
Sterically Stabilised Polymeric Mesoporous Silica Nanoparticles Improve Doxorubicin Efficiency: Tailored Cancer Therapy



Abstract
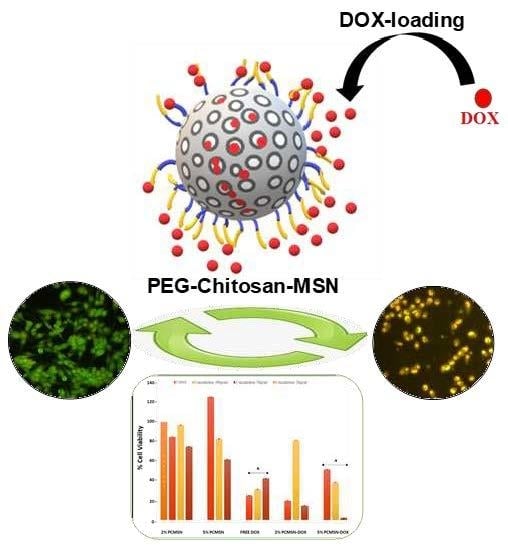
1. Introduction
2. Results
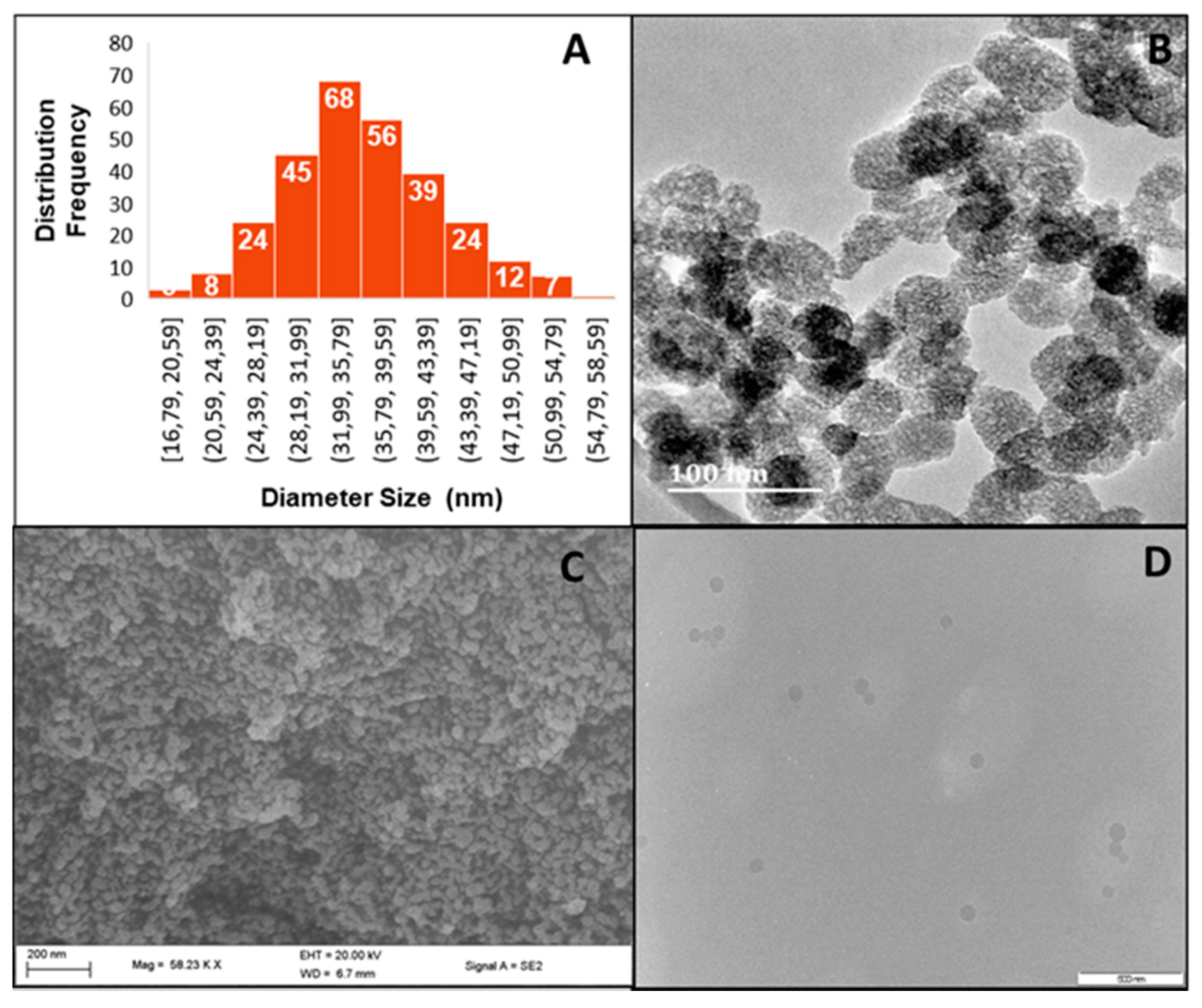
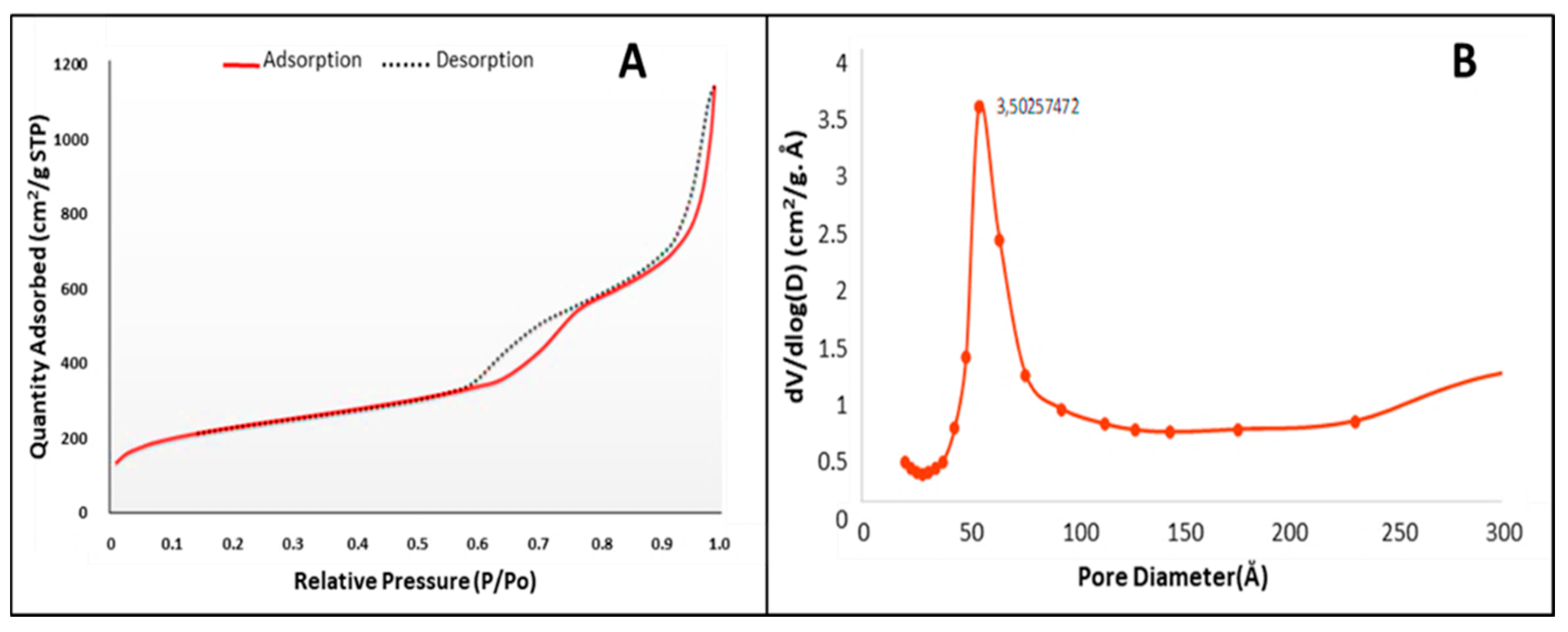
2.1. Size and Morphology
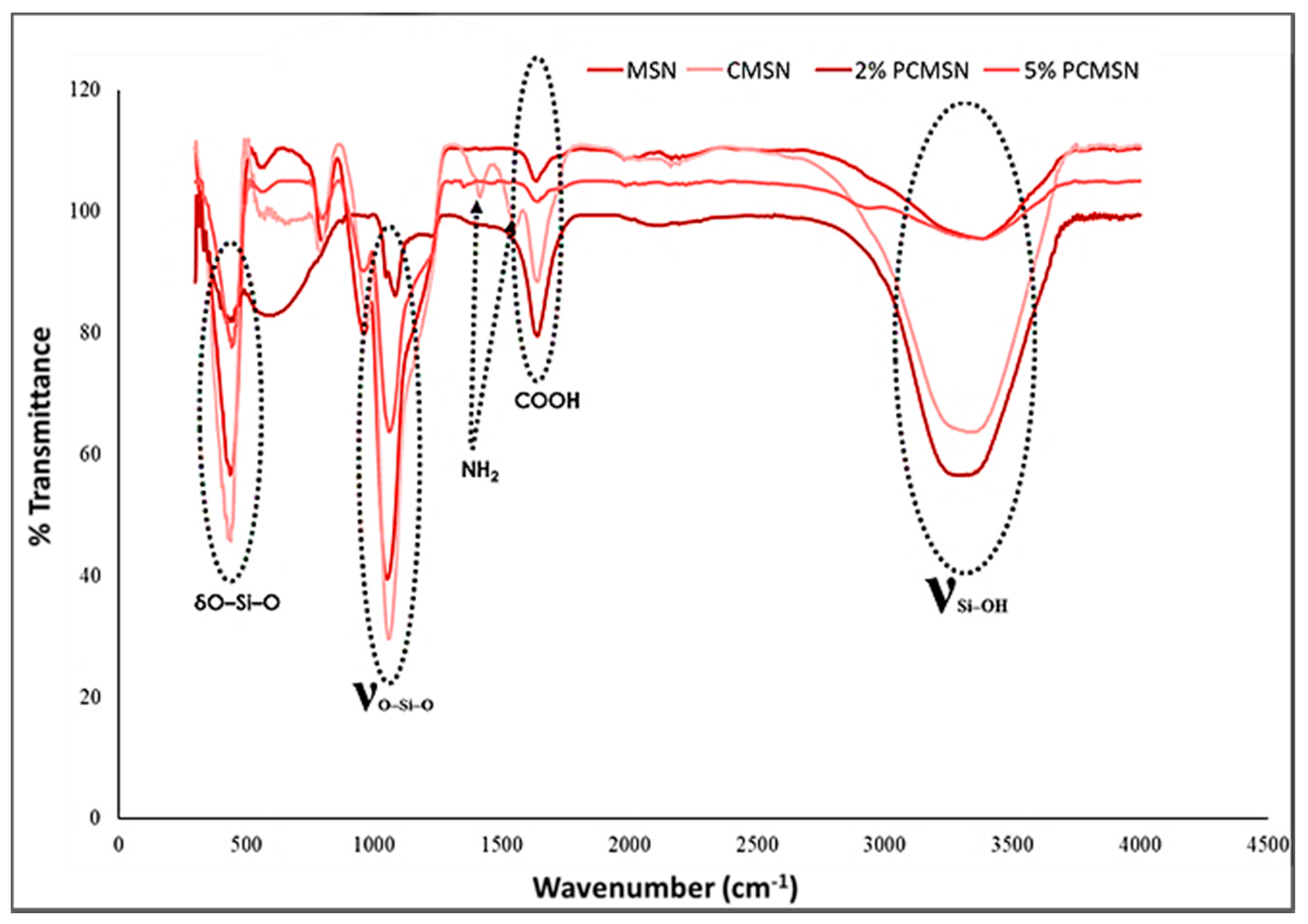
2.2. MSN Surface Modification
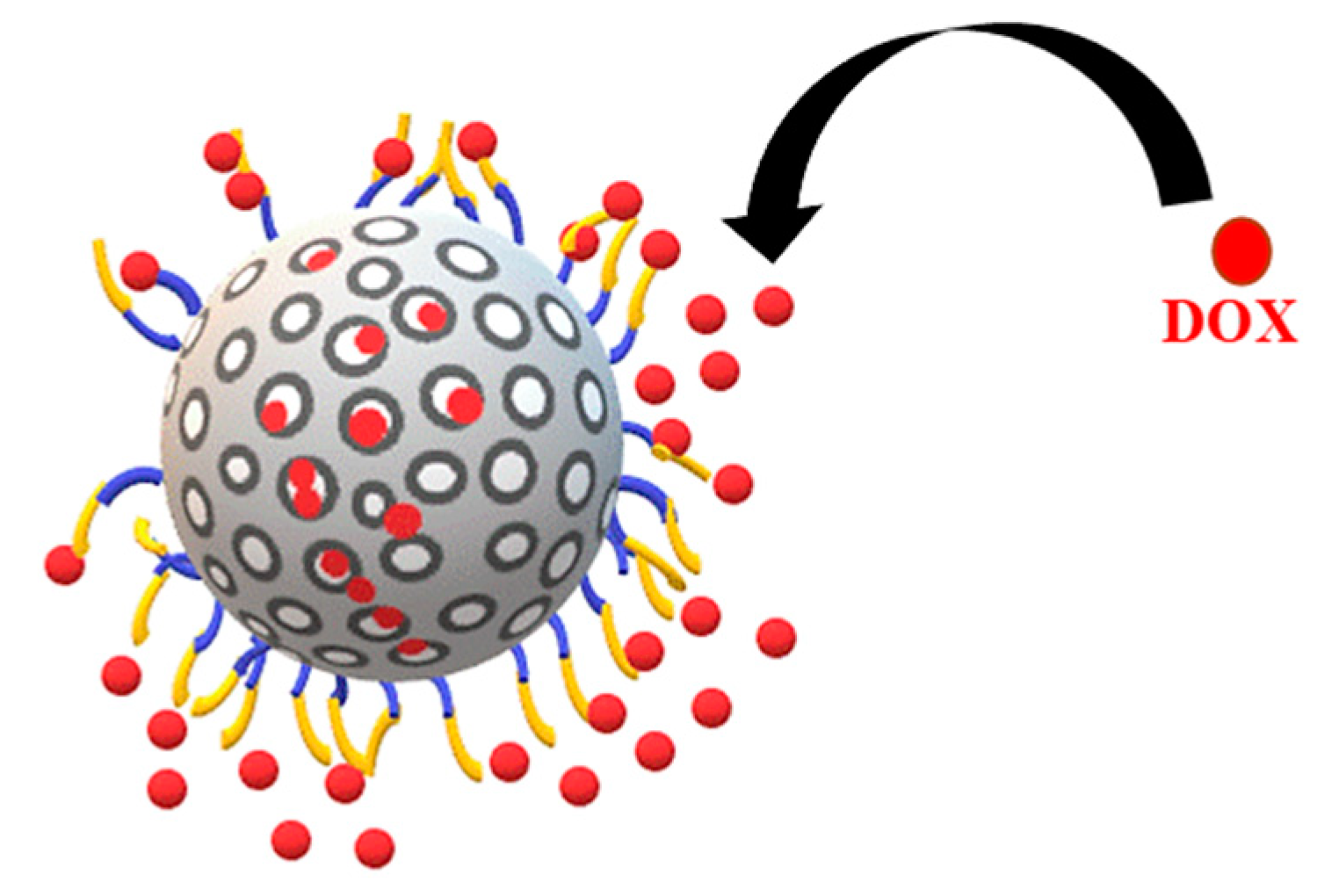
2.3. Doxorubicin Loading
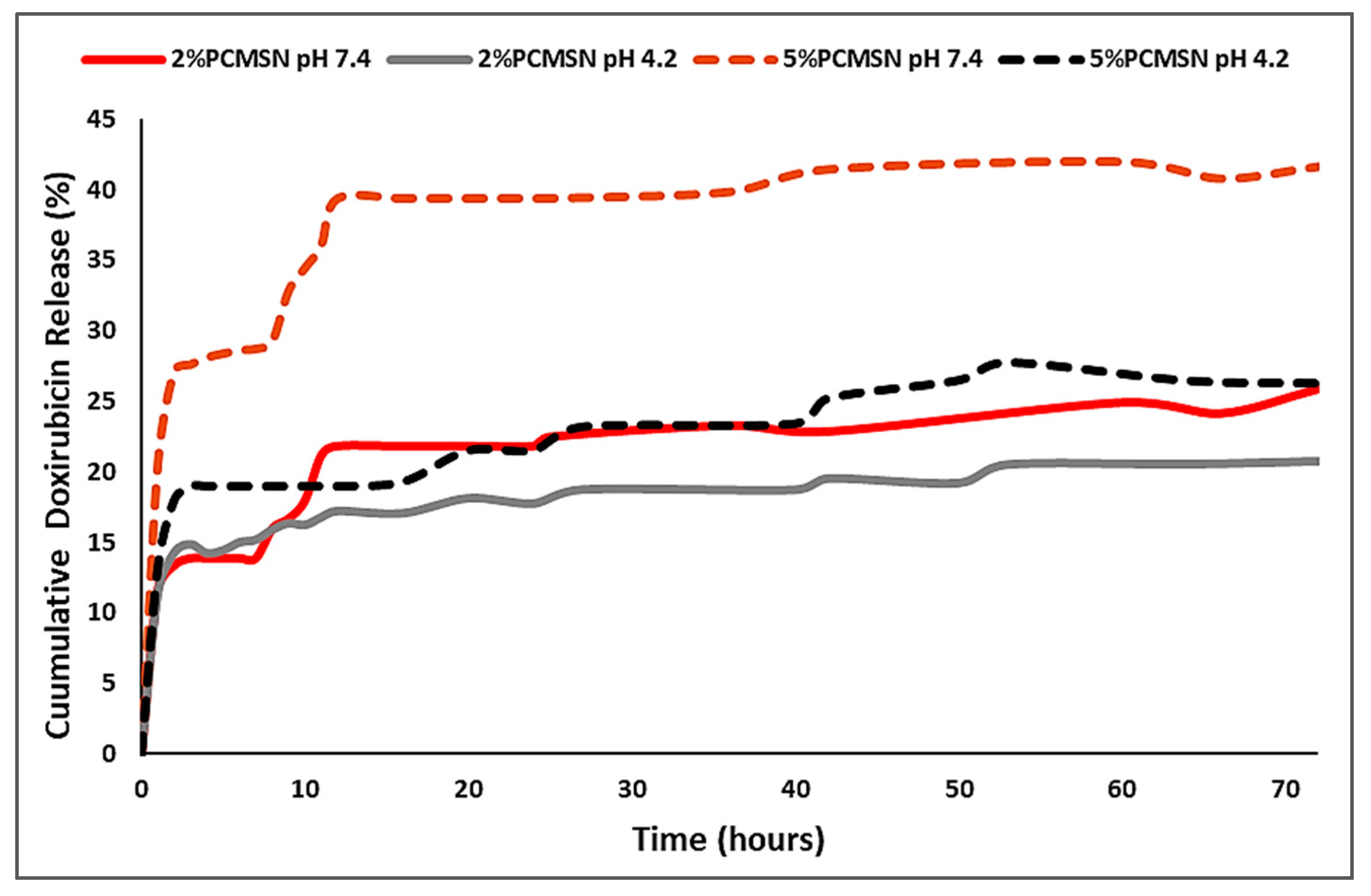
2.4. Doxorubicin Release and Pharmacokinetic Modelling
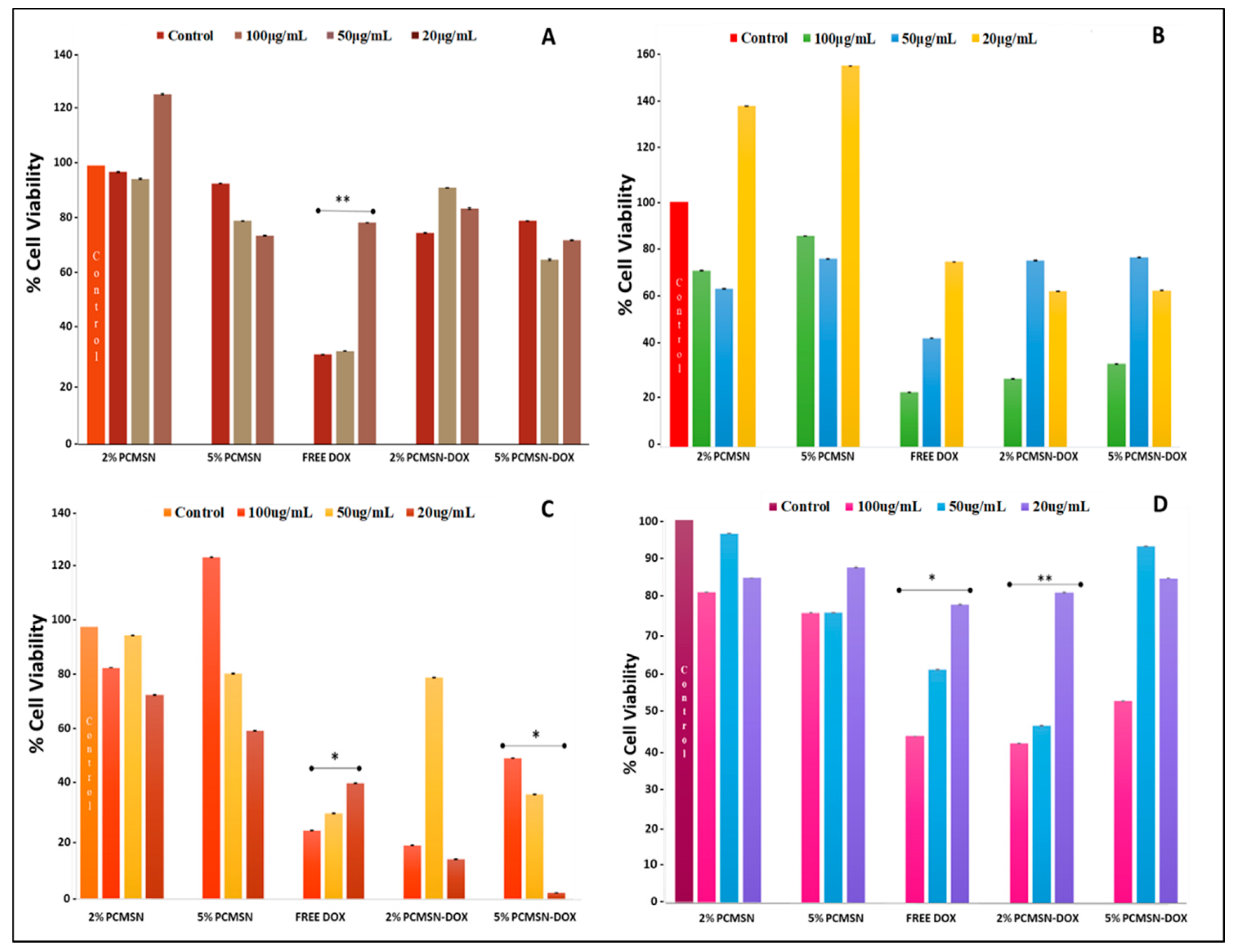
2.5. Cell-Based Cytotoxicity Studies
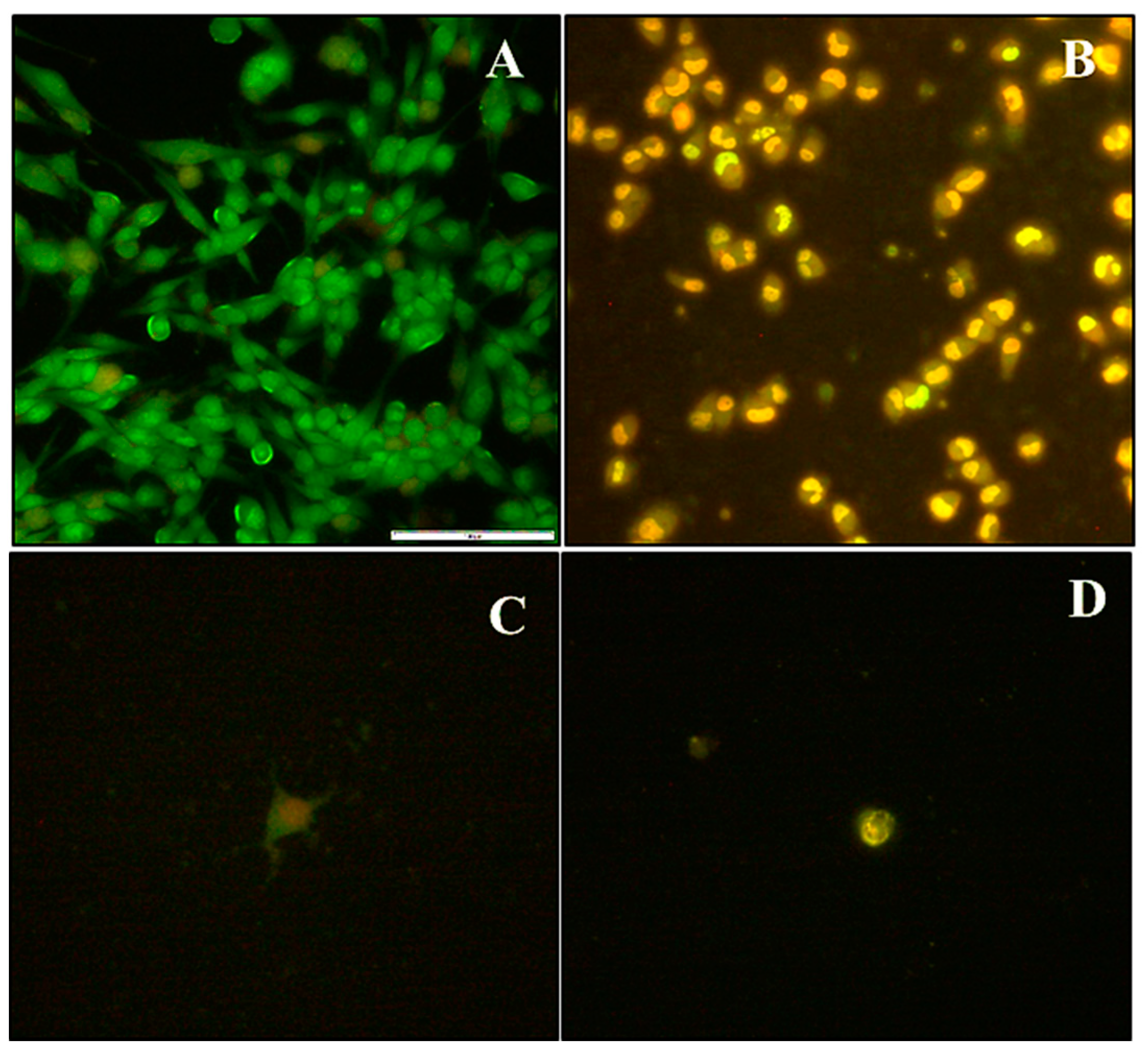
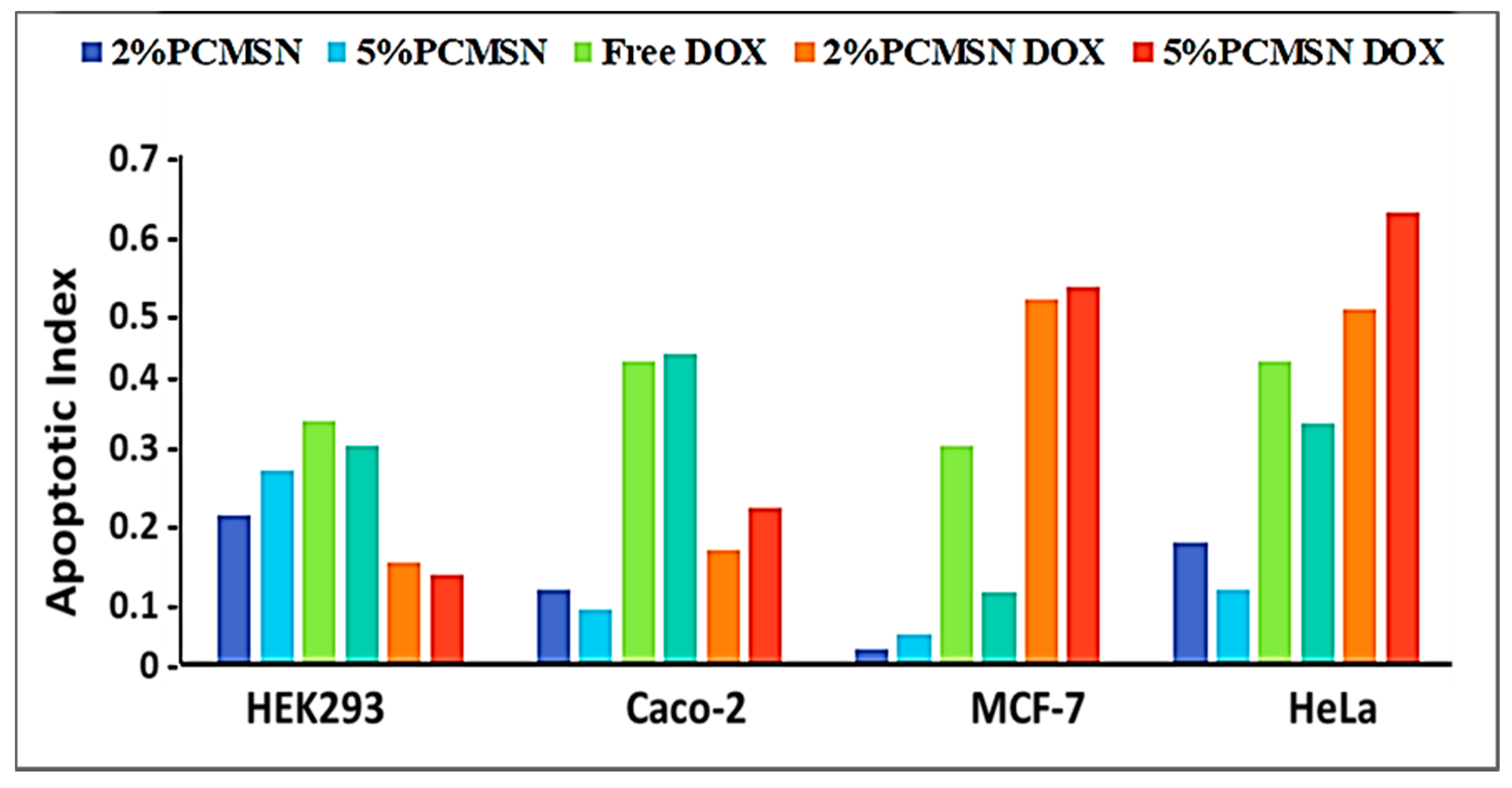
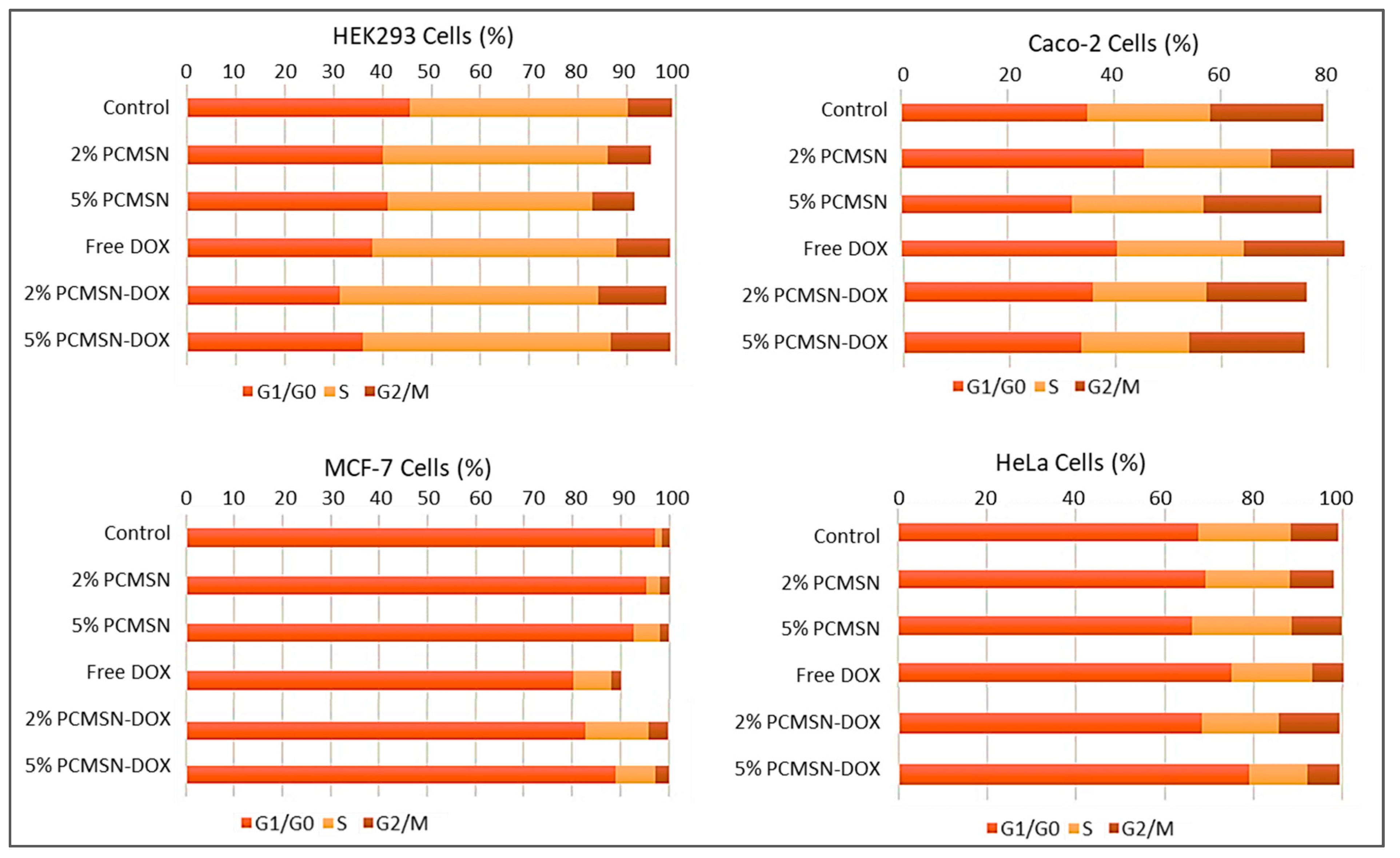
Apoptosis and Cell Cycle Analysis
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of Mesoporous Silica Nanoparticles (MSNs)
4.3. MSN modification with Chitosan and Polyethyleneglycol
4.4. Doxorubicin (DOX) Loading
4.5. Electron Microscopy and Energy Dispersive X-Ray Spectroscopy
4.6. Nanoparticle Tracking Analysis (NTA)
4.7. Nitrogen Adsorption and Desorption
4.8. Fourier Transform Infrared Spectrometer (FTIR)
4.9. Doxorubicin Release
4.10. MTT Cytotoxicity Assay
4.11. Acridine orange/Ethidium bromide (AO/EB) Apoptosis Assay
4.12. Cell cycle Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moorthi, C.; Manavalan, R.; Kathiresan, K. Nanotherapeutics to overcome conventional cancer chemotherapy limitations. J. Pharm. Pharm. Sci. 2011, 14, 67–77. [Google Scholar]
- Cheng, R.; Meng, F.; Deng, C.; Zhong, Z. Bioresponsive polymeric nanotherapeutics for targeted cancer chemotherapy. Nano Today 2015, 10, 656–670. [Google Scholar] [CrossRef]
- Wang, A.Z.; Langer, R.; Farokhzad, O.C. Nanoparticle Delivery of Cancer Drugs. Annu. Rev. Med. 2012, 63, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T. Clinical Application of Drug Delivery Systems in Cancer Chemotherapy: Review of the Efficacy and Side Effects of Approved Drugs. Biol. Pharm. Bull. 2013, 36, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Jahangirian, H.; Lemraski, E.G.; Webster, T.J.; Rafiee-Moghaddam, R.; Abdollahi, Y. A review of drug delivery systems based on nanotechnology and green chemistry: Green nanomedicine. Int. J. Nanomedicine 2017, 12, 2957–2978. [Google Scholar] [CrossRef] [PubMed]
- Souris, J.S.; Lee, C.-H.; Cheng, S.-H.; Chen, C.-T.; Yang, C.-S.; Ho, J.A.; Mou, C.-Y.; Lo, L.-W. Surface charge-mediated rapid hepatobiliary excretion of mesoporous silica nanoparticles. Biomaterials 2010, 31, 5564–5574. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S.A. Review of Clinical Translation of Inorganic Nanoparticles. AAPS J. 2015, 17, 1041–1054. [Google Scholar] [CrossRef]
- Yang, M.; Lai, S.K.; Wang, Y.-Y.; Zhong, W.; Happe, C.; Zhang, M.; Fu, J.; Hanes, J. Biodegradable Nanoparticles Composed Entirely of Safe Materials that Rapidly Penetrate Human Mucus. Angew. Chemie Int. Ed. 2011, 50, 2597–2600. [Google Scholar] [CrossRef]
- Croissant, J.G.; Fatieiev, Y.; Khashab, N.M. Degradability and Clearance of Silicon, Organosilica, Silsesquioxane, Silica Mixed Oxide, and Mesoporous Silica Nanoparticles. Adv. Mater. 2017, 29, 1604634. [Google Scholar] [CrossRef]
- Li, Z.; Barnes, J.C.; Bosoy, A.; Stoddart, J.F.; Zink, J.I.; Strebhardt, K.; Ullrich, A.; Peer, D.; Karp, J.M.; Hong, S.; et al. Mesoporous silica nanoparticles in biomedical applications. Chem. Soc. Rev. 2012, 41, 2590–2605. [Google Scholar] [CrossRef]
