Metronidazole and Secnidazole Carbamates: Synthesis, Antiprotozoal Activity, and Molecular Dynamics Studies

 ,
,  , ,
, ,  , , and
, , and 
Abstract
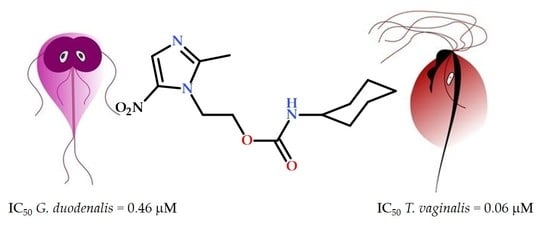
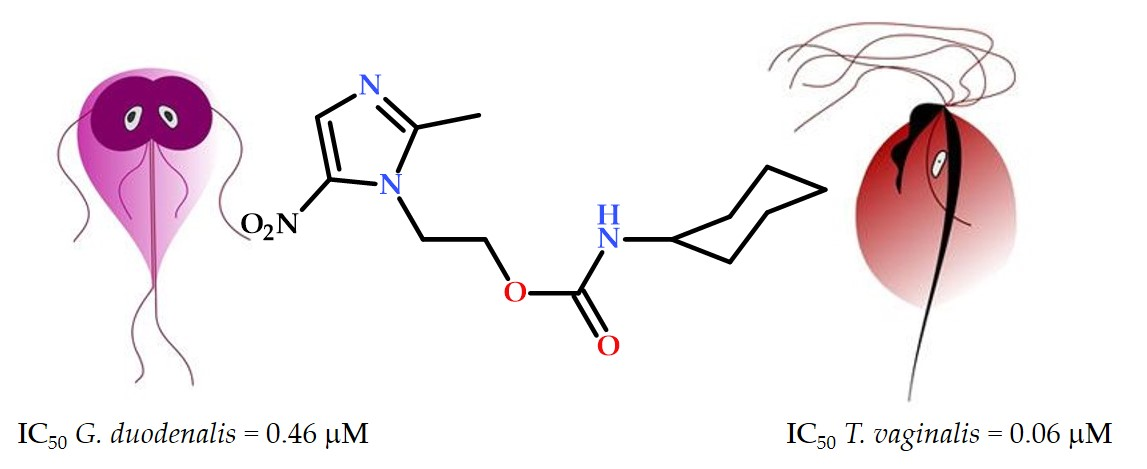
1. Introduction
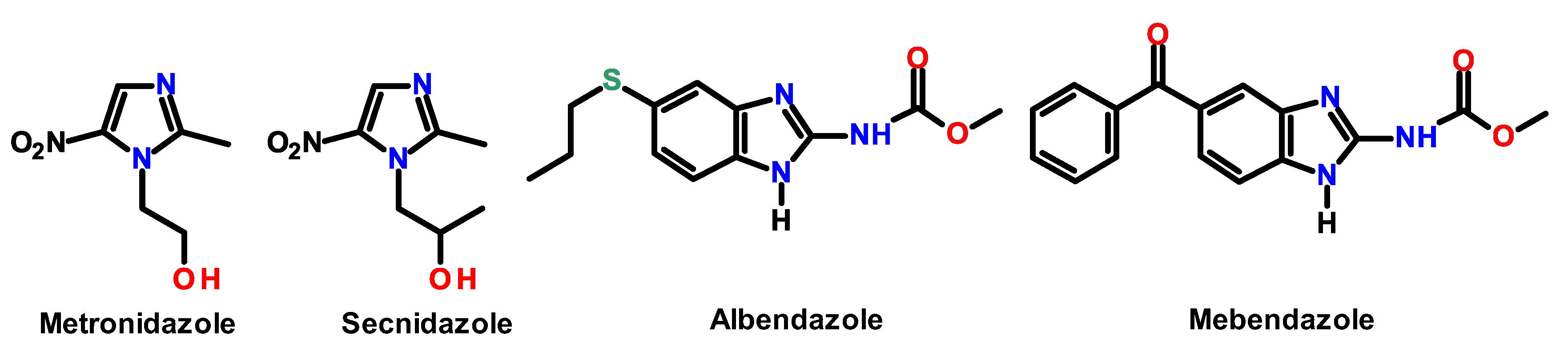
2. Results and Discussion
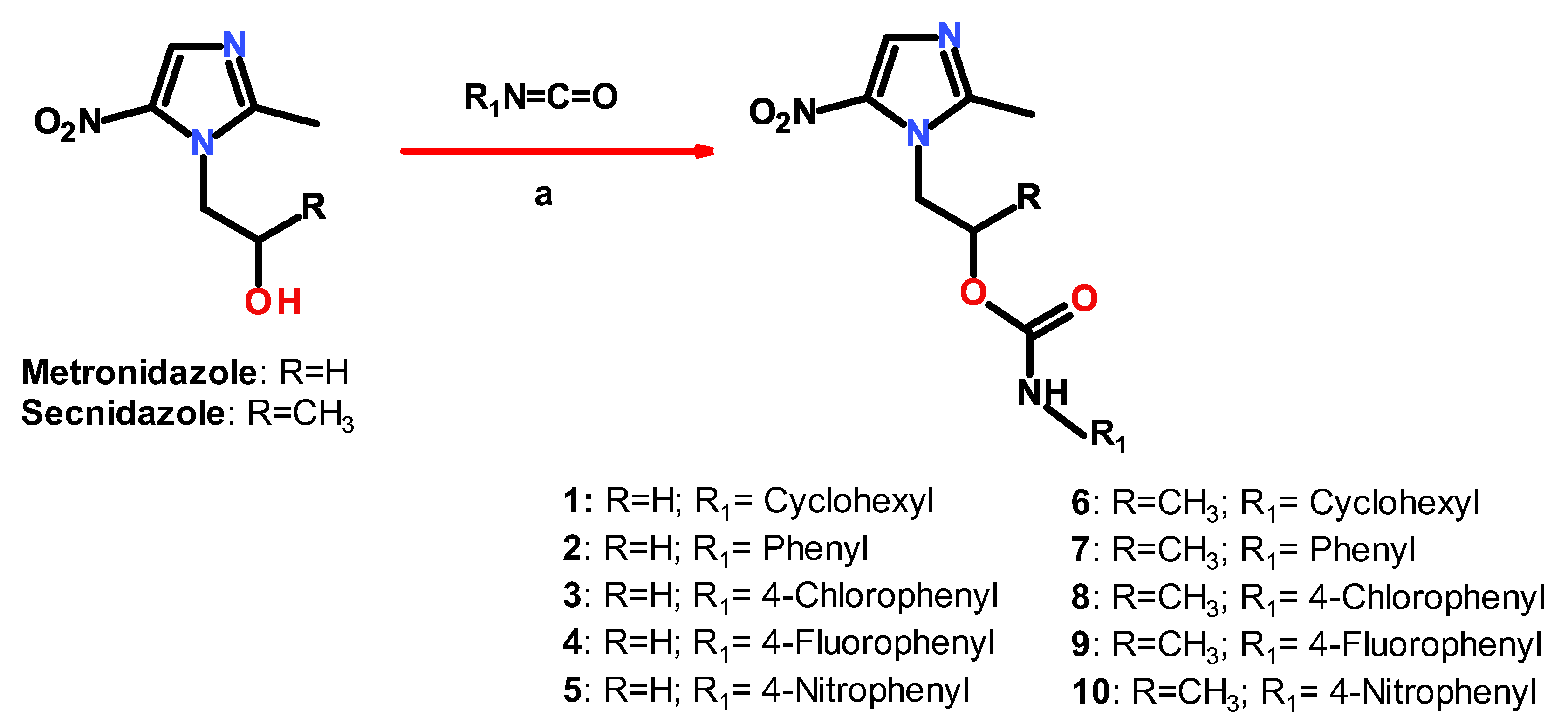
2.1. Chemistry
2.2. In Vitro Antiprotozoal Activity
2.3. In Vitro Cytotoxicity Assay
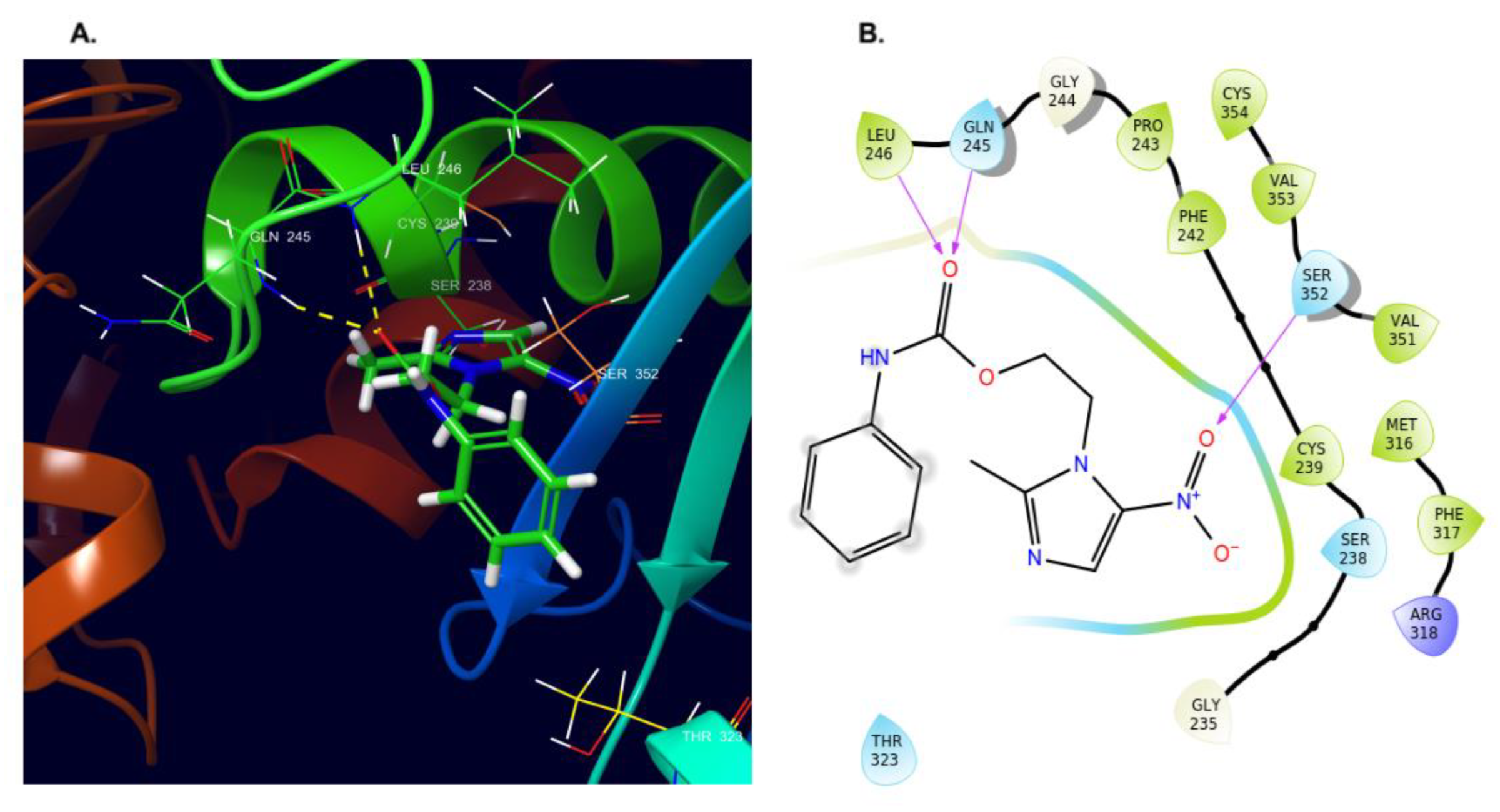
2.4. Molecular Docking and Dynamics Studies
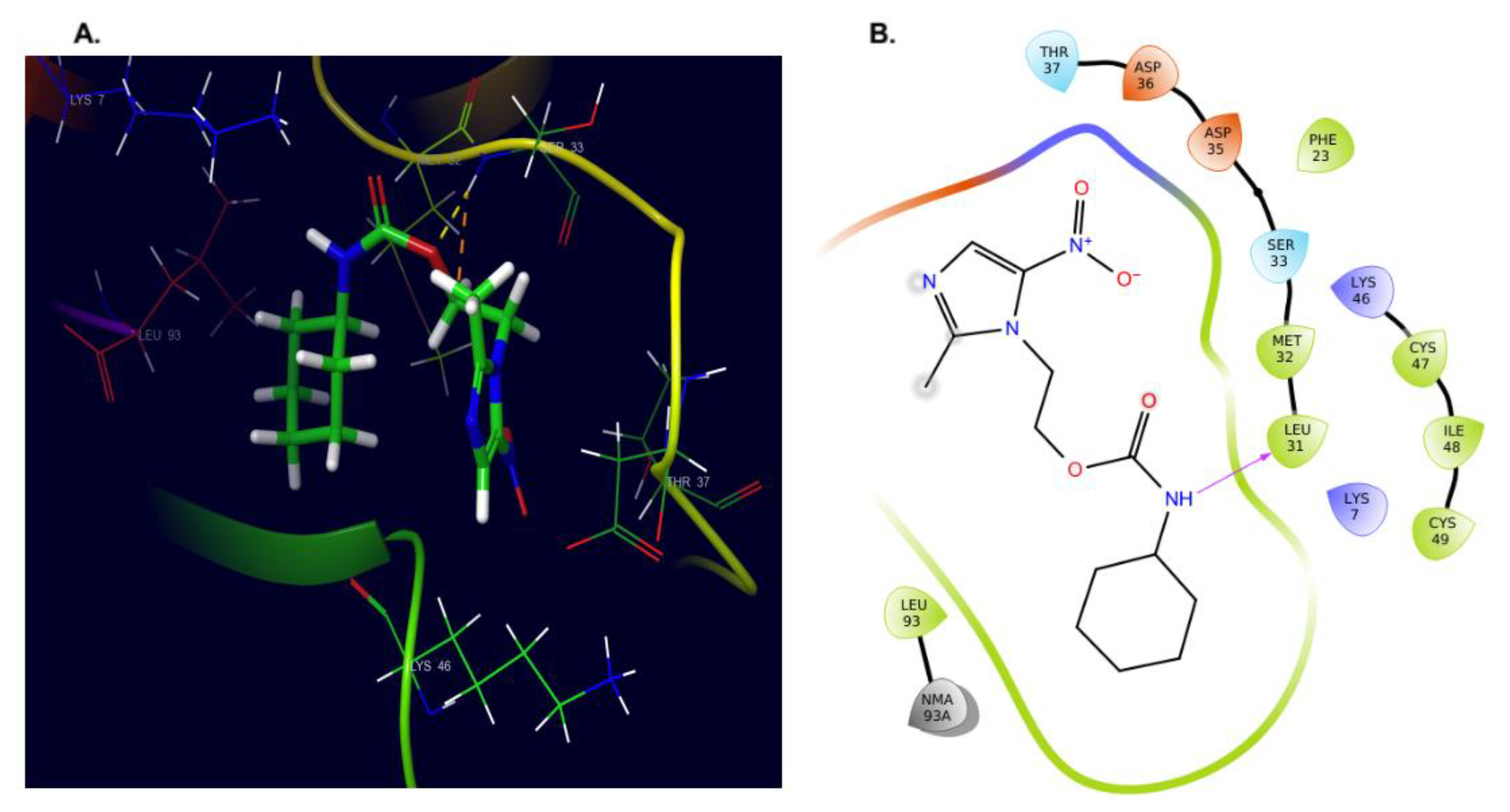
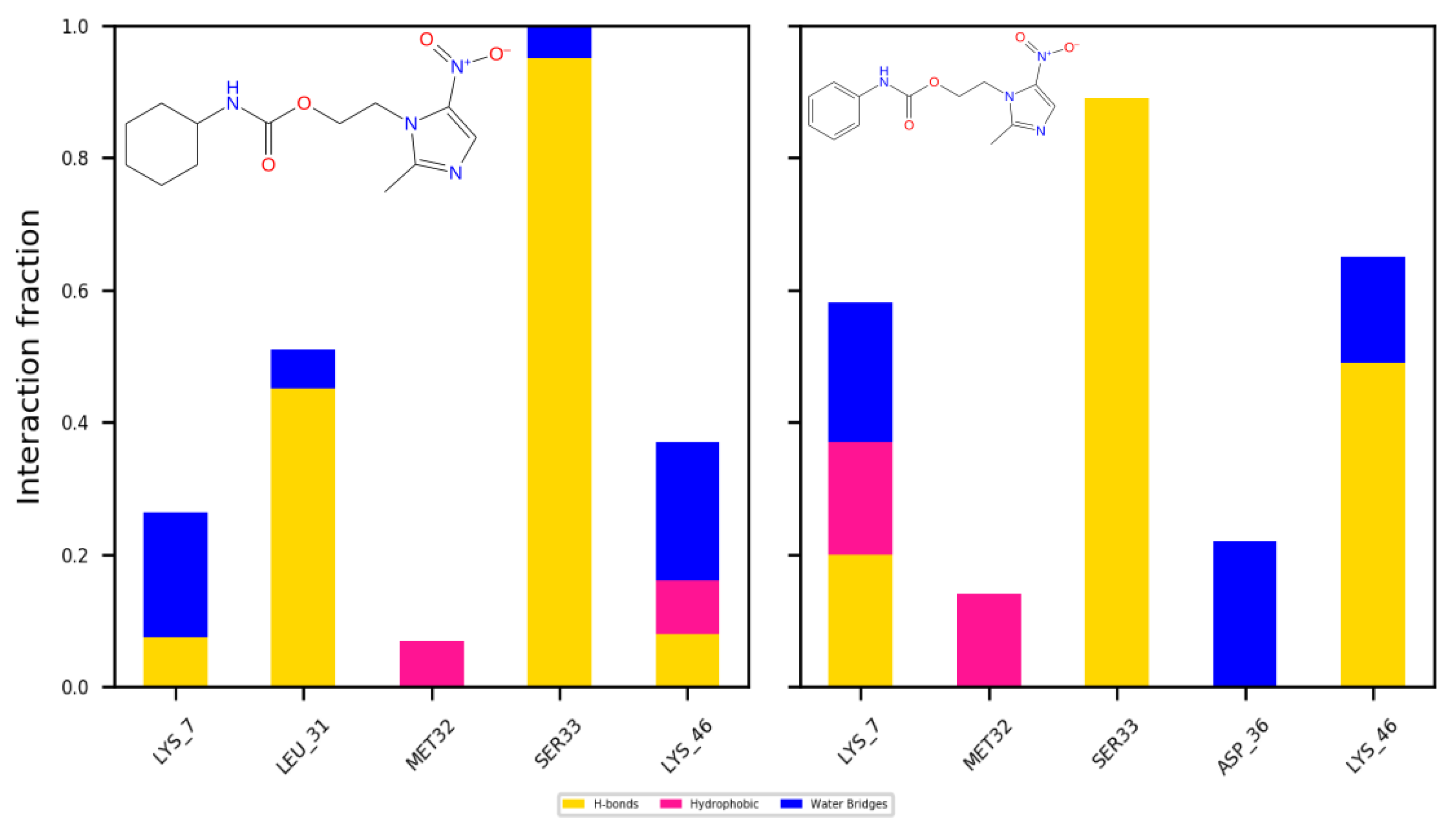
2.4.1. Docking
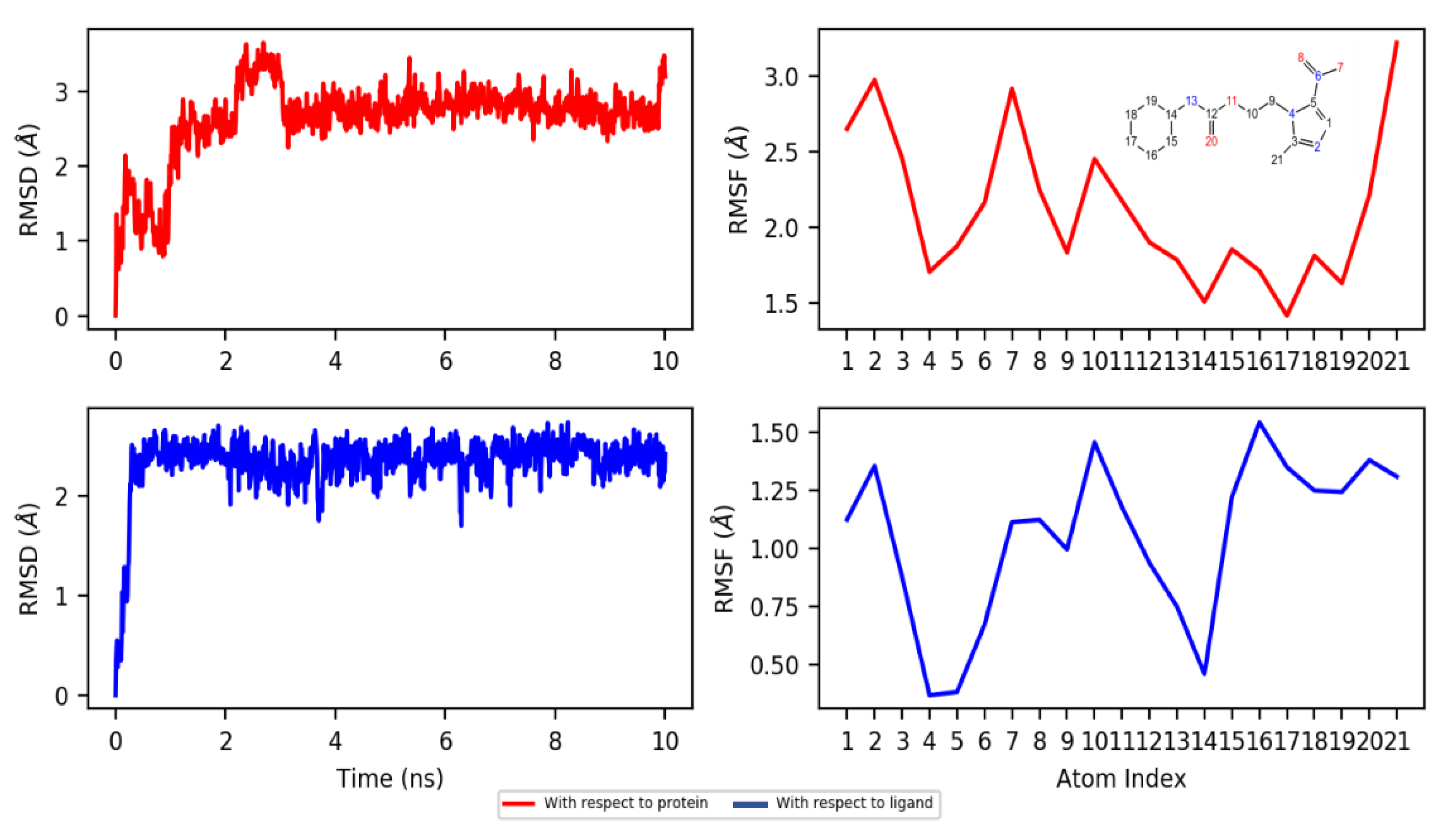
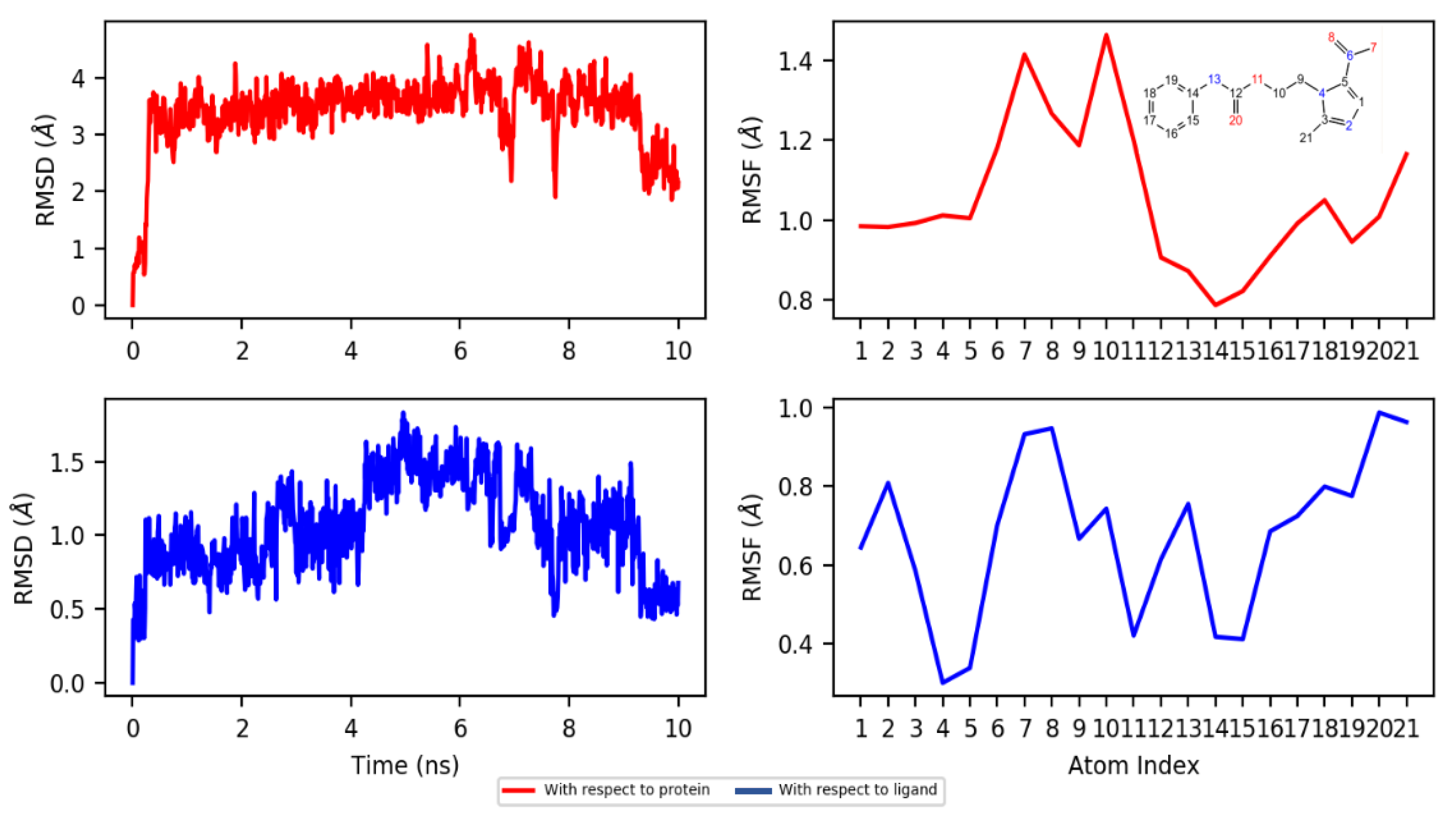
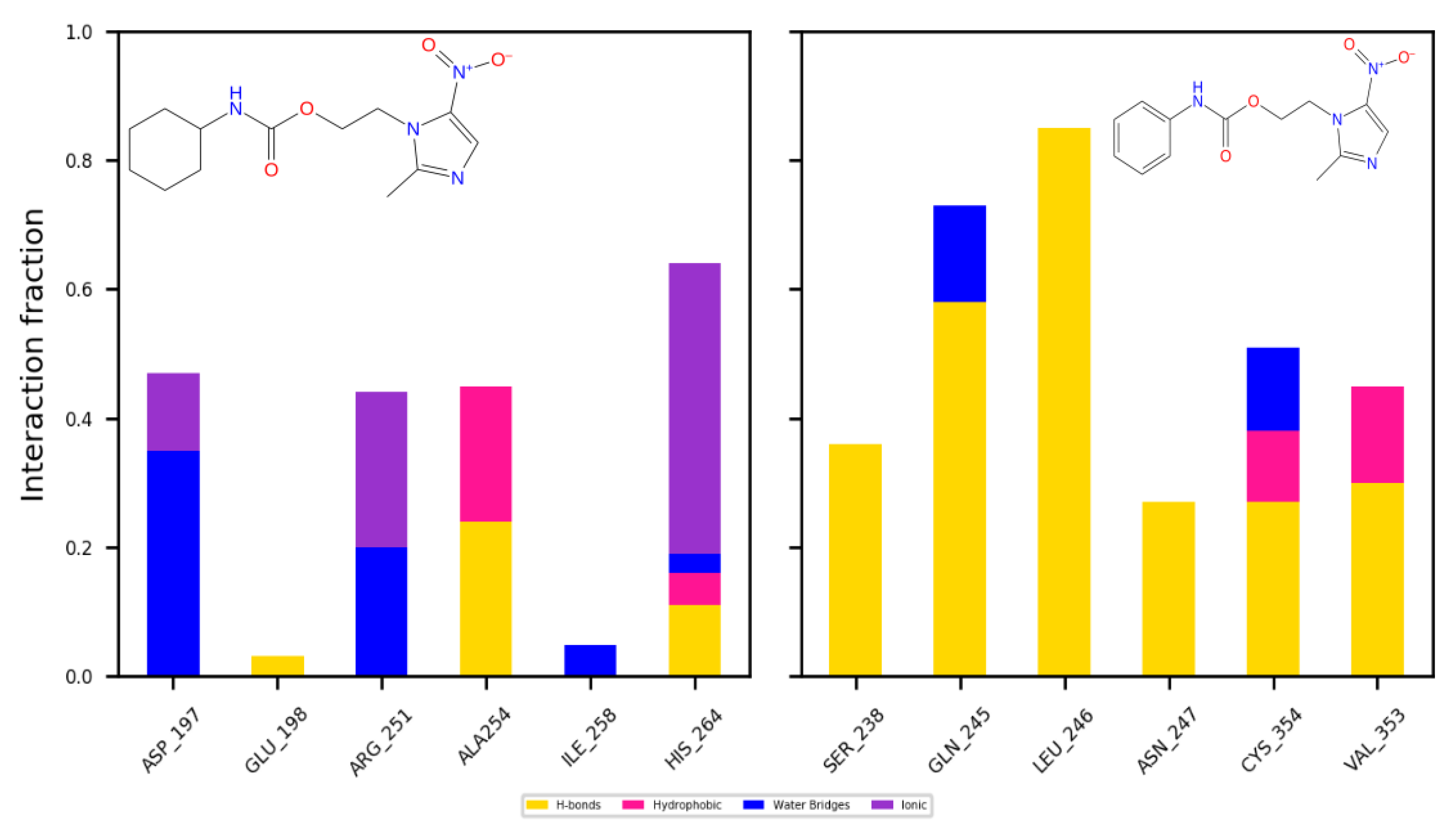
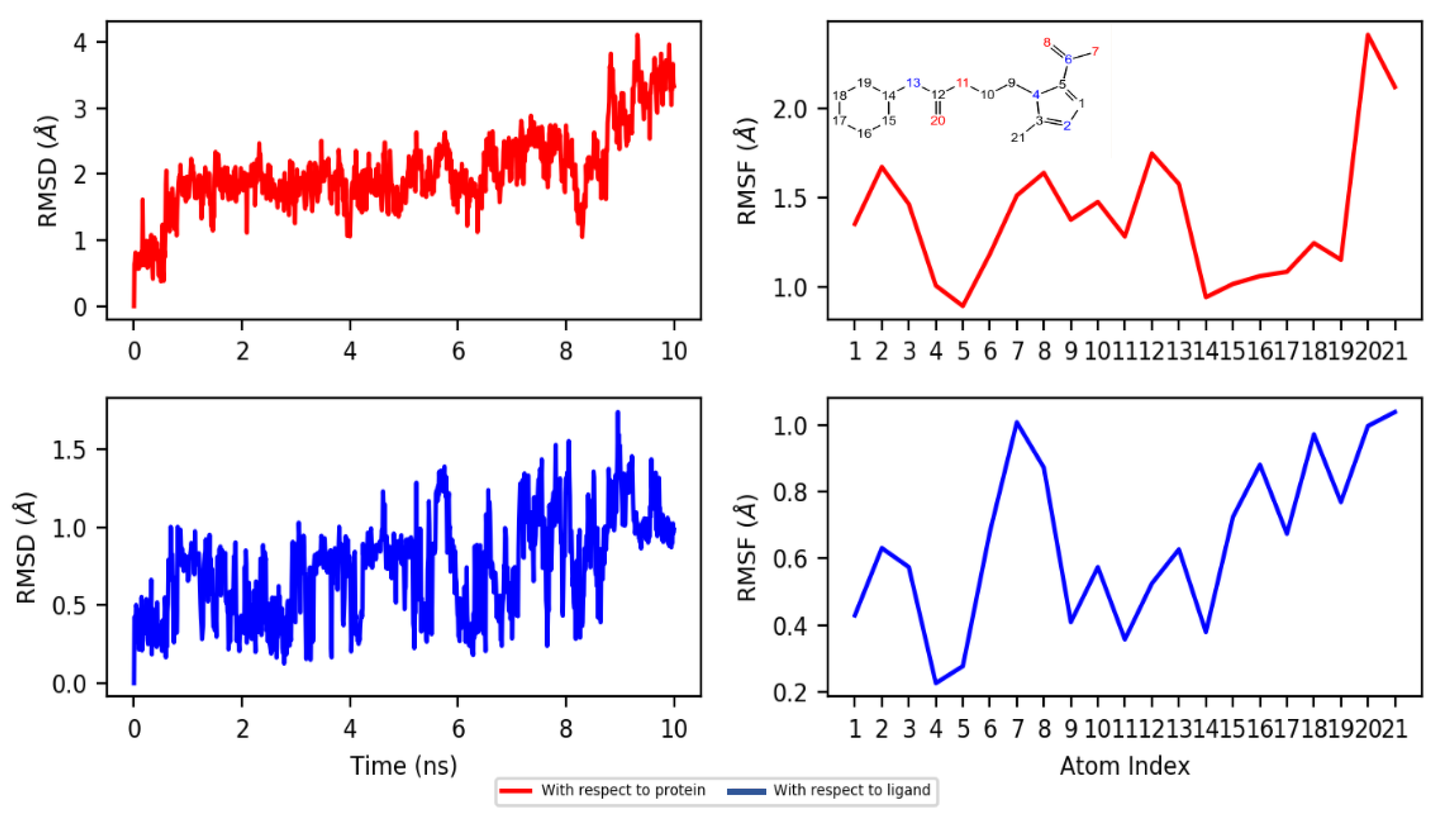
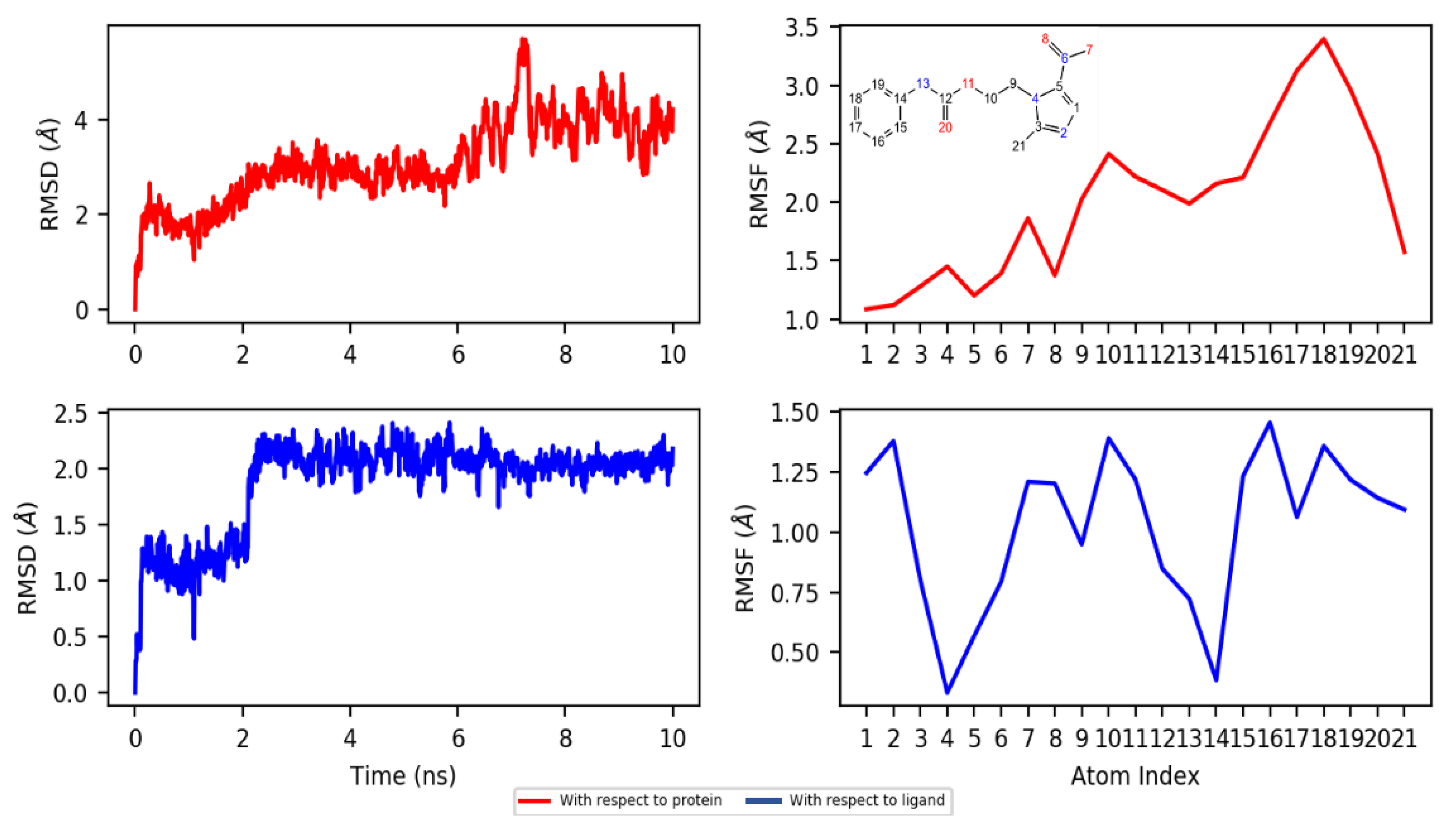
2.4.2. Dynamics
3. Materials and Methods
3.1. Chemistry
