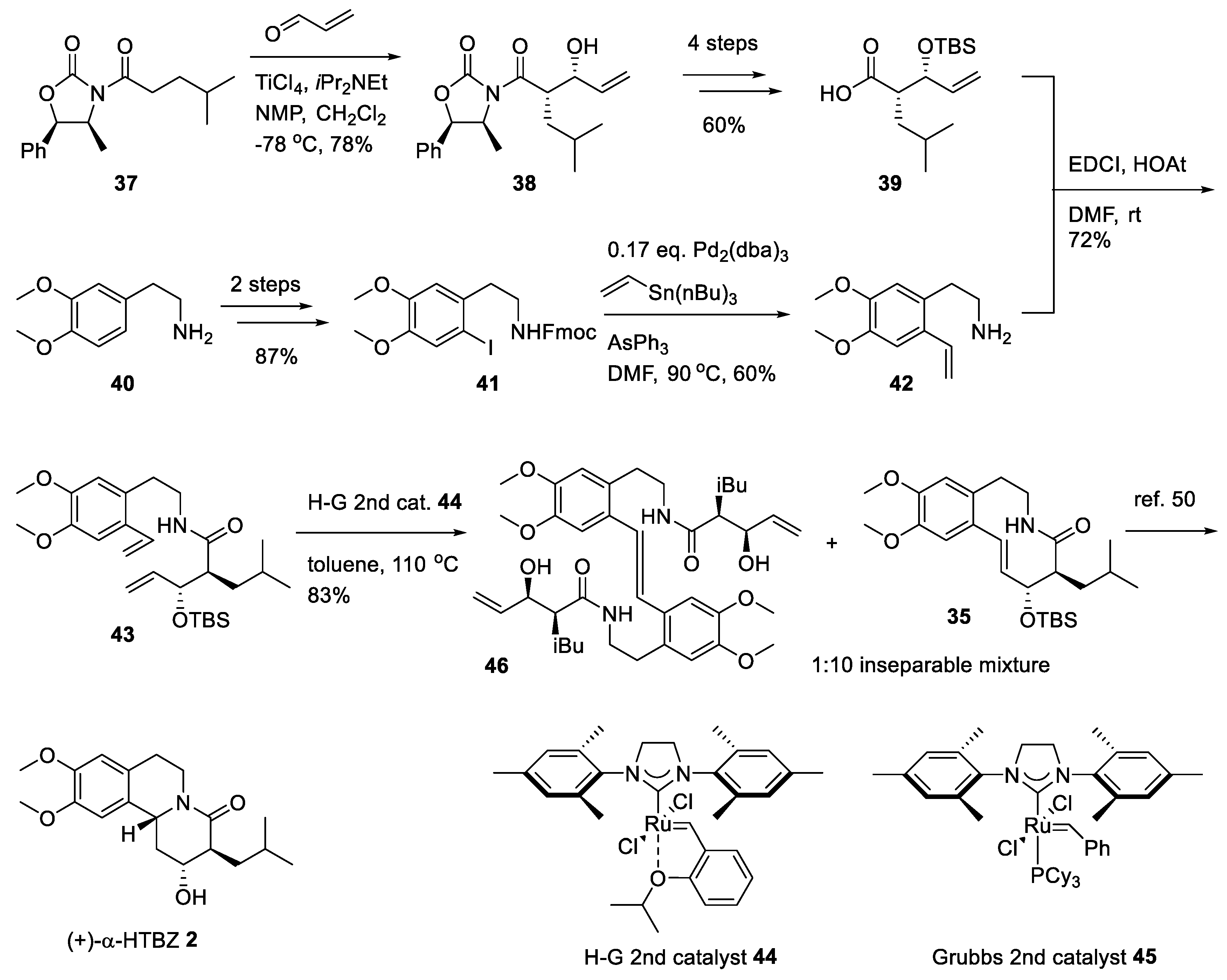
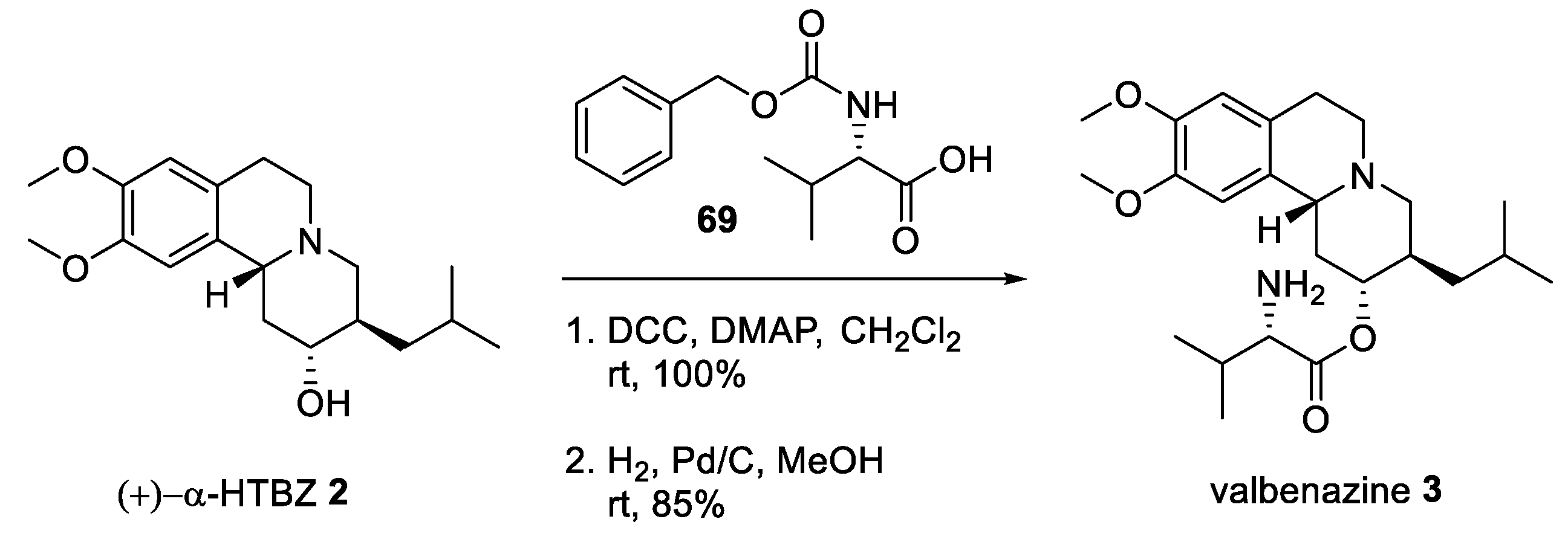
2.5. Preparation of a TBZ Prodrug
3. Conclusions
Funding
Conflicts of Interest
References
- Volkow, N.D.; Wise, R.A.; Baler, R. The dopamine motive system: Implications for drug and food addiction. Nat. Rev. Neurosci. 2017, 18, 741–752. [Google Scholar] [CrossRef]
- Niemann, N.; Jankovic, J. Treatment of Tardive Dyskinesia: A General Overview with Focus on the Vesicular Monoamine Transporter 2 Inhibitors. Drugs 2018, 78, 525–541. [Google Scholar] [CrossRef]
- Lohr, K.M.; Bernstein, A.I.; Stout, K.; Dunn, A.; Lazo, C.R.; Alter, S.P.; Wang, M.; Li, Y.; Fan, X.; Hess, E.J.; et al. Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 9977–9982. [Google Scholar] [CrossRef]
- Huntington Study Group. Tetrabenazine as Antichorea Therapy in Huntington Disease: A Randomized Controlled Trial. Neurology 2006, 66, 3663–3672. [Google Scholar]
- Zubieta, J.-K.; Taylor, S.F.; Huguelet, P.; Koeppe, R.A.; Kilbourn, M.; Frey, K.A. Vesicular monoamine transporter concentrations in bipolar disorder type I, schizophrenia, and healthy subjects. Boil. Psychiatry 2001, 49, 110–116. [Google Scholar] [CrossRef]
- A Frey, K.; A Koeppe, R.; Kilbourn, M.R. Imaging the vesicular monoamine transporter. Adv. Neurol. 2001, 86, 237–247. [Google Scholar]
- Kaur, N.; Kumar, P.; Jamwal, S.; Deshmukh, R.; Gauttam, V. Tetrabenazine: Spotlight on Drug Review. Ann. Neurosci. 2016, 23, 176–185. [Google Scholar] [CrossRef]
- Guay, D.R.P. Tetrabenazine, a monoamine-depleting drug used in the treatment of hyperkinetic movement disorders. Am. J. Geriatr. Pharmacother. 2010, 8, 331–373. [Google Scholar] [CrossRef]
- Chen, J.J.; Ondo, W.G.; Dashtipour, K.; Swope, D. Tetrabenazine for the Treatment of Hyperkinetic Movement Disorders: A Review of the Literature. Clin. Ther. 2012, 34, 1487–1504. [Google Scholar] [CrossRef]
- Scott, L.J. Tetrabenazine. CNS drugs 2011, 25, 1073–1085. [Google Scholar] [CrossRef]
- Pletscher, A.; Schachter, D. Release of 5-Hydroxytryptamine by Benzoquinolizine Derivatives with Sedative Action. Science 1957, 126, 507. [Google Scholar] [CrossRef]
- Brodie, B.A.; Shore, P.A.; Pletscher, A. Serotonin-Releasing Activity Limited to Rauwolfia Alkaloids with Tranquilizing Action. Science 1956, 123, 992–993. [Google Scholar] [CrossRef]