- Zhu, J.; Niu, Y.; Li, Y.; Gong, Y.; Shi, H.; Huo, Q.; Liu, Y.; Xu, Q. Stimuli-responsive delivery vehicles based on mesoporous silica nanoparticles: Recent advances and challenges. J. Mater. Chem. B 2017, 5, 1339–1352. [Google Scholar] [CrossRef]
- Lai, C.-Y.; Trewyn, B.G.; Jeftinija, D.M.; Jeftinija, K.; Xu, S.; Jeftinija, S.; Lin, V.S.-Y. A Mesoporous Silica Nanosphere-Based Carrier System with Chemically Removable CdS Nanoparticle Caps for Stimuli-Responsive Controlled Release of Neurotransmitters and Drug Molecules. J. Am. Chem. Soc. 2003, 125, 4451–4459. [Google Scholar] [CrossRef] [PubMed]
- Wang, S. Ordered mesoporous materials for drug delivery. Microporous Mesoporous Mater. 2009, 117, 1–9. [Google Scholar] [CrossRef]
- Slowing, I.I.; Trewyn, B.G.; Giri, S.; Lin, V.S.-Y. Mesoporous Silica Nanoparticles for Drug Delivery and Biosensing Applications. Adv. Funct. Mater. 2007, 17, 1225–1236. [Google Scholar] [CrossRef]
- Rosenholm, J.M.; Sahlgren, C.; Lindén, M. Towards multifunctional, targeted drug delivery systems using mesoporous silica nanoparticles - Opportunities & challenges. Nanoscale 2010, 2, 1870–1883. [Google Scholar] [PubMed]
- Baudino, T. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef]
- O’Shaughnessy, J. Extending survival with chemotherapy in metastatic breast cancer. Oncologist 2005, 10, 20–29. [Google Scholar] [CrossRef]
- Carey, M.P.; Burish, T.G. Etiology and Treatment of the Psychological Side Effects Associated With Cancer Chemotherapy: A Critical Review and Discussion. Psychol. Bull. 1988, 104, 307–325. [Google Scholar] [CrossRef]
- Eckford, P.D.W.; Sharom, F.J. ABC Efflux Pump-Based Resistance to Chemotherapy Drugs. Chem. Rev. 2009, 109, 2989–3011. [Google Scholar] [CrossRef]
- Citron, M.L.; Berry, D.A.; Cirrincione, C.; Hudis, C.; Winer, E.P.; Gradishar, W.J.; Davidson, N.E.; Martino, S.; Livingston, R.; Ingle, J.N.; et al. Randomized Trial of Dose-Dense Versus Conventionally Scheduled and Sequential Versus Concurrent Combination Chemotherapy as Postoperative Adjuvant Treatment of NodePositive Primary Breast Cancer: First Report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J Clin. Oncol. 2003, 21, 1431–1439. [Google Scholar]
- Abraham, S.A.; Waterhouse, D.N.; Mayer, L.D.; Cullis, P.R.; Madden, T.D.; Bally, M.B. The liposomal formulation of doxorubicin. Methods Enzymol. 2005, 391, 71–97. [Google Scholar] [PubMed]
- Martin, M.; Villar, A.; Sole-Calvo, A.; Gonzalez, R.; Massuti, B.; Lizon, J.; Camps, C.; Carrato, A.; Casado, A.; Candel, M.T.; et al. Doxorubicin in combination with fluorouracil and cyclophosphamide (i.v. FAC regimen, day 1, 21) versus methotrexate in combination with fluorouracil and cyclophosphamide (i.v. CMF regimen, day 1, 21) as adjuvant chemotherapy for operable breast cancer: A study by the GEICAM group. Ann. Oncol. 2003, 14, 833–842. [Google Scholar] [PubMed]
- Bray, J.; Sludden, J.; Griffin, M.J.; Cole, M.; Verrill, M.; Jamieson, D.; Boddy, A.V. Influence of pharmacogenetics on response and toxicity in breast cancer patients treated with doxorubicin and cyclophosphamide. Br. J. Cancer 2010, 102, 1003–1009. [Google Scholar] [CrossRef]
