Measuring Sulforaphane and Its Metabolites in Human Plasma: A High Throughput Method

 , and
, and
Abstract
1. Introduction
2. Results
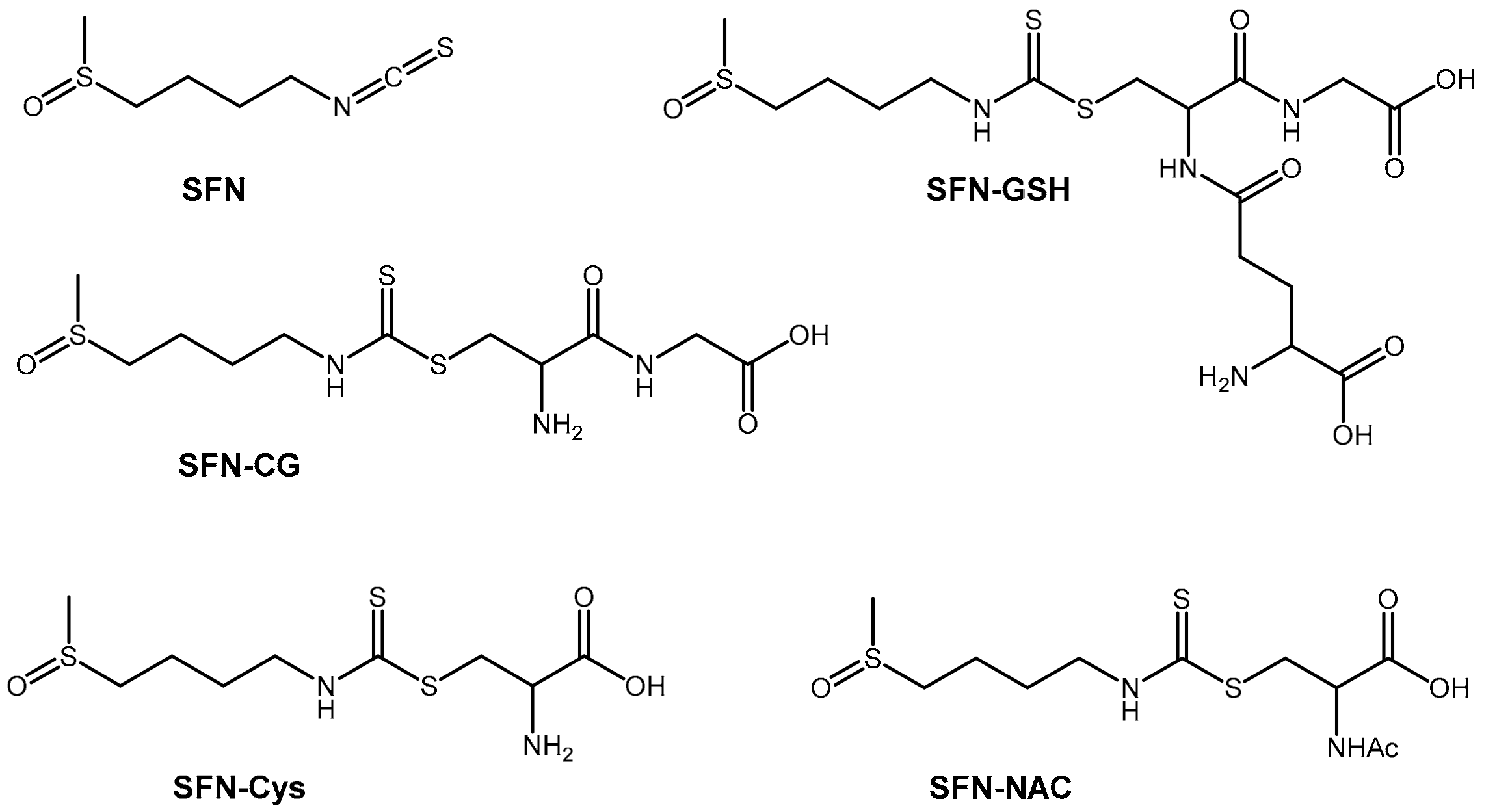
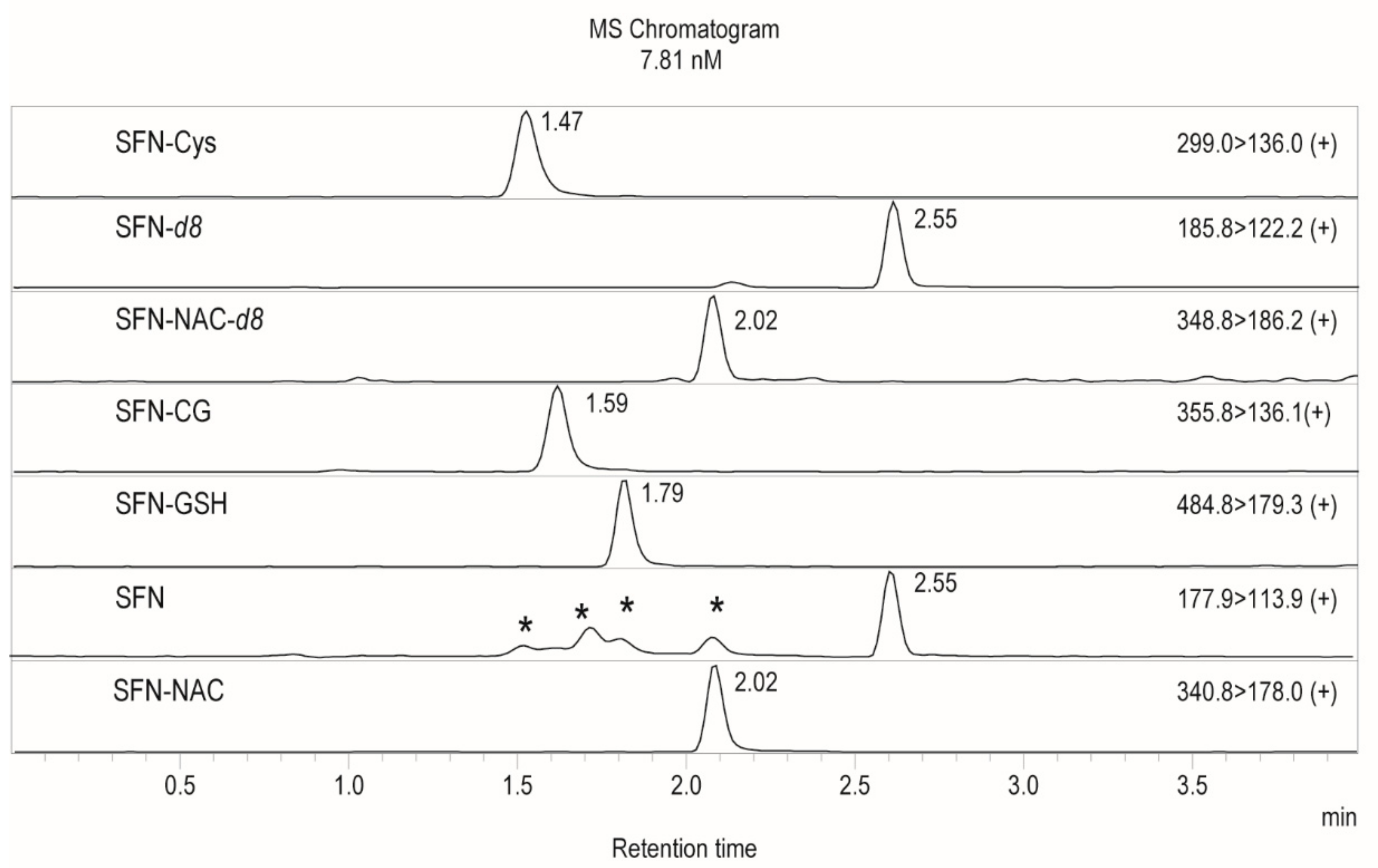
2.1. Method Development
2.2. Sample Preparation
2.3. Accuracy and Linearity
2.4. Limit of Quantification
2.5. Precision
2.6. Recovery
2.7. Sample Stability in Autosampler
2.8. Matrix Effects
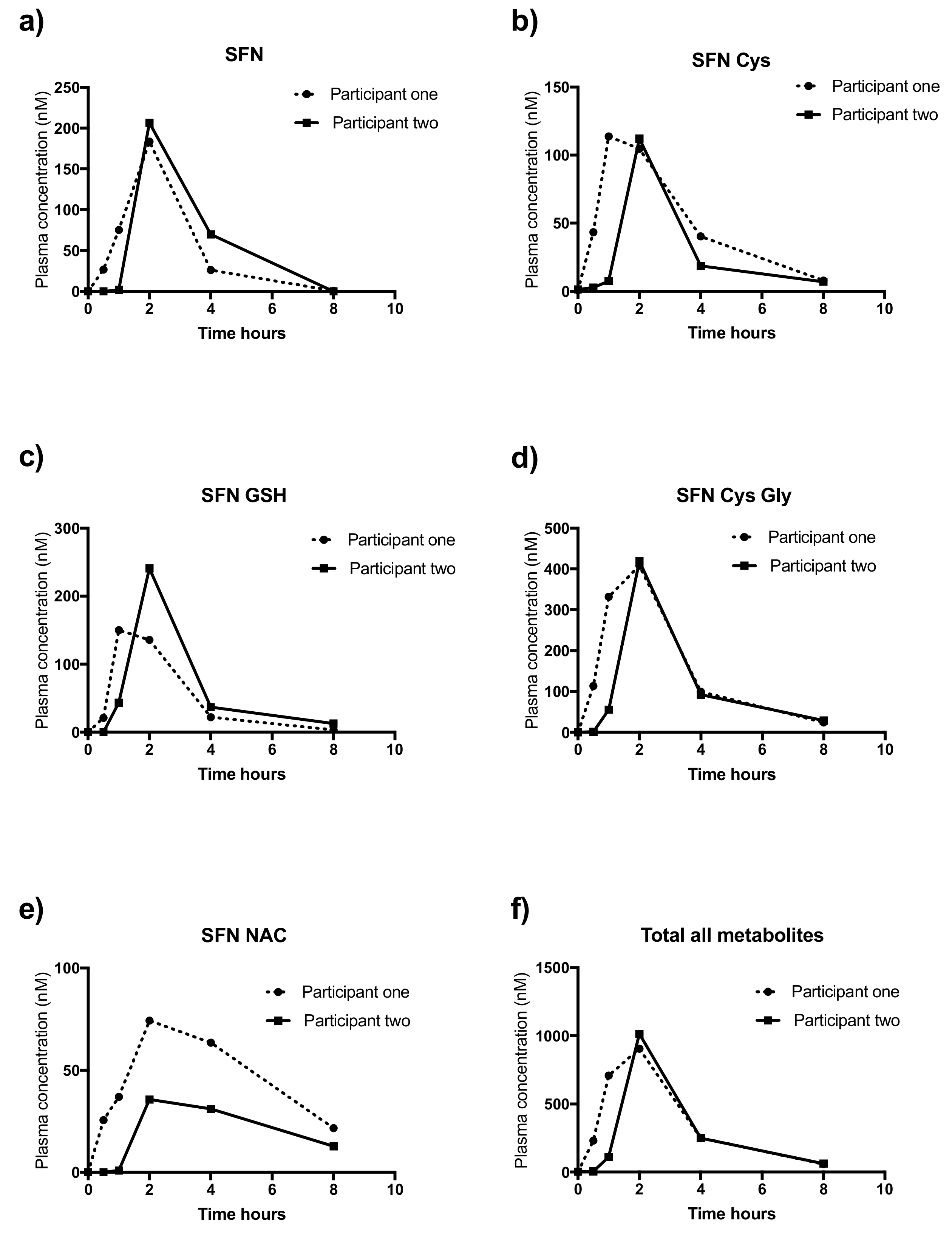
2.9. Application of Study Method to Human Samples
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Application of Study Methods
4.3. Chemical Synthesis of SFN-CG
4.4. Spectroscopic Data of Standard
4.5. Preparation of Stock Solutions
4.6. Preparation of Plasma Samples for LC–MS
4.7. Liquid Chromatography–Mass Spectrometry
4.8. Method Validation
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Ouyang, Y.; Liu, J.; Zhu, M.; Zhao, G.; Bao, W.; Hu, F.B. Fruit and vegetable consumption and mortality from all causes, cardiovascular disease, and cancer: Systematic review and dose-response meta-analysis of prospective cohort studies. BMJ 2014, 349, g4490. [Google Scholar]
- O’Mealey, G.B.; Berry, W.L.; Plafker, S.M. Sulforaphane is a Nrf2-independent inhibitor of mitochondrial fission. Redox. Biol. 2017, 11, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Riboli, E.; Norat, T. Epidemiologic evidence of the protective effect of fruit and vegetables on cancer risk. Am. J. Clin. Nutr. 2003, 78, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Kosmeder, J.W.; Pezzuto, J.M. Biological Effects of Resveratrol. Antioxid. Redox Signal. 2001, 3, 1041–1064. [Google Scholar] [CrossRef]
- Gurusinghe, S.; Cox, A.G.; Rahman, R.; Chan, S.T.; Muljadi, R.; Singh, H.; Leaw, B.; Mockler, J.C.; Murthi, P.; Lim, R.; et al. Resveratrol mitigates trophoblast and endothelial dysfunction partly via activation of nuclear factor erythroid 2-related factor-2. Placenta 2017, 60, 74–85. [Google Scholar] [CrossRef]
