
Reduced-Dimensionality Quantum Dynamics Study of the 3Fe(CO)4 + H2 → 1FeH2(CO)4 Spin-inversion Reaction


Abstract
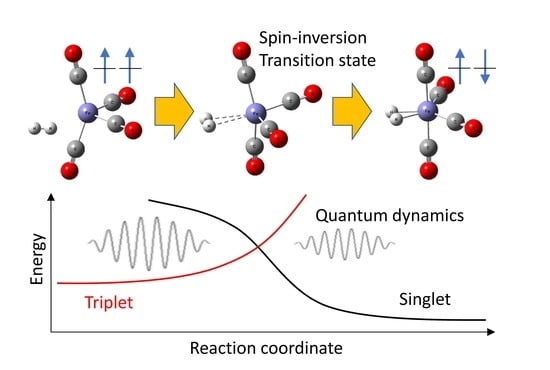
{kind=link}
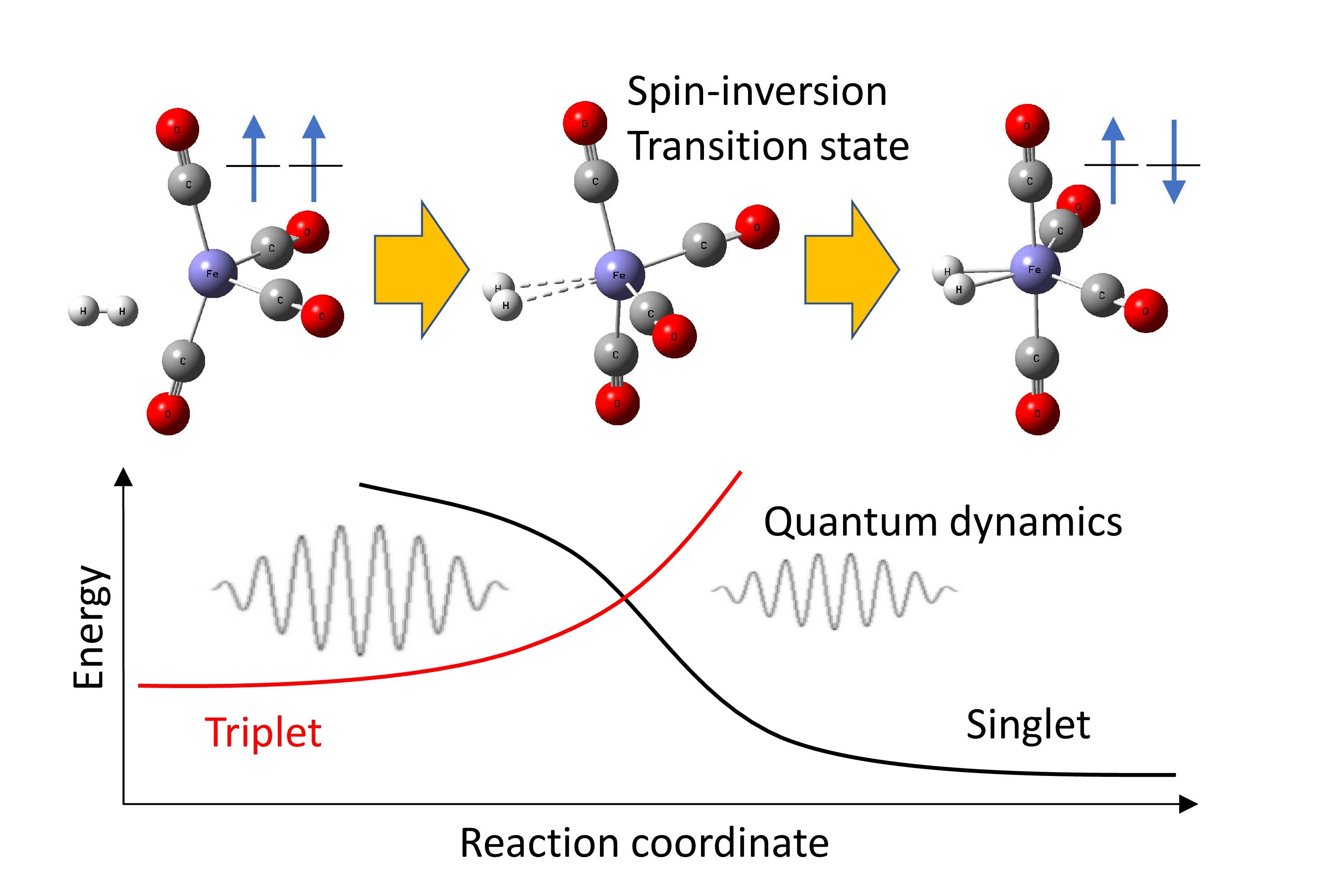
{kind=link}
{kind=link}
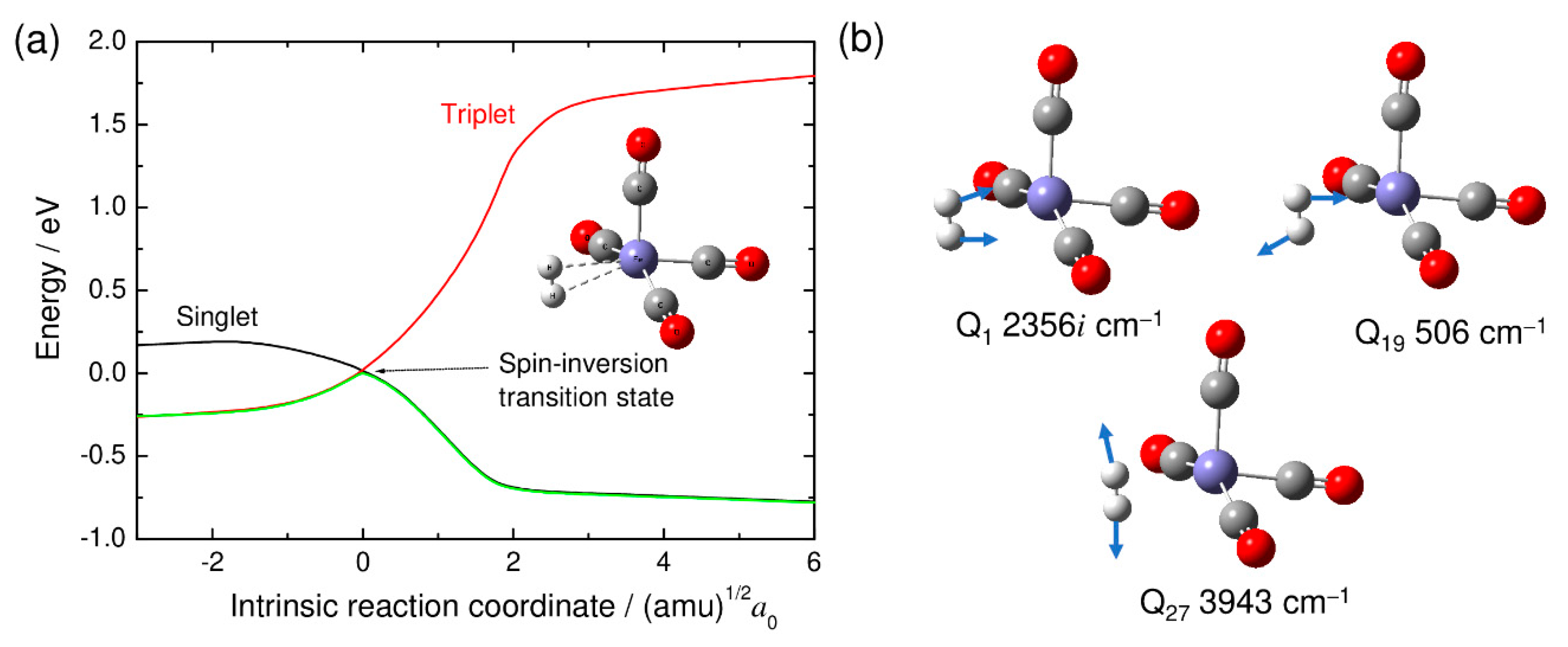
{kind=link}
{kind=link}
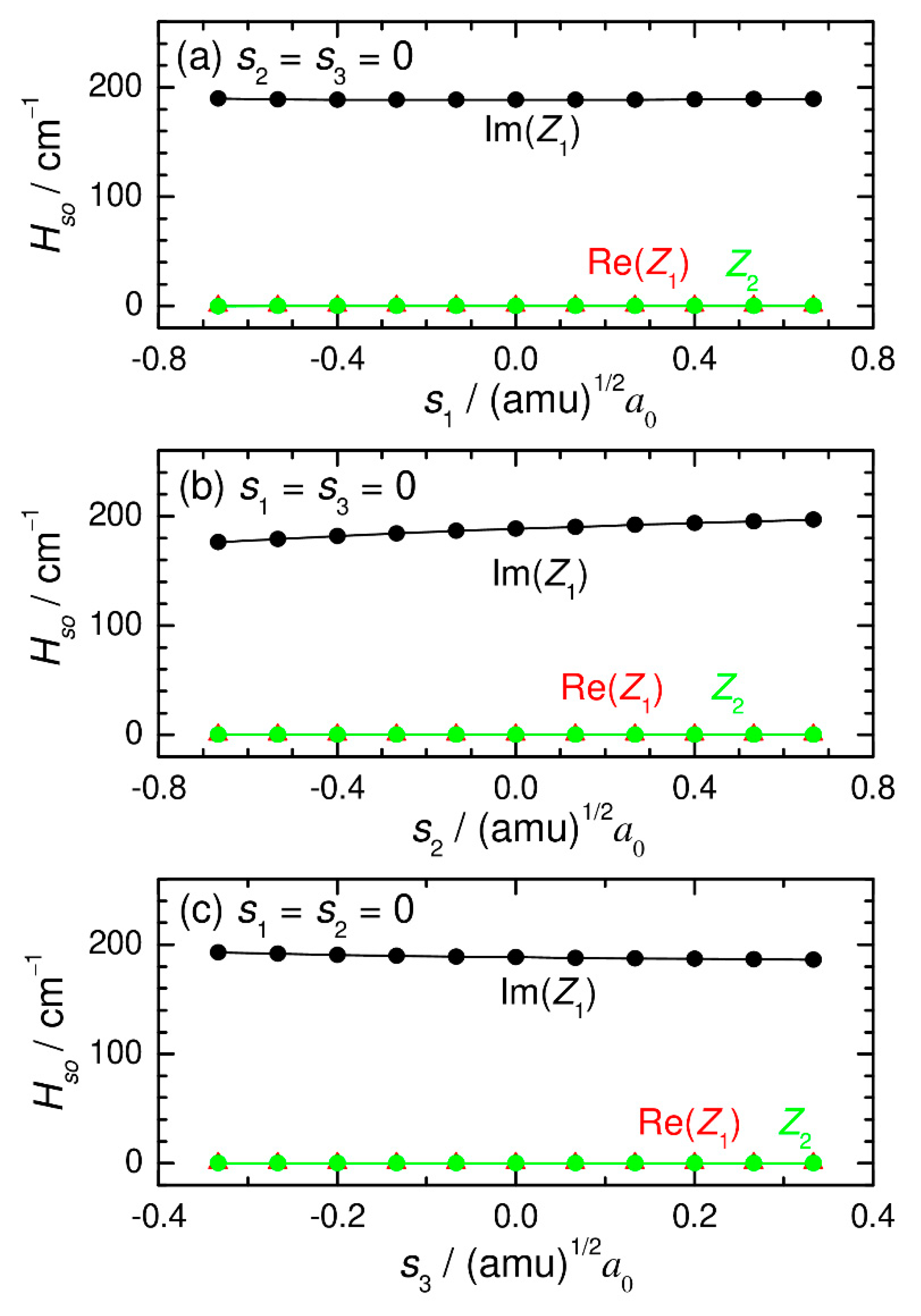
{kind=link}
{kind=link}
1. Introduction
2. Computational Procedure
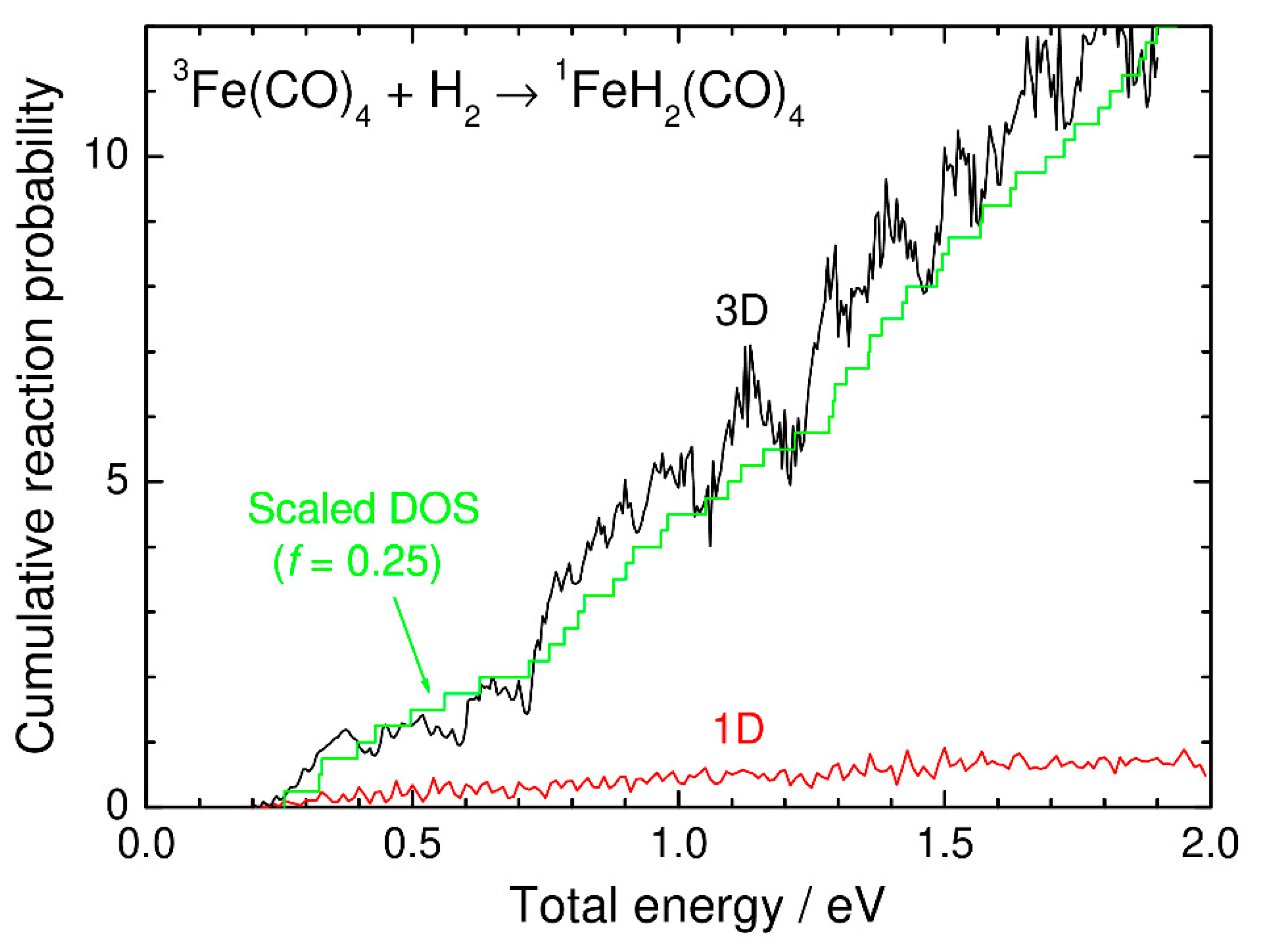
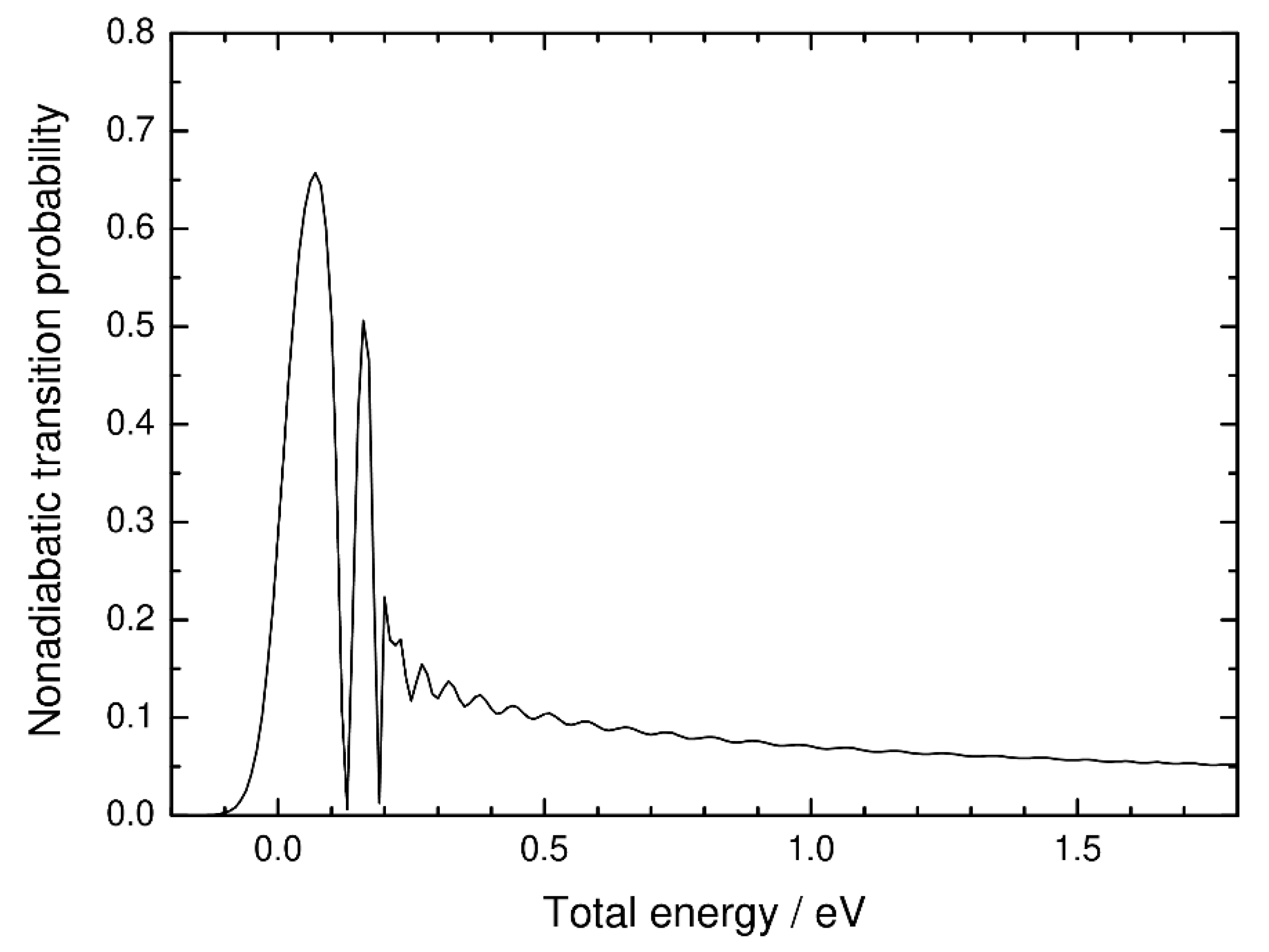
3. Numerical Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Harvey, J.N.; Poli, R.; Smith, K.M. Understanding the Reactivity of Transition Metal Complexes Involving Multiple Spin States. Coord. Chem. Rev. 2003, 238, 347–361. [Google Scholar] [CrossRef]
- Poli, R.; Harvey, J.N. Spin Forbidden Chemical Reactions of Transition Metal Compounds. New Ideas and New Computational Challenges. Chem. Soc. Rev. 2003, 32, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N. Understanding the Kinetics of Spin-Forbidden Reactions. Phys. Chem. Chem. Phys. 2007, 9, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Marian, C.M. Spin-Orbit Coupling and Intersystem Crossing in Molecules. WIREs Comp. Mol. Sci. 2012, 2, 187–203. [Google Scholar] [CrossRef]