- Kim, M.S.; Jeon, J.B.; Chang, J.Y. Selectively functionalized mesoporous silica particles with the PEGylated outer surface and the doxorubicin-grafted inner surface: Improvement of loading content and solubility. Microporous Mesoporous Mater. 2013, 172, 118–124. [Google Scholar] [CrossRef]
- Mayer, L.D.; Tai, L.C.L.; Bally, M.B.; Mitilenes, G.N.; Ginsberg, R.S.; Cullis, P.R. Characterization of liposomal systems containing doxorubicin entrapped in response to pH gradients. Biochim. Biophys. Acta Biomembr. 1990, 1025, 143–151. [Google Scholar] [CrossRef]
- Shahabi, S.; Döscher, S.; Bollhorst, T.; Treccani, L.; Maas, M.; Dringen, R.; Rezwan, K. Enhancing Cellular Uptake and Doxorubicin Delivery of Mesoporous Silica Nanoparticles via Surface Functionalization: Effects of Serum. ACS Appl. Mater. Interfaces 2015, 7, 26880–26891. [Google Scholar] [CrossRef]
- Vanhoefer, U.; Rougier, P.; Wilke, H.; Ducreux, M.P.; Lacave, A.J.; Van Cutsem, E.; Planker, M.; Santos, J.G. Dos; Piedbois, P.; Paillot, B.; et al. Final Results of a Randomized Phase III Trial of Sequential High-Dose Methotrexate, Fluorouracil, and Doxorubicin Versus Etoposide, Leucovorin, and Fluorouracil Versus Infusional Fluorouracil and Cisplatin in Advanced Gastric Cancer: A Trial of the European Organization for Research and Treatment of Cancer Gastrointestinal Tract Cancer Cooperative Group. J. Clin. Oncol. 2000, 18, 2648–2657. [Google Scholar]
- Mostafa, M.G.; Mima, T.; Ohnishi, S.T.; Mori, K. S-Allylcysteine Ameliorates Doxorubicin Toxicity in the Heart and Liver in Mice. Planta Med. 2000, 66, 148–151. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Rifai, N.; Dalton, V.M.; Levy, D.E.; Silverman, L.B.; Lipsitz, S.R.; Colan, S.D.; Asselin, B.L.; Barr, R.D.; Clavell, L.A.; et al. The Effect of Dexrazoxane on Myocardial Injury in Doxorubicin-Treated Children with Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2004, 351, 145–153. [Google Scholar] [CrossRef]
- Lin, H.; She, Y.H.; Cassileth, B.R.; Sirotnak, F.; Rundles, S.C. Maitake beta-glucan MD-fraction enhances bone marrow colony formation and reduces doxorubicin toxicity in vitro. Int. Immunopharmacol. 2004, 4, 91–99. [Google Scholar] [CrossRef]
- Moertel, C.G.; Lefkopoulo, M.; Lipsitz, S.; Hahn, R.G.; Klaassen, D. Streptozocin–Doxorubicin, Streptozocin–Fluorouracil, or Chlorozotocin in the Treatment of Advanced Islet-Cell Carcinoma. N. Engl. J. Med. 1992, 326, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Tursz, T.; Mouridsen, H.; Verweij, J.; Steward, W.; SomersJ Buesa, R.; Casali, P.; Spooner, D.; Rankin, E. Doxorubicin versusCYVADIC versus doxorubicin plus ifosfamide in first-linetreatment of advanced soft tissue sarcomas: A randomizedstudy of the European Organization for Research and Treat-ment of Cancer, Soft Tissue and Bone Sarcoma Group. J. Clin. Oncol. 1995, 13, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.K.; Park, Y.S.; Heo, D.S.; Suh, C.; Kim, S.Y.; Park, K.C.; Kang, Y.K.; Shin, D.B.; Kim, H.T.; Kim, H.J.; et al. A phase III randomized study of 5--fluorouracil and cisplatin versus 5--fluorouracil, doxorubicin, and mitomycin C versus 5--fluorouracil alone in the treatment of advanced gastric cancer. Cancer 1993, 71, 3813–3818. [Google Scholar] [CrossRef]
- Judson, I.; Radford, J.A.; Harris, M.; Blay, J.Y.; Van Hoesel, Q.; Le Cesne, A.; Van Oosterom, A.T.; Clemons, M.J.; Kamby, C.; Hermans, C.; et al. Randomised phase II trial of pegylated liposomal doxorubicin (DOXIL®/CAELYX®) versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: A study by the EORTC Soft Tissue and Bone Sarcoma Group. Eur. J. Cancer 2001, 37, 870–877. [Google Scholar] [CrossRef]