- Poston, L.; Briley, A.; Seed, P.; Kelly, F.; Shennan, A. Vitamin C and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): Randomised placebo-controlled trial. Lancet. 2006, 367, 1145–1154. [Google Scholar] [CrossRef]
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Radic. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef]
- Battin, E.E.; Brumaghim, J.L. Antioxidant activity of sulfur and selenium: A review of reactive oxygen species scavenging, glutathione peroxidase, and metal-binding antioxidant mechanisms. Cell Biochem. Biophys. 2009, 55, 1–23. [Google Scholar] [CrossRef]
- Zhang, Y.; Talalay, P.; Chot, C.-G.; Posnert, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Med. Sci. 1992, 89, 2399–2403. [Google Scholar] [CrossRef]
- Riedl, M.A.; Saxon, A.; Diaz-Sanchez, D. Oral sulforaphane increases Phase II antioxidant enzymes in the human upper airway. Clin. Immunol. 2009, 130, 244–251. [Google Scholar] [CrossRef]
- Wu, L.; Noyan Ashraf, M.H.; Facci, M.; Wang, R.; Paterson, P.G.; Ferrie, A.; Juurlink, B.H.J. Dietary approach to attenuate oxidative stress, hypertension, and inflammation in the cardiovascular system. Proc. Natl. Acad. Sci. 2004, 101, 7094–7099. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.Y.; Kang, N.I.; Lee, H.K.; Jang, K.Y.; Park, J.W.; Park, B.H. Sulforaphane protects kidneys against ischemia-reperfusion injury through induction of the Nrf2-dependent phase 2 enzyme. Biochem. Pharm. 2008, 75, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, P.; Kim, J.S. Anti-carcinogenic glucosinolates in cruciferous vegetables and their antagonistic effects on prevention of cancers. Molecules 2018, 23, 2983. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, N.A.; Wilkins, H.M.; Linseman, D.A. Nutraceutical antioxidants as novel neuroprotective agents. Molecules 2010, 15, 7792–7814. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, M. Isothiocyanates from Cruciferous Vegetables: Kinetics, Biomarkers and Effects; Wageningen University: Wageningen, The Netherlands, 2009. [Google Scholar]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or sulforaphane: Is it the source or dose that matters? Molecules 2019, 24, 3593. [Google Scholar] [CrossRef]