- Goswami, R.; Ponde, D.E.; Kung, M.-P.; Hou, C.; Kilbourn, M.R.; Kung, H.F. Fluoroalkyl derivatives of dihydrotetrabenazine as positron emission tomography imaging agents targeting vesicular monoamine transporters. Nucl. Med. Boil. 2006, 33, 685–694. [Google Scholar] [CrossRef]
- Kenney, C.; Jankovic, J. Tetrabenazine in the treatment of hyperkinetic movement disorders. Expert Rev. Neurother. 2006, 6, 7–17. [Google Scholar] [CrossRef]
- Frank, S. Tetrabenazine: The first approved drug for the treatment of chorea in US patients with Huntington disease. Neuropsychiatr. Dis. Treat. 2010, 6, 657–665. [Google Scholar] [CrossRef]
- Yu, Q.-S.; Luo, W.; Deschamps, J.; Holloway, H.W.; Kopajtic, T.; Katz, J.; Brossi, A.; Greig, N.H. Preparation and Characterization of Tetrabenazine Enantiomers against Vesicular Monoamine Transporter 2. ACS Med. Chem. Lett. 2010, 1, 105–109. [Google Scholar] [CrossRef]
- Kilbourn, M.R.; Lee, L.C.; Heeg, M.J.; Jewett, D.M. Absolute Configuration of (+)-α-dihydrotetrabenazine, an Active Metabolite of Tetrabenazine. Chirality 1997, 9, 59–62. [Google Scholar] [CrossRef]
- Boldt, K.G.; Biggers, M.S.; Phifer, S.S.; Brine, G.A.; Rehder, K.S. Synthesis of (+)-and (−)-Tetrabenazine from the Resolution of A-Dihydrotetrabenazine. Synth. Commun. 2009, 39, 3574–3585. [Google Scholar] [CrossRef]
- Rishel, M.J.; Amarasinghe, K.K.D.; Dinn, S.R.; Johnson, B.F. Asymmetric Synthesis of Tetrabenazine and Dihydrotetrabenazine. J. Org. Chem. 2009, 74, 4001–4004. [Google Scholar] [CrossRef] [PubMed]
- Deveaugh-Geiss, J.; Clarence-Smith, K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev. Neurother. 2011, 11, 1509–1523. [Google Scholar]
- Yao, Z.; Wei, X.; Wu, X.; Katz, J.; Kopajtic, T.; Greig, N.H.; Sun, H. Preparation and evaluation of tetrabenazine enantiomers and all eight stereoisomers of dihydrotetrabenazine as VMAT2 inhibitors. Eur. J. Med. Chem. 2011, 46, 1841–1848. [Google Scholar] [CrossRef]
- Grigoriadis, D.E.; Smith, E.; Hoare, S.R.J.; Madan, A.; Bozigian, H. Pharmacologic Characterization of Valbenazine (NBI-98854) and Its Metabolites. J. Pharmacol. Exp. Ther. 2017, 361, 454–461. [Google Scholar] [CrossRef]
- Yero, T.; Rey, J.A. Tetrabenazine (Xenazine), An FDA-Approved Treatment Option For Huntington’s Disease–Related Chorea. Pharm. Ther. 2008, 33, 690–694. [Google Scholar]