3.2. General Procedure for the Synthesis of Compounds 1–10
3.3. Biological Assays
3.3.1. Giardicidal and Trichomonicidal Assays
3.3.2. Cytotoxicity on VERO Cell Line
3.4. In Silico Methods
3.4.1. Homology Modeling
3.4.2. Molecular Docking
3.4.3. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ortega-Pierres, M.G.; Argüello-García, R. Giardia duodenalis: Role of secreted molecules as virulent factors in the cytotoxic effect on epithelial cells. Adv. Parasitol. 2019, 106, 129–169. [Google Scholar] [CrossRef]
- Bala, V.; Chhonker, Y.S. Recent developments in anti-Trichomonas research: An update review. Eur. J. Med. Chem. 2018, 143, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Leitsch, D.; Schlosser, S.; Burgess, A.; Duchêne, M. Nitroimidazole drugs vary in their mode of action in the human parasite Giardia lamblia. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Dingsdag, S.A.; Hunter, N. Metronidazole: An update on metabolism, structure-cytotoxicity and resistance mechanisms. J. Antimicrob. Chemother. 2018, 73, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Braga, S.; Heller, M.; Müller, N. Resistance formation to nitro drugs in Giardia lamblia: No common markers identified by comparative proteomics. Int. J. Parasitol. Drugs Drug Resist. 2019, 9, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Núñez, E.; Tlahuext, H.; Moo-Puc, R.; Torres-Gómez, H.; Reyes-Martínez, R.; Cedillo-Rivera, R.; Nava-Zuazo, C.; Navarrete-Vazquez, G. Synthesis and in vitro trichomonicidal, giardicidal and amebicidal activity of N-acetamide(sulfonamide)-2-methyl-4-nitro-1H-imidazoles. Eur. J. Med. Chem. 2009, 44, 2975–2984. [Google Scholar] [CrossRef]
- Leitsch, D.; Burgess, A.G.; Dunn, L.A.; Krauer, K.G.; Tan, K.; Ducĥne, M.; Upcroft, P.; Eckmann, L.; Upcroft, J.A. Pyruvate: Ferredoxin oxidoreductase and thioredoxin