- Giudice, A.; Arra, C.; Turco, M.C. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol. Biol. 2010, 647, 37–74. [Google Scholar]
- Zhang, D.D.; Hannink, M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef]
- Yang, S.H.; Yu, L.H.; Li, L.; Guo, Y.; Zhang, Y.; Long, M.; Li, P.; Bin He, J. Protective mechanism of sulforaphane on cadmium-induced sertoli cell injury in mice testis via Nrf2/ARE signaling pathway. Molecules 2018, 23, 1774. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Chu, X.Y.; Jiang, L.H.; Liu, M.Y.; Mei, Z.L.; Zhang, H.Y. Identification of non-electrophilic Nrf2 activators from approved drugs. Molecules 2017, 22, 883. [Google Scholar] [CrossRef]
- Wedel, S.; Manola, M.; Cavinato, M.; Trougakos, I.P.; Jansen, D.P. Targeting protein quality control mechanisms by natural products to promote healthy ageing. Molecules 2018, 23, 1219. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Su, Z.Y.; Kong, A.N.T. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Campas-Baypoli, O.N.; Sánchez-Machado, D.I.; Bueno-Solano, C.; Ramírez-Wong, B.; López-Cervantes, J. HPLC method validation for measurement of sulforaphane level in broccoli by-products. Biomed. Chromatogr. 2010, 24, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Yuan, Q.P.; Dong, H.R.; Liu, Y.M. Determination of sulforaphane in broccoli and cabbage by high-performance liquid chromatography. J. Food Compos. Anal. 2006, 19, 473–476. [Google Scholar] [CrossRef]
- Hauder, J.; Winkler, S.; Bub, A.; Rüfer, C.E.; Pignitter, M.; Somoza, V. LC-MS/MS quantification of sulforaphane and indole-3-carbinol metabolites in human plasma and urine after dietary intake of selenium-fortified broccoli. J. Agric. Food Chem. 2011, 59, 8047–8057. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.; Youngman, L.D.; Palmer, A.; Parish, S.; Peto, R.; Collins, R. Stability of plasma analytes after delayed separation of whole blood: Implications for epidemiological studies. Int. J. Epidemiol. 2003, 32, 125–130. [Google Scholar] [CrossRef]