- Patel, R.S.; Mansuri, Z.; Motiwala, F.; Saeed, H.; Jannareddy, N.; Patel, H.; Zafar, M.K. A systematic review on treatment of tardive dyskinesia with valbenazine and deutetrabenazine. Ther. Adv. Psychopharmacol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Niemann, N.; Jimenez-Shahed, J. Deutetrabenazine in the treatment of tardive dyskinesia. Neurodegener. Dis. Manag. 2019, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Hewitt, M.C. The kinetic isotope effect in the search for deuterated drugs. Drug News Perspect. 2010, 23, 398. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Sung, V. Review of deutetrabenazine: A novel treatment for chorea associated with Huntington’s disease. Drug Des. Dev. Ther. 2018, 12, 313–319. [Google Scholar]
- Kilbourn, M.; DaSilva, J.N.; Frey, K.A.; Koeppe, R.A.; Kuhl, D.E. In Vivo Imaging of Vesicular Monoamine Transporters in Human Brain Using [11C]Tetrabenazine and Positron Emission Tomography. J. Neurochem. 1993, 60, 2315–2318. [Google Scholar] [CrossRef]
- Jankovic, J.; Beach, J. Long-term effects of tetrabenazine in hyperkinetic movement disorders. Neurology 1997, 48, 358–362. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, Y.; Xu, S.; Kung, H.F.; Qiao, H.; Jiang, L.; Zhu, L.; Guo, Q.; Yi, C.; Luo, G.; et al. Decreased Striatal Vesicular Monoamine Transporter Type 2 Correlates With the Nonmotor Symptoms in Parkinson Disease. Clin. Nucl. Med. 2019, 44, 707–713. [Google Scholar] [CrossRef]
- DaSilva, J.N.; Kilbourn, M.R.; Mangner, T.J. Synthesis of [11C] Tetrabenazine, a Vesicular Monoamine Uptake Inhibitor, for PET Imaging Studies. Appl. Radiat. Isot. 1993, 44, 673–676. [Google Scholar] [CrossRef]
- Kung, H.F.; Lieberman, B.P.; Zhuang, Z.-P.; Oya, S.; Kung, M.-P.; Choi, S.R.; Poessl, K.; Blankemeyer, E.; Hou, C.; Skovronsky, D.; et al. In vivo imaging of vesicular monoamine transporter 2 in pancreas using an 18F epoxide derivative of tetrabenazine. Nucl. Med. Boil. 2008, 35, 825–837. [Google Scholar] [CrossRef][Green Version]
- Hefti, F.F.; Kung, H.F.; Kilbourn, M.R.; Carpenter, A.P.; Clark, C.M.; Skovronsky, D.M. 18F-AV-133: A Selective VMAT2-binding Radiopharmaceutical for PET Imaging of Dopaminergic Neurons. PET Clin. 2010, 5, 75–82. [Google Scholar] [CrossRef]