- Harvey, J.N. Spin-Forbidden Reactions: Computational Insight into Mechanism and Kinetics. WIREs Comp. Mol. Sci. 2014, 4, 1–14. [Google Scholar] [CrossRef]
- Penfold, T.J.; Gindensperger, E.; Daniel, C.; Marian, C.M. Spin-Vibronic Mechanism for Intersystem Crossing. Chem. Rev. 2018, 118, 6975–7025. [Google Scholar] [CrossRef]
- Lykhin, A.O.; Kaliakin, D.S.; dePolo, G.E.; Kuzubov, A.A.; Varganov, S.A. Nonadiabatic Transition State Theory: Application to Intersystem Crossings in the Active Sites of Metal-Sulfur Proteins. Int. J. Quantum Chem. 2016, 116, 750–761. [Google Scholar] [CrossRef]
- Schröder, D.; Shaik, S.; Schwarz, H. Two-State Reactivity as a New Concept in Organometallic Chemistry. Acc. Chem. Res. 2000, 33, 139–145. [Google Scholar] [CrossRef]
- Hirao, H.; Kumar, D.; Que, L.; Shaik, S. Two-State Reactivity in Alkane Hydroxylation by Non-Heme Iron–Oxo Complexes. J. Am. Chem. Soc. 2006, 128, 8590–8606. [Google Scholar] [CrossRef]
- Kumar, D.; de Visser, S.P.; Shaik, S. Multistate Reactivity in Styrene Epoxidation by Compound I of Cytochrome P450: Mechanisms of Products and Side Products Formation. Chem. Eur. J. 2005, 11, 2825–2835. [Google Scholar] [CrossRef]