- Roscoe, J.A.; Morrow, G.R.; Colagiuri, B.; Heckler, C.E.; Pudlo, B.D.; Colman, L.; Hoelzer, K.; Jacobs, A. Insight in the prediction of chemotherapy-induced nausea. Support. Care Cancer 2010, 18, 869–876. [Google Scholar] [CrossRef]
- Tecza, K.; Pamula-Pilat, J.; Lanuszewska, J.; Butkiewicz, D.; Grzybowska, E. Pharmacogenetics of toxicity of 5-fluorouracil, doxorubicin and cyclophosphamide chemotherapy in breast cancer patients. Oncotarget 2018, 9, 9114–9136. [Google Scholar] [CrossRef]
- Lemieux, J.; Maunsell, E.; Provencher, L. Chemotherapy-induced alopecia and effects on quality of life among women with breast cancer: A literature review. Psychooncology 2008, 17, 317–328. [Google Scholar] [CrossRef]
- Nakanishi, T.; Fukushima, S.; Okamoto, K.; Suzuki, M.; Matsumura, Y.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Kataoka, K. Development of the polymer micelle carrier system for doxorubicin. J. Control. Release 2001, 74, 295–302. [Google Scholar] [CrossRef]
- Barenholz, Y. (Chezy) Doxil® -The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Moodley, T.; Singh, M. Polymeric Mesoporous Silica Nanoparticles for Enhanced Delivery of 5-Fluorouracil In Vitro. Pharmaceutics 2019, 11, 288. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, X.; Zhang, G.; Trewyn, B.G.; Slowing, I.I.; Lin, V.S.-Y. Interaction of Mesoporous Silica Nanoparticles with Human Red Blood Cell Membranes: Size and Surface Effects. ACS Nano 2011, 5, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Hudson, S.P.; Padera, R.F.; Langer, R.; Kohane, D.S. The biocompatibility of mesoporous silicates. Biomater. 2008, 29, 4045–4055. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhang, Z.; Gao, F.; Li, Y.; Shi, J. In vivo biodistribution and urinary excretion of mesoporous silica nanoparticles: Effects of particle size and PEGylation. Small 2011, 7, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, N.I.; Gonzalez, Z.; Ferrari, B.; Castro, Y. Synthesis of mesoporous silica nanoparticles by sol–gel as nanocontainer for future drug delivery applications. Boletín la Soc. Española Cerámica y Vidr. 2017, 56, 139–145. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- He, Q.; Shi, J.; Cui, X.; Zhao, J.; Chen, Y.; Zhou, J. Rhodamine B-co-condensed spherical SBA-15 nanoparticles: Facile co-condensation synthesis and excellent fluorescence features. J. Mater. Chem. 2009, 19, 3395. [Google Scholar] [CrossRef]
- Agudelo, D.; Bourassa, P.; Bérubé, G.; Tajmir-Riahi, H.-A. Intercalation of antitumor drug doxorubicin and its analogue by DNA duplex: Structural features and biological implications. Int. J. Biol. Macromol. 2014, 66, 144–150. [Google Scholar] [CrossRef]
- Beganskiene, A.; Sirutkaitis, V.; Kurtinaitiene, M.; Juskenas, R.; Kareiva, A. FTIR, TEM, and NMR Investigations of Stober Silica Nanoparticles. Mater. Sci. 2004, 10, 287–290. [Google Scholar]
- Kumar, B.; Kulanthaivel, S.; Banerjee, B.; Mondal, A.; Mishra, S.; Bhaumik, A.; Banerjee, I.; Giri, S. Mesoporous Silica Nanoparticle Based Enzyme Responsive System for Colon Specific Drug Delivery through Guar Gum Capping. Colloids Surf B Biointerfaces 2017, 150, 352–361. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol Pharm. 2010, 67, 217–223. [Google Scholar]