- Brossi, A.; Schnider, O.; Walter, M. Quinolizine Derivatives. U.S. Patent 2,830,993, 15 April 1958. [Google Scholar]
- Orgren, L.R.; Maverick, E.E.; Marvin, C.C. Synthesis of (±)-Tetrabenazine by Visible Light Photoredox Catalysis. J. Org. Chem. 2015, 80, 12635–12640. [Google Scholar] [CrossRef]
- Openshaw, H.T.; Whittaker, N. The Preparation of Quinolizine Derivatives. GB 999095, 21 July 1965. [Google Scholar]
- Duffield, A.J. Desmethyl Derivatives of Tetrabenazine and Pharmaceutical Compositions Thereof. GB2463452A, 17 March 2010. [Google Scholar]
- Burns, N.Z.; Baran, P.S.; Hoffmann, R.W. Redox economy in organic synthesis. Angew. Chem. Int. Ed. 2009, 48, 2854–2867. [Google Scholar] [CrossRef]
- Xuan, J.; Xiao, W.-J. Visible-Light Photoredox Catalysis. Angew. Chem. Int. Ed. 2012, 51, 6828–6838. [Google Scholar] [CrossRef]
- Son, Y.W.; Kwon, T.H.; Lee, J.K.; Pae, A.N.; Lee, J.Y.; Cho, Y.S.; Min, S.-J. A Concise Synthesis of Tetrabenazine: An Intramolecular Aza-Prins-Type Cyclization via Oxidative C–H Activation∥. Org. Lett. 2011, 13, 6500–6503. [Google Scholar] [CrossRef]
- Lee, T.V.; Channon, J.A.; Cregg, C.; Porter, J.R.; Roden, F.S.; Yeoh, H.T.-L. The cerium (III) mediated reaction of trimethylsilylmethyl magnesium chloride with esters and lactones: The efficient synthesis of some functionalised allylsilanes of use in annulation reactions. Tetrahedron 1989, 45, 5877–5886. [Google Scholar] [CrossRef]
- Comins, D.L.; Dehghani, A. Pyridine-Derived Triflating Reagents: An Improved Preparation of Vinyl Triflates from Metallo Enolates. Tetrahedron Lett. 1992, 33, 6299–6302. [Google Scholar] [CrossRef]
- Limmert, M.E.; Roy, A.H.; Hartwig, J.F. Kumada Coupling of Aryl and Vinyl Tosylates Under Mild Conditions. J. Org. Chem. 2005, 70, 9364–9370. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Cheng, X. Enantioselective Total Synthesis of (-)-Zampanolide, a Potent Microtubule-Stabilizing Agent. Org. Lett. 2011, 13, 4108–4111. [Google Scholar] [CrossRef]
- Sasamoto, N.; Dubs, C.; Hamashima, Y.; Sodeoka, M. Pd (II)-Catalyzed Asymmetric Addition of Malonates to Dihydroisoquinolines. J. Am. Chem. Soc. 2006, 128, 14010–14011. [Google Scholar] [CrossRef]