- Shaik, S.; Hirao, H.; Kumar, D. Reactivity of High-Valent Iron–Oxo Species in Enzymes and Synthetic Reagents: A Tale of Many States. Acc. Chem. Res. 2007, 40, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Gao, J.; Liu, Y.; Liu, C. Water-Dependent Reaction Pathways: An Essential Factor for the Catalysis in HEPD Enzyme. J. Phys. Chem. B 2012, 116, 11837–11844. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Geng, C.Y.; Shaik, S.; Neese, F. Electronic Structure Analysis of Multistate Reactivity in Transition Metal Catalyzed Reactions: The Case of C–H Bond Activation by Non-Heme Iron(IV)-Oxo Cores. Phys. Chem. Chem. Phys. 2013, 15, 8017–8030. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, T.; Nakatomi, T. Automated Reaction Path Searches for Spin-Forbidden Reactions. J. Comp. Chem. 2018, 39, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Koido, S.; Nakatomi, T.; Watabe, Y.; Takayanagi, T. Automated Reaction Path Search Calculations of Spin-Inversion Mechanisms in the 6,4,2Nb + C2H4 Reaction. Comp. Theo. Chem. 2019, 1115, 31–37. [Google Scholar] [CrossRef]
- Nakatomi, T.; Koido, S.; Watabe, Y.; Takayanagi, T. Spin-Inversion Mechanisms in the Reactions of Transition Metal Cations (Sc+, Ti+, V+, Cr+, Mn+, Fe+, Co+, Ni+, and Cu+) with OCS in the Gas Phase: A Perspective from Automated Reaction Path Search Calculations. Int. J. Quantum Chem. 2019, 119, e25908. [Google Scholar] [CrossRef]
- Takayanagi, T.; Saito, K.; Suzuki, H.; Watabe, Y.; Fujihara, T. Computational Analysis of Two-State Reactivity in β-Hydride Elimination Mechanisms of Fe(II)- and Co(II)-Alkyl Complexes Supported by β-Diketiminate Ligand. Organometallics 2019, 38, 3582–3589. [Google Scholar] [CrossRef]
- Yang, B.; Gagliardi, L.; Truhlar, D.G. Transition States of Spin-Forbidden Reactions. Phys. Chem. Chem. Phys. 2018, 20, 4129–4136. [Google Scholar] [CrossRef]
- Jonas, V.; Thiel, W. Theoretical Study of the Vibrational Spectra of the Transition-Metal Carbonyl Hydrides HM(CO)5 (M=Mn, Re), H2M(CO)4 (M=Fe, Ru, Os), and HM(CO)4 (M=Co, Rh, Ir). J. Chem. Phys. 1996, 105, 3636–3648. [Google Scholar] [CrossRef]
- Wang, W.; Weitz, E. A theoretical Study of the Reaction H2 + Fe(CO)4 ⇆ H2Fe(CO)4. J. Phys. Chem. A 1997, 101, 2358–2363. [Google Scholar] [CrossRef]