- Higuchi, T. Mechanism of sustained--action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Hixson, A.W.; Crowell, J.H. Dependence of Reaction Velocity upon surface and Agitation. Ind. Eng. Chem. 1931, 23, 923–931. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Lustig, S.R.; Peppas, N.A. Solute and penetrant diffusion in swellable polymers. I. Mathematical modeling. J. Polym. Sci. Part B Polym. Phys. 1986, 24, 395–408. [Google Scholar] [CrossRef]
- Kopcha, M.; Tojo, K.; Lordi, N.J. Evaluation of methodology for assessing release characteristics of thermosoftening vehicles. J. Pharm. Pharmacol. 1990, 42, 745–751. [Google Scholar] [CrossRef]
- Sankalia, J.M.; Sankalia, M.G.; Mashru, R.C. Drug release and swelling kinetics of directly compressed glipizide sustained-release matrices: Establishment of level A IVIVC. J. Control. Release 2008, 129, 49–58. [Google Scholar] [CrossRef]
- Mulye, N. V.; Turco, S.J. A Simple Model Based on First Order Kinetics to Explain Release of Highly Water Soluble Drugs from Porous Dicalcium Phosphate Dihydrate Matrices. Drug Dev. Ind. Pharm. 1995, 21, 943–953. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Von Meerwall, E.; Peppas, N.A. Solute and penetrant diffusion in swellable polymers. II. Verification of theoretical models. J. Polym. Sci. Part B Polym. Phys. 1986, 24, 409–434. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Evaluation of mathematical models describing drug release from estradiol transdermal systems. Drug Dev. Ind. Pharm. 2003, 29, 89–97. [Google Scholar] [CrossRef]
- Barzegar-Jalali, M.; Adibkia, K.; Valizadeh, H.; Reza, M.; Shadbad, S.; Nokhodchi, A.; Omidi, Y.; Mohammadi, G.; Nezhadi, S.H.; Hasan, M. Kinetic Analysis of Drug Release From Nanoparticles. J. Pharm. Sci. 2008, 11, 167–177. [Google Scholar] [CrossRef]
- Higuchi, W.I. Diffusional models useful in biopharmaceutics. Drug release rate processes. J. Pharm. Sci. 1967, 56, 315–324. [Google Scholar] [CrossRef]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Yin, Y.; Shang, L.; Wu, T.; Zhang, D.; Kong, M.; Zhao, Y.; He, Y.; Tan, S.; Guo, Y.; et al. Tumor Microenvironment Responsive Nanogel for the Combinatorial Antitumor Effect of Chemotherapy and Immunotherapy. Nano Lett. 2017, 17, 6366–6375. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. EPR effect: The unique characteristics of tumor blood vessels for drug delivery, factors involved, its limitation and augmentation. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Langer, R. Drug Delivery and Targeting. Nature 1998, 392, 5–10. [Google Scholar]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef]
- Wang, M.; Thanou, M. Targeting nanoparticles to cancer. Pharmacol. Res. 2010, 62, 90–99. [Google Scholar] [CrossRef]
- Ishida, T.; Harada, M.; Wang, X.Y.; Ichihara, M.; Irimura, K.; Kiwada, H. Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: Effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J. Control. Release 2005, 105, 305–317. [Google Scholar] [CrossRef]
- Johnstone, S.A.; Masin, D.; Mayer, L.; Bally, M.B. Surface-associated serum proteins inhibit the uptake of phosphatidylserine and poly(ethylene glycol) liposomes by mouse macrophages. Biochim. Biophys. Acta 2001, 1513, 25–37. [Google Scholar] [CrossRef]
