
Identifying Novel ATX Inhibitors via Combinatory Virtual Screening Using Crystallography-Derived Pharmacophore Modelling, Docking Study, and QSAR Analysis

Abstract
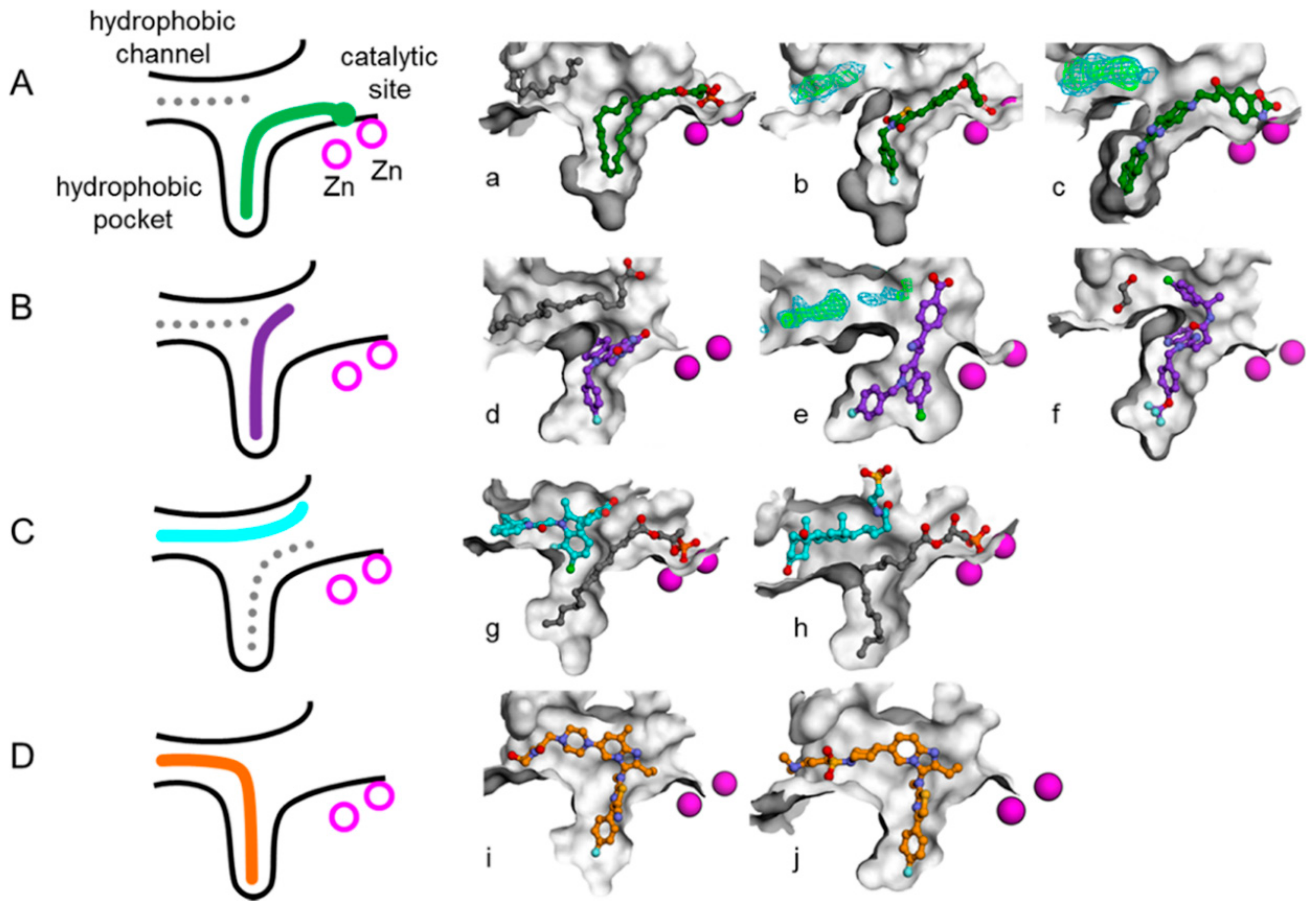
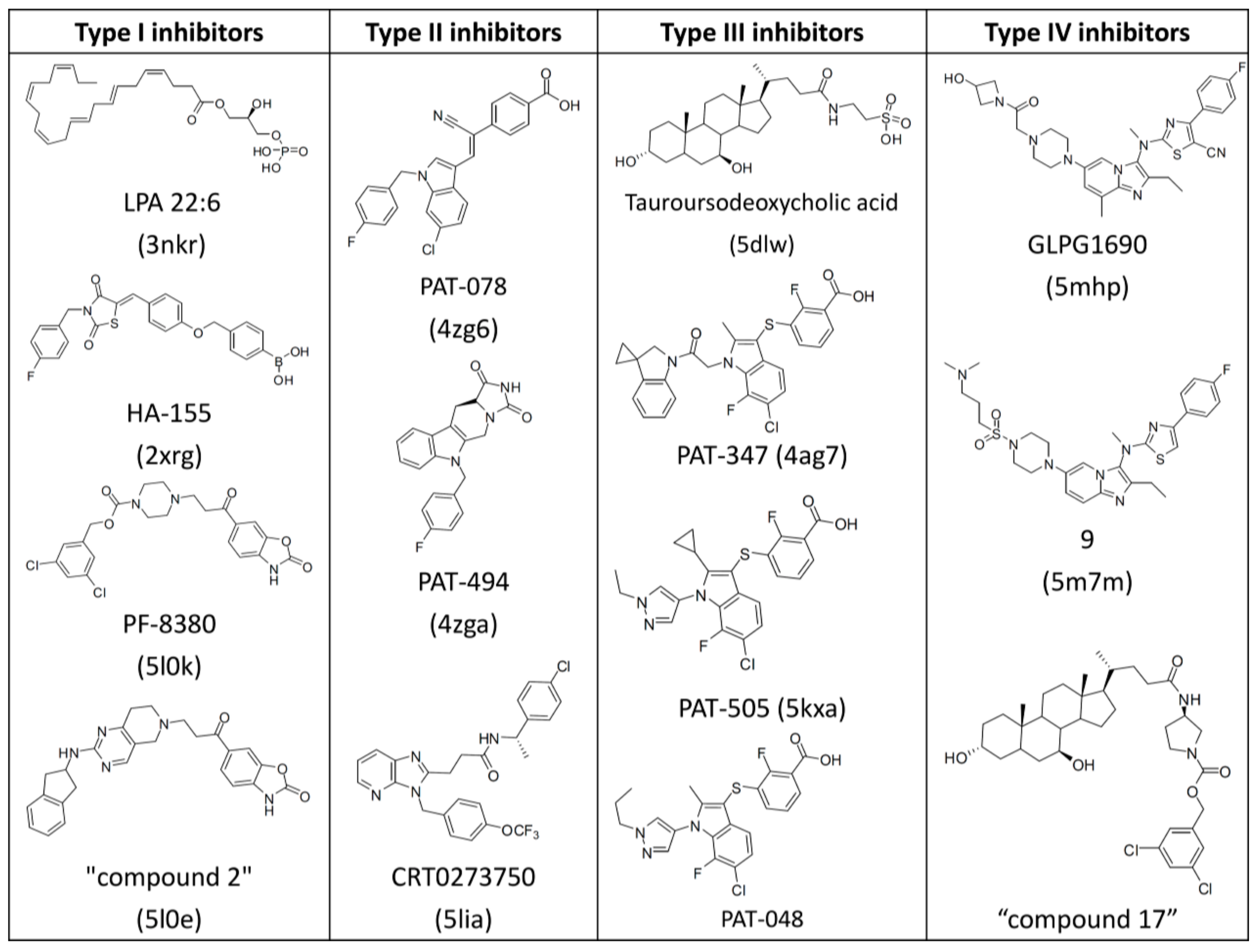
1. Introduction
2. Materials and Methods
2.1. Development of Pharmacophore Models and PB-VS
2.2. Molecular Docking Calculation
2.3. Development of 3D QSAR Model and QSARB-VS
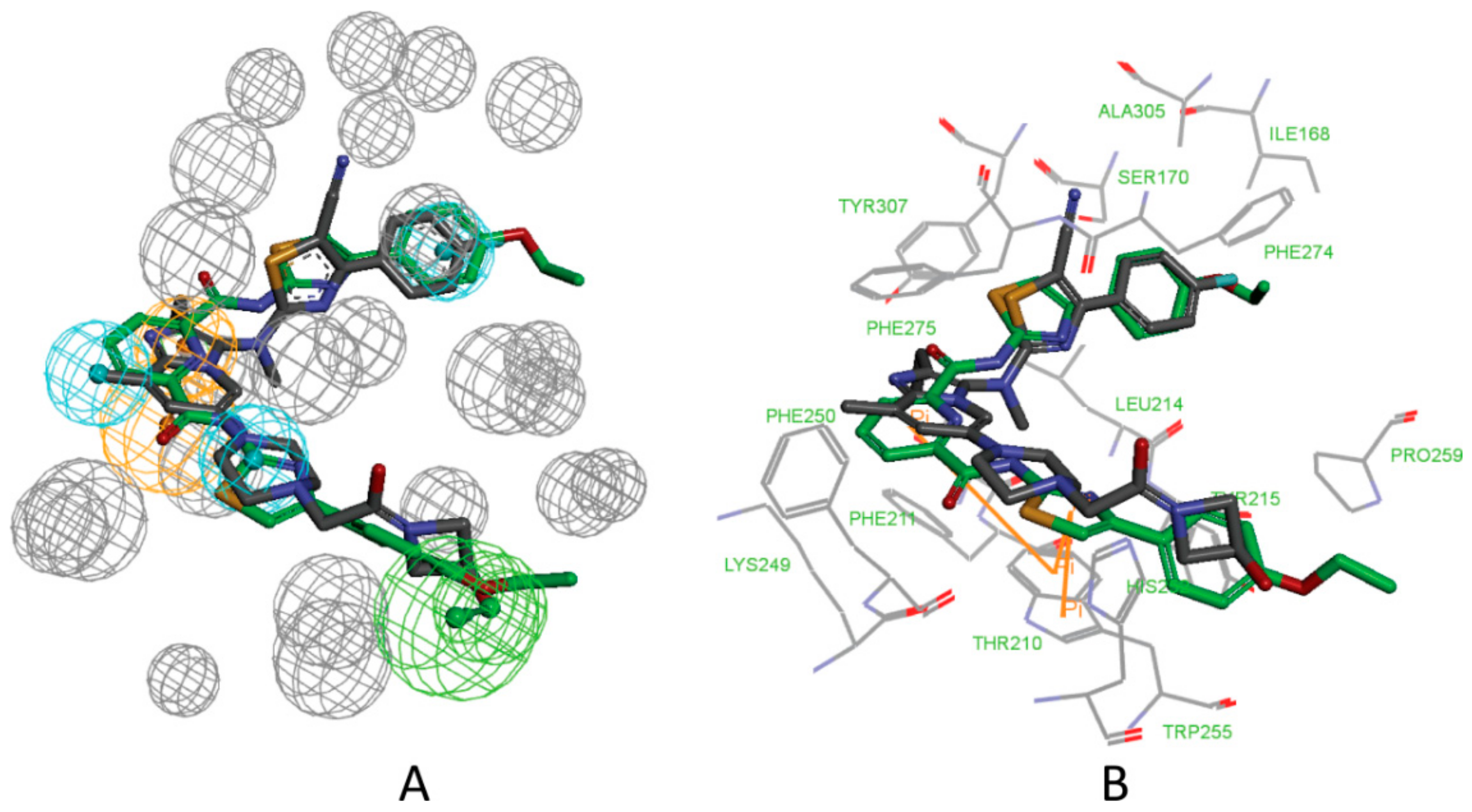
3. Results and Discussion
3.1. Establishment of Pharmacophore Models
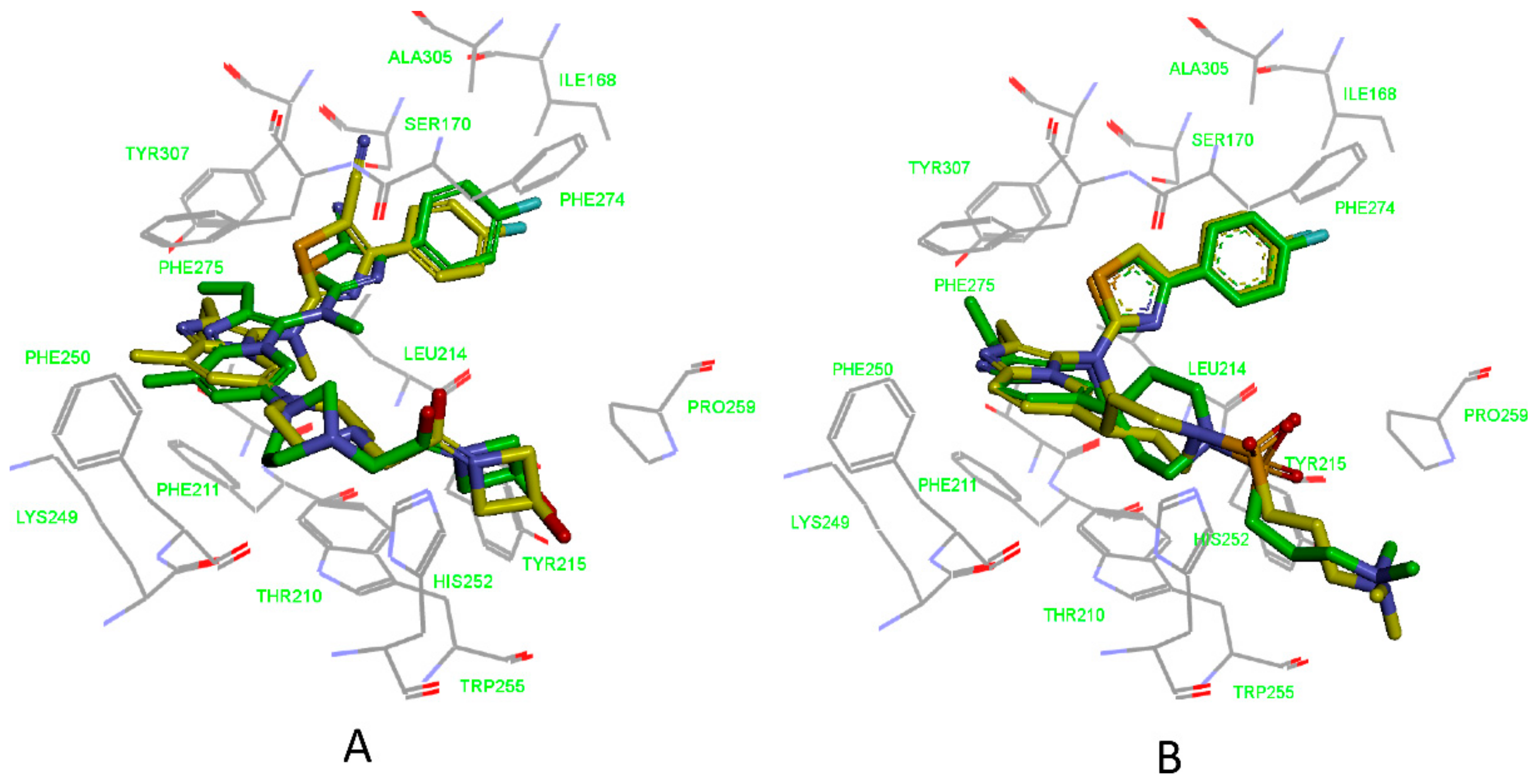
3.2. Validation of Pharmacophore Models
3.3. Determination of Parameters and Scoring Functions
3.4. Development of the 3D QSAR Model
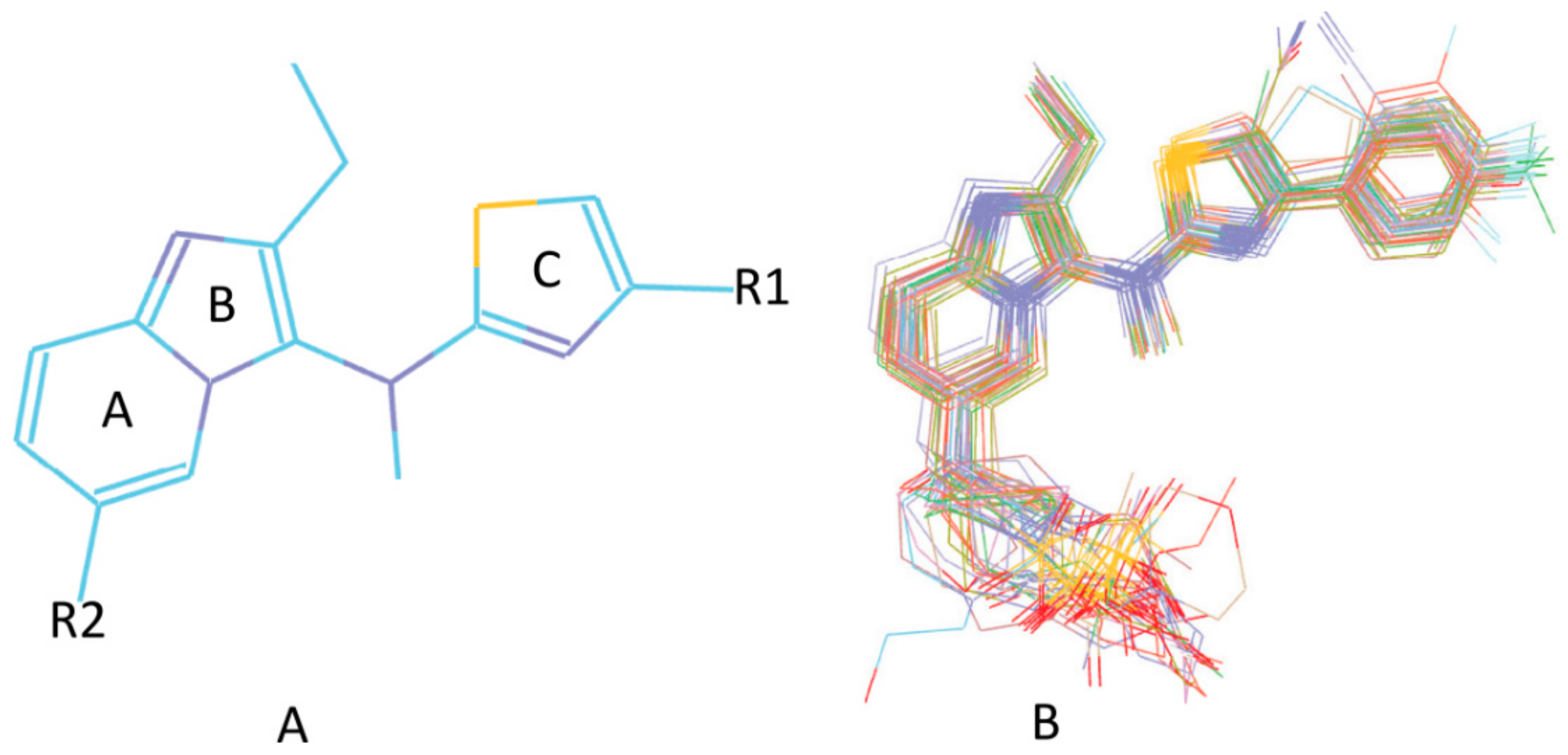
3.5. Searching for New ATX Inhibitors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethical Approval
References
- Perrakis, A.; Moolenaar, W.H. ATX: Structure-function and signaling. J. Lipid Res. 2014, 55, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Nakanaga, K.; Hama, K.; Aoki, J. ATX-An LPA producing enzyme with diverse functions. J. Biochem. 2010, 148, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Barbayianni, E.; Kaffe, E.; Aidinis, V.; Kokotos, G. ATX, a secreted lysophospholipase D, as a promising therapeutic target in chronic inflammation and cancer. Prog. Lipid Res. 2015, 58, 76–96. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, S. The ATX-lysophosphatidic acid–lysophosphatidic acid receptor cascade: Proposal of a novel potential therapeutic target for treating glioblastoma multiforme. Lipids Health Dis. 2015, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Lopane, C.; Agosti, P.; Gigante, I.; Sabbà, C.; Mazzocca, A. Implications of the lysophosphatidic acid signaling axis in liver cancer. BBA-Rev. Cancer 2017, 1868, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, R.; Peyruchaud, O. New insights into the ATX/LPA axis in cancer development and metastasis. Exp. Cell Res. 2015, 333, 183–189. [Google Scholar] [CrossRef]
- Thirunavukkarasu, K.; Swearingen, C.A.; Oskins, J.L.; Lin, C.; Bui, H.H.; Jones, S.B.; Pfeifer, L.A.; Norman, B.H.; Mitchell, P.G.; Chambers, M.G. Identification and pharmacological characterization of a novel inhibitor of ATX in rodent models of joint pain. Osteoarthr. Cartilage 2017, 25, 935–942. [Google Scholar] [CrossRef]
- Inoue, M.; Rashid, M.H.; Fujita, R.; Contos, J.J.A.; Chun, J.; Ueda, H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat. Med. 2004, 10, 712–718. [Google Scholar] [CrossRef]
