Novel Peptide CM 7 Targeted c-Met with Antitumor Activity

 , , ,
, , ,
Abstract
1. Introduction
2. Results
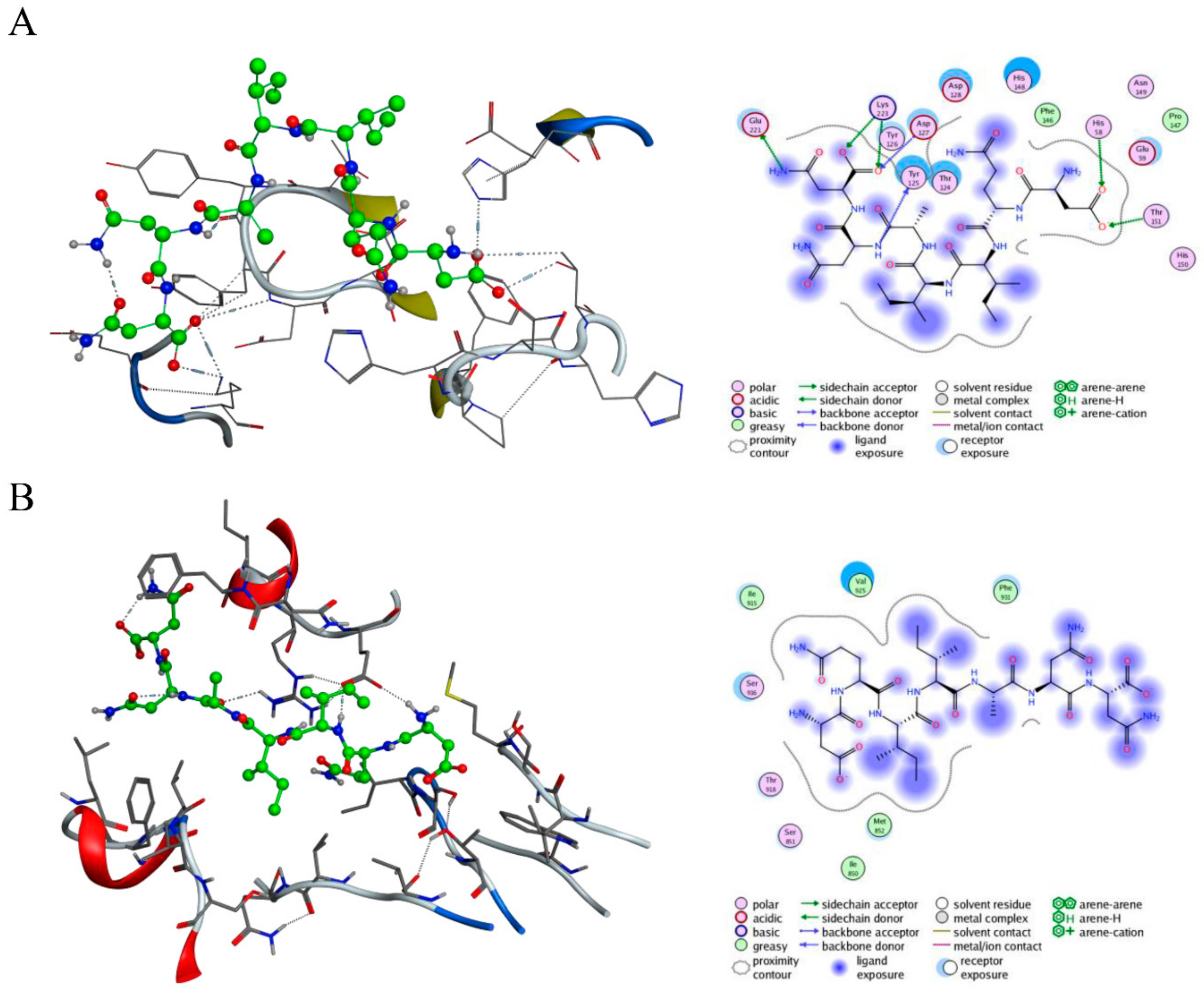
2.1. Design and Virtual Screening of c-Met-Targeting Peptides and Their Interactions with c-Met
2.2. Synthesis and Detection of the c-Met-Targeting Peptide CM 7 and the Random Sequence CM 14
2.3. Affinity of the c-Met-Targeting Peptide CM 7 or CM 14 with Its Target
2.4. The c-Met-Targeting Peptide CM 7 Regulates c-Met-Driven Cell Proliferation
2.5. Peptide CM-7 Inhibits c-Met-Dependent Proliferation by Inducing S Arrest
2.6. The c-Met-Targeting Peptide CM 7 Regulates c-Met-Dependent Cell Invasion, Migration, and Scattering
2.7. Peptide CM-7 Potently Inhibits c-Met Activation and Signaling in Cancer Cells
2.8. Peptide CM7 Inhibits c-Met-Mediated Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Design and Screening of Peptides Targeting c-Met
4.2. Cell Culture
4.3. Peptide Preparation
4.4. Flow Cytometry Analysis
4.5. Confocal Microscope Imaging Analysis
4.6. Cell Proliferation Assays
4.7. Cell Invasion and Wound Healing Assays
4.8. Cell Scatter Assay
4.9. Cell Cycle Distribution Assay
4.10. Western Blot Analysis
4.11. In Vivo Antitumor Activity Assay
4.12. Immunohistochemistry Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.L.; Kmiecik, T.E.; Woude, G.F.V.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Ponzetto, C.; Direnzo, M.F.; Cooper, C.S.; Comoglio, P.M. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 1989, 339, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Tempest, P.R.; Stratton, M.R.; Cooper, C.S. Structure of the met protein and variation of met protein kinase activity among human tumour cell lines. Br. J. Cancer 1988, 58, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.; Lazarus, R.A.; Yao, X.; Kirchhofer, D.; Wiesmann, C. Crystal structure of the HGF b-chain in complex with the Sema domain of the Met receptor. EMBO J. 2004, 23, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Vigna, E.; Chiriaco, C.; Cignetto, S.; Fontani, L.; Basilico, C.; Petronzelli, F.; Comoglio, P.M. Inhibition of ligand-independent constitutive activation of the Met oncogenic receptor by the engineered chemically-modified antibody DN30. Mol. Oncol. 2015, 9, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Basilico, C.; Arnesano, A.; Galluzzo, M.; Comoglio, P.M.; Michieli, P. A high affinity hepatocyte growth factor-binding site in the immunoglobulin-like region of Met. J. Biol. Chem. 2008, 283, 21267–21277. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef]
- Ma, W.W.; Adjei, A.A. Novel agents on the horizon for cancer therapy. CA Cancer J. Clin. 2009, 59, 111–137. [Google Scholar] [CrossRef]