- Verhoef, J.J.F.; Anchordoquy, T.J. Questioning the Use of PEGylation for Drug Delivery. Drug Deliv. Transl. Res. 2013, 3, 499–503. [Google Scholar] [CrossRef]
- Chen, T.; Wu, W.; Xiao, H.; Chen, Y.; Chen, M.; Li, J. Intelligent Drug Delivery System Based on Mesoporous Silica Nanoparticles Coated with an Ultra-pH-Sensitive Gatekeeper and Poly(ethylene glycol). ACS Macro Lett. 2016, 5, 55–58. [Google Scholar] [CrossRef]
- He, Q.; Zhang, J.; Shi, J.; Zhu, Z.; Zhang, L.; Bu, W.; Guo, L.; Chen, Y. The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses. Biomaterials 2010, 31, 1085–1092. [Google Scholar] [CrossRef]
- Feng, W.; Zhou, X.; He, C.; Qiu, K.; Nie, W.; Chen, L.; Wang, H.; Mo, X.; Zhang, Y. Polyelectrolyte multilayer functionalized mesoporous silica nanoparticles for pH-responsive drug delivery: Layer thickness-dependent release profiles and biocompatibility. J. Mater. Chem. B 2013, 1, 5886–5898. [Google Scholar] [CrossRef]
- Rosenholm, J.M.; Mamaeva, V.; Sahlgren, C.; Lindén, M. Nanoparticles in targeted cancer therapy: Mesoporous silica nanoparticles entering preclinical development stage. Nanomedicine 2012, 7, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Hadjesfandiari, N. Stealth coatings for nanoparticles: Polyethylene glycol alternatives. Eng. Biomater. Drug Deliv. Syst. 2018, 345–361. [Google Scholar]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R.H. “Stealth” corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B. Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Ngamcherdtrakul, W.; Morry, J.; Gu, S.; Castro, D.J.; Goodyear, S.M.; Sangvanich, T.; Reda, M.M.; Lee, R.; Mihelic, S.A.; Beckman, B.L.; et al. Cationic Polymer Modified Mesoporous Silica Nanoparticles for Targeted siRNA Delivery to HER2 + Breast Cancer. Adv. Funct. Mater. 2015, 25, 2646–2659. [Google Scholar] [CrossRef] [PubMed]
- Maney, V.; Singh, M. An in vitro assessment of novel chitosan/bimetallic PtAu Nanocomposites as delivery vehicles for doxorubicin. Nanomedicine 2017, 12, 2625–2640. [Google Scholar] [CrossRef]
- Dhopeshwarkar, V.; Zatz, J.L. Evaluation of Xanthan Gum in the Preparation of Sustained Release Matrix Tablets. Drug Dev. Ind. Pharm. 1993, 19, 999–1017. [Google Scholar] [CrossRef]
- Kim, S.W.; Bae, Y.H.; Okano, T. Hydrogels: Swelling, Drug Loading, and Release. Pharm. Res. 1992, 9, 283–290. [Google Scholar] [CrossRef]
- Zhao, W.; Lang, M.; Li, Y.; Li, L.; Shi, J. Fabrication of uniform hollow mesoporous silica spheres and ellipsoids of tunable size through a facile hard-templating route. J. Mater. Chem. 2009, 19, 2778–2783. [Google Scholar] [CrossRef]
- She, X.; Chen, L.; Velleman, L.; Li, C.; Zhu, H.; He, C.; Wang, T.; Shigdar, S.; Duan, W.; Kong, L. Fabrication of high specificity hollow mesoporous silica nanoparticles assisted by Eudragit for targeted drug delivery. J. Colloid Interface Sci. 2015, 445, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, P.; Upadhyay, S. Evaluation of drug release kinetics from ibuprofen matrix tablets using HPMC. J. Appl. Pharm. Sci. 2011, 1, 186–190. [Google Scholar]
- Liu, K.; Liu, P.; Liu, R.; Wu, X. Dual AO/EB staining to detect apoptosis in osteosarcoma cells compared with flow cytometry. Med. Sci. Monit. Basic Res. 2015, 21, 15–20. [Google Scholar]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef]