- Jones, S.B.; Pfeifer, L.A.; Bleisch, T.J.; Beauchamp, T.J.; Durbin, J.D.; Klimkowski, V.J.; Hughes, N.E.; Rito, C.J.; Dao, Y.; Gruber, J.M. Novel ATX Inhibitors for the Treatment of Osteoarthritis Pain: Lead Optimization via Structure-Based Drug Design. ACS Med. Chem. Lett. 2016, 7, 857–861. [Google Scholar] [CrossRef]
- Wunsch, E.; Krawczyk, M.; Milkiewicz, M.; Trottier, J.; Barbier, O.; Neurath, M.F.; Lammert, F.; Kremer, A.E.; Milkiewicz, P. Serum ATX is a Marker of the Severity of Liver Injury and Overall Survival in Patients with Cholestatic Liver Diseases. Sci. Rep. 2016, 6, 30847. [Google Scholar] [CrossRef] [PubMed]
- Kremer, A.E.; Van Dijk, R.; Leckie, P.; Schaap, F.G.; Kuiper, E.M.; Mettang, T.; Reiners, K.S.; Raap, U.; Van Buuren, H.R.; Van Erpecum, K.J.; et al. Serum ATX is increased in pruritus of cholestasis, but not of other origin, and responds to therapeutic interventions. Hepatology 2012, 56, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Castelino, F.V.; Bain, G.; Pace, V.A.; Black, K.E.; George, L.; Probst, C.K.; Goulet, L.; Lafyatis, R.; Tager, A.M. An ATX/lysophosphatidic acid/interleukin-6 amplification loop drives scleroderma fibrosis. Arthritis Rheumatol. 2016, 68, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Desroy, N.; Housseman, C.; Bock, X.; Joncour, A.; Bienvenu, N.; Cherel, L.; Labeguere, V.; Rondet, E.; Peixoto, C.; Grassot, J.M. Discovery of 