reductase are involved in 5-nitroimidazole activation while flavin metabolism is linked to 5-nitroimidazole resistance in Giardia lamblia. J. Antimicrob. Chemother. 2011, 66, 1756–1765. [Google Scholar] [CrossRef]
- Hernández-Ochoa, B.; Navarrete-Vázquez, G.; Nava-Zuazo, C.; Castillo-Villanueva, A.; Méndez, S.T.; Torres-Arroyo, A.; Gómez-Manzo, S.; Marcial-Quino, J.; Ponce-Macotela, M.; Rufino-González, Y.; et al. Novel giardicidal compounds bearing proton pump inhibitor scaffold proceeding through triosephosphate isomerase inactivation. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Hernández-Núñez, E.; Tlahuext, H.; Moo-Puc, R.; Moreno, D.; González-Díaz, M.O.; Vázquez, G.N. Design, synthesis and biological evaluation of 2-(2-amino-5(6)-nitro-1H-benzimidazol-1-yl)-narylacetamides as antiprotozoal agents. Molecules 2017, 22, 579. [Google Scholar] [CrossRef]
- Aguayo-Ortiz, R.; Méndez-Lucio, O.; Romo-Mancillas, A.; Castillo, R.; Yépez-Mulia, L.; Medina-Franco, J.L.; Hernández-Campos, A. Molecular basis for benzimidazole resistance from a novel β-tubulin binding site model. J. Mol. Graph. Model. 2013, 45, 26–37. [Google Scholar] [CrossRef]
- Valladares-Méndez, A.; García-Flores, M.; Navarrete-Vázquez, G.; Orozco-Castellanos, L.M.; Hernandez-Nuñez, E.; Rivera-Leyva, J.C. Physicochemical characterization of two new Nitazoxanide analogs with antiparasitic activity. Med. Chem. Res. 2017, 26, 9–18. [Google Scholar] [CrossRef]
- Colín-Lozano, B.; León-Rivera, I.; Chan-Bacab, M.J.; Ortega-Morales, B.O.; Moo-Puc, R.; López-Guerrero, V.; Hernández-Núñez, E.; Argüello-Garcia, R.; Scior, T.; Barbosa-Cabrera, E.; et al. Synthesis, in vitro and in vivo giardicidal activity of nitrothiazole-NSAID chimeras displaying broad antiprotozoal spectrum. Bioorg. Med. Chem. Lett. 2017, 27, 3490–3494. [Google Scholar] [CrossRef]