- Heitz, M.-C.; Daniel, C. Photodissociation Dynamics of Organometallics: Quantum Simulation for the Dihydrogen Complex H2Fe(CO)4. J. Am. Chem. Soc. 1997, 119, 8269–8275. [Google Scholar] [CrossRef]
- Drouin, B.J.; Kukolich, S.G. Molecular Structure of Tetracarbonyldihydroiron: Microwave Measurements and Density Functional Theory Calculations. J. Am. Chem. Soc. 1998, 120, 6774–6780. [Google Scholar] [CrossRef]
- Harvey, J.N.; Poli, R. Computational Study of the Spin-Forbidden H2 Oxidative Addition to 16-Electron Fe(0) Complexes. Dalton Trans. 2003, 4100–4106. [Google Scholar] [CrossRef]
- Vallet, V.; Bossert, J.; Strich, A.; Daniel, C. The Electronic Spectroscopy of Transition metal di-hydrides H2M(CO)4 (M = Fe,Os): A Theoretical Study Based on CASSCF/MS-CASPT2 and TD-DFT. Phys. Chem. Chem. Phys. 2003, 5, 2948–2953. [Google Scholar] [CrossRef]
- Carreón-Macedo, J.-L.; Harvey, J.N. Computational Study of the Energetics of 3Fe(CO)4, 1Fe(CO)4 and 1Fe(CO)4(L), L = Xe, CH4, H2 and CO. Phys. Chem. Chem. Phys. 2006, 8, 93–100. [Google Scholar] [CrossRef]