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300. [Google Scholar] [CrossRef]
- Suzuki, Y.; Sakai, K.; Ueki, J.; Xu, Q.; Nakamura, T.; Shimada, H.; Nakamura, T.; Matsumoto, K. Inhibition of Met/HGF receptor and angiogenesis by NK4 leads to suppression of tumor growth and migration in malignant pleural mesothelioma. Int. J. Cancer 2010, 127, 1948–1957. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Borowiak, M.; Garratt, A.N.; Wüstefeld, T.; Strehle, M.; Trautwein, C.; Birchmeier, C. Met provides essential signals for liver regeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 10608–10613. [Google Scholar] [CrossRef]
- Gavine, P.R.; Ren, Y.; Han, L.; Lv, J.; Fan, S.; Zhang, W.; Xu, W.; Liu, Y.J.; Zhang, T.; Fu, H.; et al. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol. Oncol 2015, 9, 323–333. [Google Scholar] [CrossRef]
- Burggraaf, J.; Kamerling, I.M.C.; Gordon, P.B.; Schrier, L.; de Kam, M.L.; Kales, A.J.; Bendiksen, R.; Indrevoll, B.; Bjerke, R.M.; Moestue, S.A.; et al. Detection of colorectal polyps in humans using an intravenously administered fluorescent peptide targeted against c-Met. Nat. Med. 2015, 21, 955–961. [Google Scholar] [CrossRef]
- Zhang, H.; Feng, Q.; Chen, W.D.; Wang, Y.D. HGF/c-MET: A Promising Therapeutic Target in the Digestive System Cancers. Int. J. Mol. Sci. 2018, 19, 3295. [Google Scholar] [CrossRef]
- Wu, Y.; Fan, Q.; Zeng, F.; Zhu, J.; Chen, J.; Fan, D.; Li, X.; Duan, W.; Guo, Q.; Cao, Z.; et al. Peptide-Functionalized Nanoinhibitor Restrains Brain Tumor Growth by Abrogating Mesenchymal-Epithelial Transition Factor (MET) Signaling. Nano. Lett. 2018, 18, 5488–5498. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Y.; Liu, Y.; Zhang, W.; Liu, Y.; Yang, X.; Cao, Y.; Wang, S. C7 peptide inhibits hepatocellular carcinoma metastasis by targeting the HGF/c-Met signaling pathway. Cancer Biol. Ther. 2019, 20, 1430–1442. [Google Scholar] [CrossRef]
- Pockley, A.G.; Lindsay, J.O.; Foulds, G.A.; Rutella, S.; Gribben, J.G.; Alexander, T.; Snowden, J.A. Immune Reconstitution After Autologous Hematopoietic Stem Cell Transplantation in Crohn’s Disease: Current Status and Future Directions. A Review on Behalf of the EBMT Autoimmune Diseases Working Party and the Autologous Stem Cell Transplantation In Refractory CD-Low Intensity Therapy Evaluation Study Investigators. Front. Immunol. 2018, 9, 646. [Google Scholar]
- Zhang, Y.; Su, Y.; Volpert, O.V.; Woude, G.F.V. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 12718–12723. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nawa, K.; Ichihara, A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem. Biophys. Res. Commun. 1984, 122, 1450–1459. [Google Scholar] [CrossRef]