- Naaz, F.; Haider, M.R.; Shafi, S.; Yar, M.S. Anti-tubulin agents of natural origin: Targeting taxol, vinca, and colchicine binding domains. Eur. J. Med. Chem. 2019, 171, 310–331. [Google Scholar] [CrossRef]
- Weksberg, T.E.; Lynch, G.C.; Krause, K.L.; Pettitt, B.M. Molecular dynamics simulations of Trichomonas vaginalis ferredoxin show a loop-cap transition. Biophys. J. 2007, 92, 3337–3345. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lü, J.T.; Hu, B.Z.; Hedegård, P.; Brandbyge, M. Semi-classical generalized Langevin equation for equilibrium and nonequilibrium molecular dynamics simulation. Prog. Surf. Sci. 2019, 94, 21–40. [Google Scholar] [CrossRef]
- Chen, J.C.; Kim, A.S. Brownian dynamics, molecular dynamics, and monte carlo modeling of colloidal systems. Adv. Colloid Interface Sci. 2004, 112, 159–173. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinforma. 2009, 77, 114–122. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Qiu, J.; Elber, R. SSALN: An alignment algorithm using structure-dependent substitution matrices and gap penalties learned from structurally aligned protein pairs. Proteins Struct. Funct. Genet. 2006, 62, 881–891. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Benkert, P.; Tosatto, S.C.E.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins Struct. Funct. Bioinforma. 2008, 71, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.L.; MacArthur, M.W.; Hutchinson, E.G.; Thornton, J.M. Stereochemical quality of protein structure coordinates. Proteins Struct. Funct. Bioinforma. 1992, 12, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Hooft, R.W.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in protein structures [3]. Nature 1996, 381, 272. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2019.01 2019.