2-[[2-Ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methylimidazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)thiazole-5-carbonitrile (GLPG1690), a First-in-Class ATX Inhibitor Undergoing Clinical Evaluation for the Treatment of Idiopathic Pulmonary Fibrosis. J. Med. Chem. 2017, 60, 3580–3590. [Google Scholar] [CrossRef] [PubMed]
- Bain, G.; Shannon, K.E.; Huang, F.; Darlington, J.; Goulet, L.; Prodanovich, P.; Ma, G.L.; Santini, A.M.; Stein, A.J.; Lonergan, D.; et al. Selective Inhibition of ATX is Efficacious in Mouse Models of Liver Fibrosis. J. Pharmacol. Exp. Ther. 2017, 363, 1–13. [Google Scholar] [CrossRef]
- Bourgoin, S.G.; Zhao, C. ATX and lysophospholipids in rheumatoid arthritis. Curr. Opin. Investig. Drugs 2010, 11, 515–526. [Google Scholar]
- Park, G.Y.; Lee, Y.G.; Berdyshev, E.; Nyenhuis, S.; Du, J.F.; Gorshkova, I.A.; Li, Y.; Chung, S.; Karpurapu, M.; Deng, J. ATX Production of Lysophosphatidic Acid Mediates Allergic Asthmatic Inflammation. Am. J. Respir. Crit. Care Med. 2013, 188, 928–940. [Google Scholar] [CrossRef]
- Orosa, B.; García, S.; Conde, C. The ATX–lysophosphatidic acid pathway in pathogenesis of rheumatoid arthritis. Eur. J. Pharmacol. 2015, 765, 228–233. [Google Scholar] [CrossRef]
- Hui, D.Y. Intestinal phospholipid and lysophospholipid metabolism in cardiometabolic disease. Curr. Opin. Lipidol. 2016, 27, 507–512. [Google Scholar] [CrossRef]