- Krapcho, A.P.; Weimaster, J.F.; Eldridge, J.M.; Jahngen, E.G.E.; Lovey, A.J.; Stephens, W.P. Synthetic applications and mechanism studies of the decarbalkoxylations of geminal diesters and related systems effected in dimethyl sulfoxide by water and/or by water with added salts. J. Org. Chem. 1978, 43, 138–147. [Google Scholar] [CrossRef]
- Jin, H.; Uenishi, J.; Christ, W.J.; Kishi, Y. Catalytic Effect of Nickel (II) Chloride and Palladium (II) Acetate on Chromium (II)-Mediated Coupling Reaction of Iodo Olefins with Aldehydes. J. Am. Chem. Soc. 1986, 108, 5644–5646. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. A Useful 12-I-5 Triacetoxyperiodinane (the Dess-Martin Periodinane) for the Selective Oxidation of Primary or Secondary Alcohols and a Variety of Related 12-I-5 Species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar] [CrossRef]
- Baldwin, J.E. Rules for ring closure. J. Chem. Soc. Chem. Commun. 1976, 734–736. [Google Scholar] [CrossRef]
- Paek, S.-M.; Kim, N.-J.; Shin, D.; Jung, J.-K.; Jung, J.-W.; Chang, D.-J.; Moon, H.; Suh, Y.-G. A Concise Total Synthesis of (+)-Tetrabenazine and (+)-α-Dihydrotetrabenazine. Chem. A Eur. J. 2010, 16, 4623–4628. [Google Scholar] [CrossRef]
- Nakamura, M.; Hirai, A.; Nakamura, E. Enantioselective Allylzincation of Cyclic Aldimines in the Presence of Anionic Bis-oxazoline Ligand. J. Am. Chem. Soc. 1996, 118, 8489–8490. [Google Scholar] [CrossRef]
- In, J.-K.; Lee, M.-S.; Lee, M.-W.; Kwak, J.-H.; Lee, H.; Hong, J.T.; Chung, Y.; Choi, Y.; Jung, J.-K. Stereoselective synthesis of (E)- and (Z)-enol ethers from β-amino aldehydes. Arch. Pharmacal Res. 2007, 30, 695–700. [Google Scholar] [CrossRef]
- Suh, Y.-G.; Lim, C.; Sim, J.; Lee, J.K.; Surh, Y.-J.; Paek, S.-M. Construction of the Azacyclic Core of Tabernaemontanine-Related Alkaloids via Tandem Reformatsky–Aza-Claisen Rearrangement. J. Org. Chem. 2017, 82, 1464–1470. [Google Scholar] [CrossRef]
- Song, B.R.; Ha, M.; Kim, D.; Park, C.; Lee, K.W.; Paek, S.-M. Investigation of Grignard Reagent as an Advanced Base for Aza-Claisen Rearrangement. Molecules 2019, 24, 4597. [Google Scholar] [CrossRef] [PubMed]
- Edstrom, E.D. New methodology for the synthesis of functionalized indolizidine and quinolizidine ring systems. J. Am. Chem. Soc. 1991, 113, 6690–6692. [Google Scholar] [CrossRef]