- Torres-Contreras, A.M.; Nair, V.; Cisneros-Zevallos, L.; Jacobo-Velázquez, D.A. Stability of bioactive compounds in broccoli as affected by cutting styles and storage time. Molecules 2017, 22, 636. [Google Scholar] [CrossRef]
- Atwell, L.L.; Hsu, A.; Wong, C.P.; Stevens, J.F.; Bella, D.; Yu, T.W.; Pereira, C.B.; Löhr, C.V.; Christensen, J.M.; Dashwood, R.H.; et al. Absorption and chemopreventive targets of sulforaphane in humans following consumption of broccoli sprouts or a myrosinase-treated broccoli sprout extract. Mol. Nutr. Food Res. 2015, 59, 424–433. [Google Scholar] [CrossRef]
- Agrawal, S.; Winnik, B.; Buckley, B.; Mi, L.; Chung, F.L.; Cook, T.J. Simultaneous determination of sulforaphane and its major metabolites from biological matrices with liquid chromatography-tandem mass spectroscopy. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 40, 99–107. [Google Scholar] [CrossRef]
- Janobi, A.A.A.; Mithen, R.F.; Gasper, A.V.; Shaw, P.N.; Middleton, R.J.; Ortori, C.A.; Barrett, D.A. Quantitative measurement of sulforaphane, iberin and their mercapturic acid pathway metabolites in human plasma and urine using liquid chromatography-tandem electrospray ionisation mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 844, 223–234. [Google Scholar] [CrossRef]
- Platz, S.; Piberger, A.L.; Budnowski, J.; Herz, C.; Schreiner, M.; Blaut, M.; Hartwig, A.; Lamy, E.; Hanske, L.; Rohn, S. Bioavailability and biotransformation of sulforaphane and erucin metabolites in different biological matrices determined by LC-MS-MS. Anal. Bioanal. Chem. 2015, 407, 1819–1829. [Google Scholar] [CrossRef]
- Dominguez-Perles, R.; Medina, S.; Moreno, D.Á.; García-Viguera, C.; Ferreres, F.; Gil-Izquierdo, Á. A new ultra-rapid UHPLC/MS/MS method for assessing glucoraphanin and sulforaphane bioavailability in human urine. Food Chem. 2014, 143, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Alumkal, J.J.; Slottke, R.; Schwartzman, J.; Cherala, G.; Munar, M.; Graff, J.N.; Beer, T.M.; Ryan, C.W.; Koop, D.R.; Gibbs, A.; et al. A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Invest. New Drugs. 