- Nikolaou, A.; Kokotou, M.G.; Limnios, D.; Psarra, A. ATX inhibitors: A patent review (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 815–829. [Google Scholar] [CrossRef]
- Castagna, D.; Budd, D.C.; Macdonald, S.J.F.; Jamieson, C.; Watson, A.J.B. Development of ATX Inhibitors: An Overview of the Patent and Primary Literature. J. Med. Chem. 2016, 59, 5604–5621. [Google Scholar] [CrossRef] [PubMed]
- Joncour, A.; Desroy, N.; Housseman, C.; Bock, X.; Bienvenu, N. Discovery, Structure-Activity Relationship and BindingMode of Imidazo[1,2-a]pyridine Series of ATX Inhibitors. J. Med. Chem. 2017, 60, 7371–7392. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.X.; Li, C.P.; Zhou, X.L.; Cao, X.S.; Xie, Y. In silico approaches to identify novel myeloid cell leukemia-1 (Mcl-1) inhibitors for treatment of cancer. J. Biomol. Struct. Dyn. 2018, 36, 2424–2435. [Google Scholar] [CrossRef] [PubMed]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef]
- Yu, M.M.; Yang, H.; Wu, K.H.; Ji, Y.; Ju, L.L.; Lu, X.Y. Novel pyrazoline derivatives asbi-inhibitor of COX-2 and B-Raf in treating cervical carcinoma. Bioorgan. Med. Chem. 2014, 22, 4109–4118. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, P.; Zhang, T.G.; Kang, Y.L.; Zhao, X.; Li, Y.Y.; He, J.H.; Zhang, J. In silico prediction of drug-induced myelotoxicity by using Naïve Bayes method. Mol. Divers. 2015, 19, 945–953. [Google Scholar] [CrossRef]