- Fedorov, D.A.; Pruitt, S.R.; Keipert, K.; Gordon, M.S.; Varganov, S.A. Ab Initio Multiple Spawning Method for Intersystem Crossing Dynamics: Spin-Forbidden Transitions between 3B1 and 1A1 States of GeH2. J. Phys. Chem. A 2016, 120, 2911–2919. [Google Scholar] [CrossRef]
- Fedorov, D.A.; Lykhin, A.O.; Varganov, S.A. Predicting Intersystem Rates with AIMS-DFT Molecular Dynamics. J. Phys. Chem. A 2018, 122, 3480–3488. [Google Scholar] [CrossRef]
- Zaari, R.R.; Varganov, S.A. Nonadiabatic Transition State Theory and Trajectory Surface Hopping Dynamics: Intersystem Crossing between 3B1 and 1A1 States of SiH2. J. Phys. Chem. A 2015, 119, 1332–1338. [Google Scholar] [CrossRef]
- Zhang, D.H.; Light, J.C. The Cumulative Reaction Probability for the H2 + OH Reaction. J. Chem. Phys. 1997, 106, 551–563. [Google Scholar] [CrossRef]
- Light, J.C.; Zhang, D.H. The Quantum Transition State Wave Packet Method. Faraday Discuss. 1998, 110, 105–118. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Granucci, G.; Persico, M.; Spighi, G. Surface Hopping Trajectory Simulations with Spin-Orbit and Dynamical Couplings. J. Chem. Phys. 2012, 137, 22A501. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, N.; Kalyanaraman, C.; Sathyamurthy, N. Time-Dependent Quantum Mechanical Approach to Reactive Scattering and Related Processes. Phys. Rep. 1997, 280, 79–144. [Google Scholar] [CrossRef]
- Nyman, G.; Yu, H.-G. Quantum Theory of Bimolecular Chemical Reactions. Rep. Prog. Phys. 2000, 63, 1001–1059. [Google Scholar] [CrossRef]
- Werner, H.-J.; Knowles, P.J.; Amos, R.D.; Bernhardsson, A.; Berning, A.; Celani, P.; Cooper, D.L.; Deegan, M.J.O.; Dobbyn, A.J.; Eckert, F. MOLPRO, 2010.1, a Package of Ab Initio Programs; Cardiff University: Cardiff, UK, 2010. [Google Scholar]
- Light, J.C.; Hamilton, I.P.; Lill, J.V. Generalized Discrete Variable Approximation in Quantum Mechanics. J. Chem. Phys. 1985, 82, 1400–1409. [Google Scholar] [CrossRef]
- Xie, T.-X.; Zhang, Y.; Zhao, M.-Y.; Han, K.-L. Calculations of the F + HD Reaction on Three Potential Energy Surfaces. Phys. Chem. Chem. Phys. 2003, 5, 2034–2038. [Google Scholar] [CrossRef]
- Chu, T.-S.; Han, K.-L. Nonadiabatic Time-Dependent Wave Packet Study of the D+ + H2 Reaction System. J. Phys. Chem. A 2005, 109, 2050–2056. [Google Scholar] [CrossRef]