2015, 32, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lin, W.; Shen, G.; Khor, T.O.; Nomeir, A.A.; Kong, A.N. Development and validation of an LC-MS-MS method for the simultaneous determination of sulforaphane and its metabolites in rat plasma and its application in pharmacokinetic studies. J. Chromatogr. Sci. 2011, 49, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Franklin, S.J.; Dickinson, S.E.; Karlage, K.L.; Bowden, G.T.; Myrdal, P.B. Stability of sulforaphane for topical formulation. Drug Dev. Ind. Pharm. 2014, 40, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.L.; Foltz, H.R.; Meng, M.; Bennett, P. Ionization enhancement in atmospheric pressure chemical ionization and suppression in electrospray ionization between target drugs and stable-isotope-labeled internal standards in quantitative liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2815–2821. [Google Scholar] [PubMed]
- Kassahun, K.; Davis, M.; Hu, P.; Martin, B.; Baillie, T. Biotransformation of the naturally occurring isothiocyanate sulforaphane in the rat: Identification of phase I metabolites and glutathione conjugates. Chem. Res. Toxicol. 1997, 10, 1228–1233. [Google Scholar] [CrossRef]
- Matusheski, N.V.; Wallig, M.A.; Juvik, J.A.; Klein, B.P.; Kushad, M.M.; Jeffery, E.H. Preparative HPLC method for the purification of sulforaphane and sulforaphane nitrile from Brassica oleracea. J. Agric. Food Chem. 2001, 49, 1867–1872. [Google Scholar] [CrossRef]
- Grosser, K.; van Dam, N.M. A straightforward method for glucosinolate extraction and analysis with high-pressure liquid chromatography (HPLC). J. Vis. Exp. 2017, e55425. [Google Scholar] [CrossRef]
- Ye, L.; Dinkova-Kostova, A.T.; Wade, K.L.; Zhang, Y.; Shapiro, T.A.; Talalay, P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: Pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin. Chim. Acta. 2002, 316, 43–53. [Google Scholar] [CrossRef]