- Al-U’datt, D.G.F.; Al-Husein, B.A.A.; Qasaimeh, G.R. A mini-review of c-Met as a potential therapeutic target in melanoma. Biomed. Pharmacother. 2017, 88, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.K.; Wu, H.C.; Shen, Y.C.; Hsieh, H.Y.; Yang, S.Y.; Chang, C.C. Kruppel-like factor 4 is involved in cell scattering induced by hepatocyte growth factor. J. Cell Sci. 2012, 125, 4853–4864. [Google Scholar] [CrossRef]
- Kong-Beltran, M.; Stamos, J.; Wickramasinghe, D. The Sema domain of Met is necessary for receptor dimerization and activation. Cancer Cell 2004, 6, 75–84. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.-H.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef]
- Turecek, P.L.; Bossard, M.J.; Schoetens, F.; Ivens, I.A. PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J. Pharm. Sci. 2016, 105, 460–475. [Google Scholar] [CrossRef]
- Milla, P.; Dosio, F.; Cattel, L. PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr. Drug Metab. 2012, 13, 105–119. [Google Scholar] [CrossRef]
- Tan, Y.S.; Lane, D.P.; Verma, C.S. Stapled peptide design: principles and roles of computation. Drug Discov. Today 2016, 21, 1642–1653. [Google Scholar] [CrossRef]
- Lau, Y.H.; Wu, Y.; Rossmann, M.; Tan, B.X.; de Andrade, P.; Tan, Y.S.; Verma, C.; McKenzie, G.J.; Venkitaraman, A.R.; Hyvonen, M.; et al. Double Strain-Promoted Macrocyclization for the Rapid Selection of Cell-Active Stapled Peptides. Angew. Chem. Int. Ed. Engl. 2015, 54, 15410–15413. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.W.; Bahng, N.; Ito, K.; Ha, S.; Kim, M.Y.; Lee, E.; Suga, H.; Lee, D.S. In vivo targeting of c-Met using a non-standard macrocyclic peptide in gastric carcinoma. Cancer Lett. 2017, 385, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.; Sakai, K.; Imamura, R.; Ito, K.; Suga, H.; Sakuma, T.; Yamamoto, T.; Matsumoto, K. MET Activation by a Macrocyclic Peptide Agonist that Couples to Biological Responses Differently from HGF in a Context-Dependent Manner. Int. J. Mol. Sci. 2018, 19, 3141. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Anderson, K.M.; Chang, C.L.; Makarewich, C.A.; Nelson, B.R.; McAnally, J.R.; Kasaragod, P.; Shelton, J.M.; Liou, J.; Bassel-Duby, R.; et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 2015, 160, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Li, C.; Zhang, N.; Zhou, J.; Ding, C.; Jin, Y.; Cui, X.; Pu, K.; Zhu, Y. Peptide Blocking of PD-1/PD-L1 Interaction for Cancer Immunotherapy. Cancer Immunol. Res. 2018, 6, 178–188. [Google Scholar] [CrossRef]