- Maney, V.; Singh, M. The synergism of Platinum-Gold bimetallic nanoconjugates enhance 5-Fluorouracil delivery in vitro. Pharmaceutics 2019, 11, 439. [Google Scholar] [CrossRef]
- Vidya Priyadarsini, R.; Murugan, S.; Maitreyi, S.; Ramalingam, K.; Karunagaran, D.; Siddavaram, N. The flavonoid quercetin induces cell cycle arrest and mitochondria-mediated apoptosis in human cervical cancer (HeLa) cells through p53 induction and NF-κB inhibition. Eur. J. Pharmacol. 2010, 649, 84–91. [Google Scholar] [CrossRef]
- Finn, R.S.; Aleshin, A.; Slamon, D.J. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016, 18, 17. [Google Scholar] [CrossRef]
- Swift, L.H.; Golsteyn, R.M. Genotoxic anti-cancer agents and their relationship to DNA damage, mitosis, and checkpoint adaptation in proliferating cancer cells. Int. J. Mol. Sci. 2014, 15, 3403–3431. [Google Scholar] [CrossRef]
- Kalsbeek, D.; Golsteyn, R.M. G2/M-Phase Checkpoint Adaptation and Micronuclei Formation as Mechanisms that contribute to Genomic Instability in Human Cells. Int. J. Mol. Sci. 2017, 18, 2344. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, Q.; Teng, X.; Pan, M.; Lin, T.; Zhou, G.; Xu, B.; Yue, Z. Ionizing radiation, but not ultraviolet radiation, induces mitotic catastrophe in mouse epidermal keratinocytes with aberrant cell cycle checkpoints. Exp. Dermatol. 2018, 27, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.-L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Eom, Y.-W.; Kim, M.A.; Park, S.S.; Goo, M.J.; Kwon, H.J.; Sohn, S.; Kim, W.-H.; Yoon, G.; Choi, K.S. Two distinct modes of cell death induced by doxorubicin: Apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 2005, 24, 4765–4777. [Google Scholar] [CrossRef]
- Zhang, B.; Hirahashi, J.; Cullere, X.; Mayadas, T.N. Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis: Cross-talk between caspase 8, reactive oxygen species, and MAPK/ERK activation. J. Biol. Chem. 2003, 278, 28443–28454. [Google Scholar] [CrossRef]
- Slowing, I.I.; Vivero-Escoto, J.L.; Wu, C.W.; Lin, V.S.Y. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv. Drug Deliv. Rev. 2008, 60, 1278–1288. [Google Scholar] [CrossRef]
- Tourne-Peteilh, C.; Bégu, S.; Lerner, D.; Galarneau, A.; Lafont, U.; Devoisselle, J.-M. Sol–gel one-pot synthesis in soft conditions of mesoporous silica materials ready for drug delivery system. J. Sol-Gel Sci. Technol. 2012, 61, 455–462. [Google Scholar] [CrossRef]
- Hu, Y.; Ke, L.; Chen, H.; Zhuo, M.; Yang, X.; Zhao, D.; Zeng, S.; Xiao, X. Natural material-decorated mesoporous silica nanoparticle container for multifunctional membrane-controlled targeted drug delivery. Int. J. Nanomedicine 2017, 12, 8411–8426. [Google Scholar] [CrossRef]
- Wang, J.; Liu, H.; Leng, F.; Zheng, L.; Yang, J.; Wang, W.; Huang, C.Z. Autofluorescent and pH-responsive mesoporous silica for cancer-targeted and controlled drug release. Microporous Mesoporous Mater. 2014, 186, 187–193. [Google Scholar] [CrossRef]
- Meng, H.; Liong, M.; Xia, T.; Li, Z.; Ji, Z.; Zink, J.I.; Nel, A.E. Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and p-glycoprotein siRNA to overcome drug resistance in a cancer cell line. ACS Nano 2010, 4, 4539–4550. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P. The determination of pore volume and area distribution in porous substances. Vol. Area Distrib. Porous Subst. 1951, 73, 373–380. [Google Scholar]