- Johannes, M.; Altmann, K.-H. A Ring-Closing Metathesis-Based Approach to the Synthesis of (+)-Tetrabenazine. Org. Lett. 2012, 14, 3752–3755. [Google Scholar] [CrossRef] [PubMed]
- Crimmins, M.T.; She, J. An Improved Procedure for Asymmetric Aldol Additions with N-Acyl Oxazolidinones, Oxazolidinethiones and Thiazolidinethiones. Synlett 2004, 2004, 1371–1374. [Google Scholar] [CrossRef]
- Espinet, P.; Echavarren, A.M. The Mechanisms of the Stille Reaction. Angew. Chem. Int. Ed. 2004, 43, 4704–4734. [Google Scholar]
- Paek, S.-M. Synthesis of Tetrasubstituted Alkenes via Metathesis. Molecules 2012, 17, 3348–3358. [Google Scholar] [CrossRef]
- Scholl, M.; Trnka, T.M.; Morgan, J.P.; Grubbs, R.H. Increased ring closing metathesis activity of ruthenium-based olefin metathesis catalysts coordinated with imidazolin-2-ylidene ligands. Tetrahedron Lett. 1999, 40, 2247–2250. [Google Scholar] [CrossRef]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar] [CrossRef]
- Reddy, N.S.S.; Reddy, A.S.; Yadav, J.S.; Reddy, B.S. The Stereoselective Total Synthesis of (−)-Dihydrotetrabenazine. Tetrahedron Lett. 2012, 53, 6916–6918. [Google Scholar] [CrossRef]
- Yamato, M.; Hashigaki, K.; Qais, N.; Ishikawa, S. Asymmetric Synthesis of 1-Alkyltetrahydroisoquinolines using Chiral Oxazolo [2, 3-a] Tetrahydroisoquinolines. Tetrahedron 1990, 46, 5909–5920. [Google Scholar] [CrossRef]
- Borg, G.; Cogan, D.A.; Ellman, J.A. One-Pot Asymmetric Synthesis of Tert-Butanesulfinyl-Protected Amines from Ketones by the in Situ Reduction of Tert-Butanesulfinyl Ketimines. Tetrahedron Lett. 1999, 40, 6709–6712. [Google Scholar] [CrossRef]
- Evans, D.A.; Bartroli, J.; Shih, T.L. Enantioselective Aldol Condensations. 2. Erythro-Selective Chiral Aldol Condensations Via Boron Enolates. J. Am. Chem. Soc. 1981, 103, 2127–2129. [Google Scholar] [CrossRef]
- Schwartz, D.; Bruderer, H.; Rieder, J.; Brossi, A. Metabolic Studies of Tetrabenazine, a Psychotropic Drug in Animals and Man. Biochem. Pharmacol. 1966, 15, 645–655. [Google Scholar] [CrossRef]
- Brown, D.; Dyke, S.; Sainsbury, M. 1,2-Dihydroisoquinolines X. Tetrahedron 1969, 25, 101–117. [Google Scholar] [CrossRef]