- Cramer, J.M.; Jeffery, E.H. Sulforaphane absorption and excretion following ingestion of a semi-purified broccoli powder rich in glucoraphanin and broccoli sprouts in healthy men. Nutr. Cancer. 2011, 63, 196–201. [Google Scholar] [CrossRef]
- Cramer, J.M.; Teran-Garcia, M.; Jeffery, E.H. Enhancing sulforaphane absorption and excretion in healthy men through the combined consumption of fresh broccoli sprouts and a glucoraphanin-rich powder. Br. J. Nutr. 2012, 107, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Egner, P.A.; Kensler, T.W.; Chen, J.G.; Gange, S.J.; Groopman, J.D.; Friesen, M.D. Quantification of sulforaphane mercapturic acid pathway conjugates in human urine by high-performance liquid chromatography and isotope-dilution tandem mass spectrometry. Chem. Res. Toxicol. 2008, 21, 1991–1996. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hamid, M.E. Comparative LC-MS and HPLC analyses of selected antiepileptics and beta-blocking drugs. Il Farm. 2000, 55, 136–145. [Google Scholar] [CrossRef]
- Egner, P.A.; Chen, J.G.; Zarth, A.T.; Ng, D.K.; Wang, J.B.; Kensler, K.H.; Jacobson, L.P.; Muñoz, A.; Johnson, J.L.; Groopman, J.D.; et al. Rapid and sustainable detoxication of airborne pollutants by broccoli sprout beverage: Results of a randomized clinical trial in China. Cancer Prev. Res. 2014, 7, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Wade, K.L.; Wehage, S.L.; Holtzclaw, W.D.; Liu, H.; Talalay, P.; Fuchs, E.; Stephenson, K.K. Stabilized sulforaphane for clinical use: Phytochemical delivery efficiency. Mol. Nutr. Food Res. 2017, 61, 1600766. [Google Scholar] [CrossRef] [PubMed]
- Langston-Cox, A.G.; Marshall, S.A.; Palmer, K.R.; Wallace, E.M. Prolong: A double-blind randomised placebo-controlled trial of broccoli sprout extract in women with early onset preeclampsia. A clinical trial protocol. Bmj Open. 2019, 9, e027493. [Google Scholar] [PubMed]
- National Statement on Ethical Conduct in Human Research. 2007. Available online: https://www.nhmrc.gov.au/research-policy/ethics/national-statement-ethical-conduct-human-research (accessed on 27 June 2019).