- Zhang, P.; Wu, T.; Kong, J.-L. In Situ Monitoring of Intracellular Controlled Drug Release from Mesoporous Silica Nanoparticles Coated with pH-Responsive Charge-Reversal Polymer. ACS Appl. Mater. Interfaces 2014, 6, 17446–17453. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Akinyelu, J.; Singh, M. Folate-tagged chitosan functionalized gold nanoparticles for enhanced delivery of 5-fluorouracil to cancer cells. Applied Nanoscience 2019, 9, 7–17. [Google Scholar] [CrossRef]
- Maiyo, F.; Moodley, R.; Singh, M. Cytotoxicity, antioxidant and apoptosis studies of quercetin-3-O glucoside and 4-(β-D-glucopyranosyl-1→4-α-L-rhamnopyranosyloxy)-benzyl isothiocyanate from Moringa oleifera. Anticancer Agents Med. Chem. 2016, 16, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Maiyo, F.; Singh, M. Folate-Targeted mRNA Delivery Using Chitosan Functionalized Selenium Nanoparticles: Potential in Cancer Immunotherapy. Pharmaceuticals 2019, 12, 164. [Google Scholar] [CrossRef]
Sample Availability: No samples are available at present. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticle | Mean Diameter (TEM) (nm ± SD) | PDI (SD/mean)2 | Hydrodynamic Diameter (NTA) (nm ± SD) | PDI (SD/mean)2 | Zeta Potential (mV) |
|---|---|---|---|---|---|
| MSN [40] | 36.09 ± 7.08 | 0.0385 | 188 ± 51.6 | 0.0753 | −9.8 ± 1 |
| CMSN [40] | 39.43 ± 7.22 | 0.0335 | 62.2 ± 16 | 0.0662 | 32.4 ± 0.4 |
| 2% PCMSN [40] | 40.75 ± 7.11 | 0.0422 | 12 ± 3.3 | 0.0756 | 17.0 ± 16.5 |
| 5% PCMSN [40] | 40.37 ± 7.70 | 0.0364 | 54.8 ± 2.1 | 0.0015 | 7.4 ± 0.7 |
| 2% PCMSN-DOX | 59.98 ± 12.44 | 0.0430 | 93.0 ± 10.9 | 0.0137 | 0.4 ± 0.7 |
| 5% PCMSN-DOX | 50.82 ± 10.40 | 0.0419 | 111.7 ± 38.2 | 0.1170 | 17.4 ± 0.1 |
| Nanoparticles | Wt% | Wt% Sigma | ||
|---|---|---|---|---|
| MSN | Si | 47.38 | Si | 0.51 |
| O | 52.62 | O | 0.51 | |
| CMSN | Si | 34.00 | Si | 1.26 |
| O | 31.74 | O | 1.44 | |
| C | 34.26 | C | 2.25 | |
| 2% PCMSN | Si | 21.89 | Si | 0.17 |
| O | 49.73 | O | 0.35 | |
| C | 28.38 | C | 0.44 | |
| 5% PCMSN | Si | 34.04 | Si | 0.27 |
| O | 47.16 | O | 0.38 | |
| C | 18.80 | C | 0.56 | |
| Doxorubicin Loaded MSNS | ||
|---|---|---|
| 5% PCMSN | 2% PCMSN | |
| Loading capacity (%) | 93.32 | 97.85 |
| Loading capacity (MGDOX /MGMSN) | 0.9332 | 0.9785 |
| Cell Line | 2% PCMSN-DOX | 5% PCMSN-DOX |
|---|---|---|
| HEK293 | - | - |
| MCF-7 | 20 μg/mL | 20 μg/mL |
| Caco-2 | 20 μg/mL | 50 μg/mL |
| HeLa | 50 μg/mL | 100 μg/mL |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moodley, T.; Singh, M. Sterically Stabilised Polymeric Mesoporous Silica Nanoparticles Improve Doxorubicin Efficiency: Tailored Cancer Therapy. Molecules 2020, 25, 742. https://doi.org/10.3390/molecules25030742
Moodley T, Singh M. Sterically Stabilised Polymeric Mesoporous Silica Nanoparticles Improve Doxorubicin Efficiency: Tailored Cancer Therapy. Molecules. 2020; 25(3):742. https://doi.org/10.3390/molecules25030742
Chicago/Turabian StyleMoodley, Thashini, and Moganavelli Singh. 2020. "Sterically Stabilised Polymeric Mesoporous Silica Nanoparticles Improve Doxorubicin Efficiency: Tailored Cancer Therapy" Molecules 25, no. 3: 742. https://doi.org/10.3390/molecules25030742
APA StyleMoodley, T., & Singh, M. (2020). Sterically Stabilised Polymeric Mesoporous Silica Nanoparticles Improve Doxorubicin Efficiency: Tailored Cancer Therapy. Molecules, 25(3), 742. https://doi.org/10.3390/molecules25030742