- Sotomayor, N.; Dominguez, E.; Lete, E. Bischler−Napieralski Cyclization−N/C-Alkylation Sequences for the Construction of Isoquinoline Alkaloids. Synthesis of Protoberberines and Benzo[c]phenanthridines via C-2‘-Functionalized 3-Arylisoquinolines. J. Org. Chem. 1996, 61, 12–4062. [Google Scholar] [CrossRef]
- Jewett, D.M.; Kilbourn, M.R.; Lee, L.C. A simple synthesis of [11C]Dihydrotetrabenazine (DTBZ). Nucl. Med. Boil. 1997, 24, 197–199. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, Y.; Plössl, K.; Lieberman, B.; Liu, J.; Kung, H.F. An improved radiosynthesis of [18F]AV-133: A PET imaging agent for vesicular monoamine transporter 2. Nucl. Med. Boil. 2010, 37, 133–141. [Google Scholar] [CrossRef]
- Canney, D.J.; Guo, Y.; Kung, M.; Kung, H.F. Synthesis and Preliminary Evaluation of an Iodovinyl-tetrabenazine Analog as a Marker for the Vesicular Monoamine Transporter. J. Label. Compd. Radiopharmaceut. 1993, 33, 355–368. [Google Scholar] [CrossRef]
- Cao, L.; Xie, M.; Zhao, C.; Tang, J.; Liu, C.; Xu, Y.; Li, X.; Liu, Y.; Chen, Z. Synthesis and Preliminary Evaluation of 131I-9-Iodovinyl-Tetrabenazine Targeting Vesicular Monoamine Transporter 2. J. Radioanal. Nucl. 2018, 317, 315–323. [Google Scholar] [CrossRef]
- Ray, P.C.; Pawar, Y.D.; Singare, D.T.; Deshpande, T.N.; Singh, G.P. Novel Process for Preparation of Tetrabenazine and Deutetrabenazine. Org. Process. Res. Dev. 2018, 22, 520–526. [Google Scholar] [CrossRef]
- Gano, K.W. Substituted 3-isobutyl-9, 10-dimethoxy-1, 3, 4, 6, 7, 11b-hexahydro-2H-pyrido [2, 1-A] isoquinolin-2-ol Compounds and Methods Relating Thereto. U.S. Patent 8,039,627, 18 October 2011. [Google Scholar]




















© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paek, S.-M. Synthesis of Tetrabenazine and Its Derivatives, Pursuing Efficiency and Selectivity. Molecules 2020, 25, 1175. https://doi.org/10.3390/molecules25051175
Paek S-M. Synthesis of Tetrabenazine and Its Derivatives, Pursuing Efficiency and Selectivity. Molecules. 2020; 25(5):1175. https://doi.org/10.3390/molecules25051175
Chicago/Turabian StylePaek, Seung-Mann. 2020. "Synthesis of Tetrabenazine and Its Derivatives, Pursuing Efficiency and Selectivity" Molecules 25, no. 5: 1175. https://doi.org/10.3390/molecules25051175
APA StylePaek, S.-M. (2020). Synthesis of Tetrabenazine and Its Derivatives, Pursuing Efficiency and Selectivity. Molecules, 25(5), 1175. https://doi.org/10.3390/molecules25051175