- Ren, J.X.; Zhang, R.T.; Zhang, H.; Cao, X.S.; Liu, L.K.; Xie, Y. Identification of novel VP35 inhibitors: Virtual screening driven new scaffolds. Biomed. Pharmacother. 2016, 84, 199–207. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacophore Abstract | |||
|---|---|---|---|
| Pharmacophore Models | Frequency of Properties | Feature Set | Selectivity Score |
| 5M7M 01 | 5 | A1H1H2H3R2 | 1.3840 |
| 5M7M 02 | 4 | A1H1H2R2 | 1.0423 |
| 5M7M 03 | 4 | A1H3R1R2 | 0.95229 |
| 5M7M 04 | 4 | A1H1H2H3 | 0.47716 |
| 5MHP 01 | 6 | A1H1H2H3H4H5 | 3.6354 |
| 5MHP 02 | 6 | A1H1H2H3H4H6 | 3.6354 |
| 5MHP 03 | 6 | A1H1H2H4H6R2 | 2.8903 |
| 5MHP 04 | 6 | A1H1H3H4H6R2 | 2.8903 |
| 5MHP 05 | 5 | A1H1H3H4H5 | 2.4170 |
| 5MHP 06 | 5 | A1H1H2H3H4 | 2.4170 |
| 5MHP 07 | 5 | A1H1H2H4H5 | 2.4170 |
| 5MHP 08 | 5 | A1H1H3H4H6 | 2.4170 |
| 5MHP 09 | 5 | A1H1H2H4H6 | 2.4170 |
| 5MHP 10 | 5 | A1H1H4H6R2 | 2.3270 |
| Verification with Known Inactives/actives | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pharmacophore | TAa | TIb | TPc | TNd | FPe | FNf | SEg | SPh | Qi | ROC |
| 5M7M 01 | 34 | 6396 | 34 | 4737 | 1659 | 0 | 1 | 0.741 | 0.742 | 0.845 |