- Food Drug Administration. Bioanalytical Method Validation Guidance for Industry; U.S. Department of Health and Human Services: Silver Spring, MD, USA, 2018. [Google Scholar]
Sample Availability: Samples are not available from authors. |




{kind=link}
{kind=link}
{kind=link}
| Target Compound | Accuracy (% bias) | LOQ (nM) | Linear Range (nM) | R2 | ||||
|---|---|---|---|---|---|---|---|---|
| 3.9 nM | 7.8 nM | 11.7 nM | 200 nM | 1000 nM | ||||
| SFN | - | −2.70 | - | −11.8 | 12.2 | 7.8 | 7.8–1000 | 0.9947 |
| SFN-GSH | −5.70 | 11.3 | 2.65 | 2.6 | 0.1 | 3.9 | 3.9–1000 | 0.9944 |
| SFN-CG | −2.30 | 8.4 | −3.15 | 3.4 | −0.60 | 3.9 | 3.9–1000 | 0.9991 |
| SFN-Cys | 3.55 | 10.3 | 2.65 | 14.8 | 7.90 | 3.9 | 3.9–1000 | 0.9962 |
| SFN-NAC | 1.85 | −4.60 | −1.85 | 11.2 | 3.20 | 3.9 | 3.9–5000 | 0.9981 |
| Compound | m/z, Transition | Collision Energy | Retention Time (min) |
|---|---|---|---|
| SFN-Cys | 299.00 > 136.00 | −11 | 1.47 |
| SFN-GSH | 484.80 > 179.30 | −25 | 1.79 |
| SFN | 177.90 > 113.90 | −12 | 2.55 |
| SFN-NAC | 340.80 > 178.00 | −14 | 2.02 |
| SFN-d8 | 185.80 > 122.20 | −10 | 2.55 |
| SFN-CG | 355.80 > 136.10 | −12 | 1.59 |
| Target Compound | Repeatability (n = 6) %RSD | Intermediate Precision (n = 3) %RSD | Recovery (% Difference) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 3.9 nM | 7.8 nM | 11.7 nM | 200 nM | 200 nM | 40 nM | 200 nM | 1000 nM | ||
| SFN | - | 8.61 | - | 0.554 | 11.9 | 10.57 | 14.1 | −1.64 | |
| SFN-GSH | 9.53 | 8.18 | 7.76 | 1.96 | 5.73 | 9.81 | 19.61 | −6.15 | |
| SFN-CG | 3.68 | 4.28 | 3.95 | 1.19 | 5.47 | 15.2 | −10.1 | 0.846 | |
| SFN-Cys | 4.01 | 5.95 | 1.03 | 1.72 | 1.85 | −5.63 | −1.97 | −0.26 | |
| SFN-NAC | 6.66 | 3.38 | 3.12 | 1.25 | 0.656 | −8.92 | −4.67 | −3.12 | |
| Metabolite | Participant One | Participant Two | ||
|---|---|---|---|---|
| AUC | Mean Peak | AUC | Mean Peak | |
| SFN | 424.9 | 183.5 | 520.8 | 206.5 |
| SFN Cys | 401 | 113.8 | 245.5 | 112.2 |
| SFN-GSH | 400 | 150.1 | 530.3 | 240.8 |
| SFN-Cys-Gly | 1264 | 408 | 1007 | 419.2 |
| SFN-NAC | 385.6 | 74.3 | 172.5 | 35.6 |
| Total all metabolites | 2876 | 906.2 | 2476 | 1014 |
| Demographics | Participant One | Participant Two |
|---|---|---|
| Age (yrs) | 23 | 20 |
| BMI (m/kg2) | 24 | 26 |
| Dietary restrictions | Nil | Nil |
| Medication | OCP1 | Nil |
| Co-morbidities | Nil | Nil |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langston-Cox, A.; Anderson, D.; Creek, D.J.; Palmer, K.; Wallace, E.M.; Marshall, S.A. Measuring Sulforaphane and Its Metabolites in Human Plasma: A High Throughput Method. Molecules 2020, 25, 829. https://doi.org/10.3390/molecules25040829
Langston-Cox A, Anderson D, Creek DJ, Palmer K, Wallace EM, Marshall SA. Measuring Sulforaphane and Its Metabolites in Human Plasma: A High Throughput Method. Molecules. 2020; 25(4):829. https://doi.org/10.3390/molecules25040829
Chicago/Turabian StyleLangston-Cox, Annie, Dovile Anderson, Darren J. Creek, Kirsten Palmer, Euan M. Wallace, and Sarah A. Marshall. 2020. "Measuring Sulforaphane and Its Metabolites in Human Plasma: A High Throughput Method" Molecules 25, no. 4: 829. https://doi.org/10.3390/molecules25040829
APA StyleLangston-Cox, A., Anderson, D., Creek, D. J., Palmer, K., Wallace, E. M., & Marshall, S. A. (2020). Measuring Sulforaphane and Its Metabolites in Human Plasma: A High Throughput Method. Molecules, 25(4), 829. https://doi.org/10.3390/molecules25040829