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated Interaction Energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.; Chow, E.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossvary, I.; Moraes, M.; Sacerdoti, F.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In ACM/IEEE SC 2006 Conference (SC’06), Tampa, FL, USA, 11–17 November 2006; Association for Computing Machinery: New York, NY, USA, 2006. [Google Scholar]
- Özen, A.; Sherman, W.; Schiffer, C.A. Improving the resistance profile of hepatitis C NS3/4A inhibitors: Dynamic substrate envelope guided design. J. Chem. Theory Comput. 2013, 9, 5693–5705. [Google Scholar] [CrossRef] [PubMed]
- Cob-Calan, N.N.; Chi-Uluac, L.A.; Ortiz-Chi, F.; Cerqueda-García, D.; Navarrete-Vázquez, G.; Ruiz-Sánchez, E.; Hernández-Núñez, E. Molecular Docking and Dynamics Simulation of Protein β-Tubulin and Antifungal Cyclic Lipopeptides. Molecules. 2019, 24, 3387. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) a | CC50 (µM) a | SI = CC50/IC50 | MLogP | ||
|---|---|---|---|---|---|---|
| G. duodenalis | T. vaginalis | VERO Cells | G. duodenalis | T. vaginalis | ||
| 1 | 0.46 ± 0.01 | 0.06 ± 0.01 | >100 | >217 | >1666 | 3.73 |
| 2 | 0.98 ± 0.01 | 0.09 ± 0.01 | >100 | >102 | >1111 | 1.72 |
| 3 | 1.14 ± 0.12 | 1.79 ± 0.12 | >100 | >87 | >55 | 2.4 |
| 4 | 2.03 ± 0.01 | 3.22 ± 0.21 | >100 | >49 | >31 | 1.89 |
| 5 | 2.43 ± 0.02 | 6.25 ± 0.12 | >100 | >41 | >16 | 1.68 |
| 6 | 2.64 ± 0.61 | 8.64 ± 0.73 | >100 | >37 | >11 | 3.87 |
| 7 | 1.67 ± 0.21 | 6.52 ± 0.71 | >100 | >59 | >15 | 1.86 |
| 8 | 4.05 ± 0.24 | 5.93 ± 0.51 | >100 | >24 | >16 | 2.54 |
| 9 | 3.23 ± 0.11 | 4.82 ± 1.08 | >100 | >30 | >20 | 2.02 |
| 10 | 0.86 ± 0.09 | 9.99 ± 0.56 | >100 | >116 | >10 | 1.82 |
| Metronidazole | 4.42 ± 0.23 | 0.93 ± 0.12 | >100 | >22 | >107 | −0.47 |
| Secnidazole | 4.11 ± 0.12 | 13.45 ± 1.23 | >100 | >24 | >7 | −0.10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha-Garduño, G.; Hernández-Martínez, N.A.; Colín-Lozano, B.; Estrada-Soto, S.; Hernández-Núñez, E.; Prieto-Martínez, F.D.; Medina-Franco, J.L.; Chale-Dzul, J.B.; Moo-Puc, R.; Navarrete-Vázquez, G. Metronidazole and Secnidazole Carbamates: Synthesis, Antiprotozoal Activity, and Molecular Dynamics Studies. Molecules 2020, 25, 793. https://doi.org/10.3390/molecules25040793
Rocha-Garduño G, Hernández-Martínez NA, Colín-Lozano B, Estrada-Soto S, Hernández-Núñez E, Prieto-Martínez FD, Medina-Franco JL, Chale-Dzul JB, Moo-Puc R, Navarrete-Vázquez G. Metronidazole and Secnidazole Carbamates: Synthesis, Antiprotozoal Activity, and Molecular Dynamics Studies. Molecules. 2020; 25(4):793. https://doi.org/10.3390/molecules25040793
Chicago/Turabian StyleRocha-Garduño, Genaro, Norma Angélica Hernández-Martínez, Blanca Colín-Lozano, Samuel Estrada-Soto, Emanuel Hernández-Núñez, Fernando Daniel Prieto-Martínez, José L. Medina-Franco, Juan Bautista Chale-Dzul, Rosa Moo-Puc, and Gabriel Navarrete-Vázquez. 2020. "Metronidazole and Secnidazole Carbamates: Synthesis, Antiprotozoal Activity, and Molecular Dynamics Studies" Molecules 25, no. 4: 793. https://doi.org/10.3390/molecules25040793
APA StyleRocha-Garduño, G., Hernández-Martínez, N. A., Colín-Lozano, B., Estrada-Soto, S., Hernández-Núñez, E., Prieto-Martínez, F. D., Medina-Franco, J. L., Chale-Dzul, J. B., Moo-Puc, R., & Navarrete-Vázquez, G. (2020). Metronidazole and Secnidazole Carbamates: Synthesis, Antiprotozoal Activity, and Molecular Dynamics Studies. Molecules, 25(4), 793. https://doi.org/10.3390/molecules25040793