| 5M7M 02 | 34 | 6396 | 34 | 4108 | 2288 | 0 | 1 | 0.642 | 0.6442 | 0.770 |
| 5M7M 03 | 34 | 6396 | 34 | 4002 | 2394 | 0 | 1 | 0.626 | 0.628 | 0.793 |
| 5M7M 04 | 34 | 6396 | 34 | 3991 | 2405 | 0 | 1 | 0.624 | 0.626 | 0.773 |
| 5MHP 01 | 34 | 6396 | 19 | 6172 | 224 | 15 | 0.559 | 0.965 | 0.963 | 0.766 |
| 5MHP 02 | 34 | 6396 | 26 | 6219 | 177 | 8 | 0.765 | 0.972 | 0.971 | 0.866 |
| 5MHP 03 | 34 | 6396 | 33 | 6093 | 303 | 1 | 0.971 | 0.953 | 0.953 | 0.967 |
| 5MHP 04 | 34 | 6396 | 28 | 6155 | 241 | 6 | 0.824 | 0.962 | 0.962 | 0.894 |
| 5MHP 05 | 34 | 6396 | 25 | 5773 | 623 | 9 | 0.735 | 0.903 | 0.902 | 0.829 |
| 5MHP 06 | 34 | 6396 | 34 | 5634 | 762 | 0 | 1 | 0.881 | 0.881 | 0.969 |
| 5MHP 07 | 34 | 6396 | 34 | 5590 | 806 | 0 | 1 | 0.874 | 0.875 | 0.943 |
| 5MHP 08 | 34 | 6396 | 32 | 5845 | 551 | 2 | 0.941 | 0.914 | 0.914 | 0.927 |
| 5MHP 09 | 34 | 6396 | 34 | 5788 | 608 | 0 | 1 | 0.905 | 0.905 | 0.959 |
| 5MHP 10 | 34 | 6396 | 34 | 5650 | 746 | 0 | 1 | 0.883 | 0.884 | 0.951 |
| Ligand IDa | PDB Entry | RMSD (Å) |
|---|---|---|
| 7HR | 5M7M | 1.8001 |
| 7NB | 5MHP | 0.6007 |
| Compound | Fit Value | Docking Score | Predicted Activity (pIC50) |
|---|---|---|---|
| cpd1 | 2.95497 | 37.0191 | 5.6312 |
| cpd2 | 2.33815 | 43.5524 | 5.63735 |
| cpd3 | 2.08654 | 34.011 | 5.687 |
| cpd4 | 2.00695 | 48.8647 | 5.62577 |