- Baloıtcha, E.; Lasorne, B.; Lauvergnat, D.; Dive, G.; Justum, Y.; Desouter-Lecomte, M. Cumulative Reaction Probability by Constrained Dynamics: H Transfer in HCN, H2CO, and H3CO. J. Chem. Phys. 2002, 117, 727–739. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takayanagi, T.; Watabe, Y.; Miyazaki, T. Reduced-Dimensionality Quantum Dynamics Study of the 3Fe(CO)4 + H2 → 1FeH2(CO)4 Spin-inversion Reaction. Molecules 2020, 25, 882. https://doi.org/10.3390/molecules25040882
Takayanagi T, Watabe Y, Miyazaki T. Reduced-Dimensionality Quantum Dynamics Study of the 3Fe(CO)4 + H2 → 1FeH2(CO)4 Spin-inversion Reaction. Molecules. 2020; 25(4):882. https://doi.org/10.3390/molecules25040882
Chicago/Turabian StyleTakayanagi, Toshiyuki, Yuya Watabe, and Takaaki Miyazaki. 2020. "Reduced-Dimensionality Quantum Dynamics Study of the 3Fe(CO)4 + H2 → 1FeH2(CO)4 Spin-inversion Reaction" Molecules 25, no. 4: 882. https://doi.org/10.3390/molecules25040882
APA StyleTakayanagi, T., Watabe, Y., & Miyazaki, T. (2020). Reduced-Dimensionality Quantum Dynamics Study of the 3Fe(CO)4 + H2 → 1FeH2(CO)4 Spin-inversion Reaction. Molecules, 25(4), 882. https://doi.org/10.3390/molecules25040882