| cpd5 | 3.63318 | 42.5693 | 5.71689 |
| cpd6 | 3.55526 | 34.2583 | 5.75721 |
| cpd7 | 3.14614 | 33.264 | 5.74321 |
| cpd8 | 3.12769 | 40.4997 | 5.68862 |
| cpd9 | 2.53449 | 35.5562 | 5.76814 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, J.-X.; Zhang, R.-T.; Zhang, H. Identifying Novel ATX Inhibitors via Combinatory Virtual Screening Using Crystallography-Derived Pharmacophore Modelling, Docking Study, and QSAR Analysis. Molecules 2020, 25, 1107. https://doi.org/10.3390/molecules25051107
Ren J-X, Zhang R-T, Zhang H. Identifying Novel ATX Inhibitors via Combinatory Virtual Screening Using Crystallography-Derived Pharmacophore Modelling, Docking Study, and QSAR Analysis. Molecules. 2020; 25(5):1107. https://doi.org/10.3390/molecules25051107
Chicago/Turabian StyleRen, Ji-Xia, Rui-Tao Zhang, and Hui Zhang. 2020. "Identifying Novel ATX Inhibitors via Combinatory Virtual Screening Using Crystallography-Derived Pharmacophore Modelling, Docking Study, and QSAR Analysis" Molecules 25, no. 5: 1107. https://doi.org/10.3390/molecules25051107
APA StyleRen, J.-X., Zhang, R.-T., & Zhang, H. (2020). Identifying Novel ATX Inhibitors via Combinatory Virtual Screening Using Crystallography-Derived Pharmacophore Modelling, Docking Study, and QSAR Analysis. Molecules, 25(5), 1107. https://doi.org/10.3390/molecules25051107