- Brockmann, M.A.; Papadimitriou, A.; Brandt, M.; Fillbrandt, R.; Westphal, M.; Lamszus, K. Inhibition of Intracerebral Glioblastoma Growth by Local Treatment with the Scatter Factor/Hepatocyte Growth Factor-Antagonist NK4. Clin. Cancer. Res. 2003, 9, 4578–4585. [Google Scholar]
- Okamoto, W.; Okamoto, I.; Arao, T.; Kuwata, K.; Hatashita, E.; Yamaguchi, H.; Sakai, K.; Yanagihara, K.; Nishio, K.; Nakagawa, K. Antitumor action of the MET tyrosine kinase inhibitor crizotinib (PF-02341066) in gastric cancer positive for MET amplification. Mol. Cancer. Ther. 2012, 11, 1557–1564. [Google Scholar] [CrossRef]
- Heigener, D.F.; Reck, M. Crizotinib. Recent Results Cancer Res. 2018, 211, 57–65. [Google Scholar]
- He, C.X.; Ai, J.; Xing, W.Q.; Chen, Y.; Zhang, H.T.; Huang, M.; Hu, Y.H.; Ding, J.; Geng, M.Y. Yhhu3813 is a novel selective inhibitor of c-Met kinase that inhibits c-Met-dependent neoplastic phenotypes of human cancer cells. Acta Pharmacol. Sin. 2014, 35, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Justus, C.R.; Leffler, N.; Ruiz-Echevarria, M.; Yang, L.V. In vitro cell migration and invasion assays. J. Vis. Exp. 2014, e51046. [Google Scholar] [CrossRef]
- Li, M.; Han, Y.; Zhou, H.; Li, X.; Lin, C.; Zhang, E.; Chi, X.; Hu, J.; Xu, H. Transmembrane protein 170B is a novel breast tumorigenesis suppressor gene that inhibits the Wnt/beta-catenin pathway. Cell Death Dis. 2018, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Longati, P.; Bardelli, A.; Ponzetto, C.; Naldini, L.; Comoglio, P.M. Tyrosines1234–1235 are critical for activation of the tyrosine kinase encoded by the MET proto-oncogene (HGF receptor). Oncogene 1994, 9, 49–57. [Google Scholar]
Sample Availability: Samples of the peptide CM 7 is available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Sequences | Scoring | |
|---|---|---|---|
| Sema | IPT | ||
| CM 1 | ADQCANRCT | −7.01 | −7.48 |
| CM 2 | VPGRGC | −5.94 | −6.77 |
| CM 3 | DQIANRC | −7 | −7.19 |
| CM 4 | DQCANR | −7.02 | −7.4 |
| CM 5 | DQIANR | −6.69 | −7.86 |
| CM 6 | DQIANN | −5.98 | −6.84 |
| CM 7 | DQIIANN | −7.07 | −7.23 |
| CM 8 | VPGRGD | −6.54 | −6.81 |
| CM 9 | VPGRGS | −6.28 | −6.68 |
| CM 10 | VPGNGS | −6.42 | −6.58 |
| CM 11 | VNGRGS | −6.26 | −7.04 |
| CM 12 | VQGRGS | −6.15 | −6.92 |
| CM 13 | VQGRGC | −6.16 | −7.64 |
| CM 14 | QTRIYWQKE | −5.81 | −6.14 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, C.; Wang, Y.; Liu, C.; Wang, L.; Gao, X.; Li, D.; Qi, W.; An, R.; Xu, H. Novel Peptide CM 7 Targeted c-Met with Antitumor Activity. Molecules 2020, 25, 451. https://doi.org/10.3390/molecules25030451
Xia C, Wang Y, Liu C, Wang L, Gao X, Li D, Qi W, An R, Xu H. Novel Peptide CM 7 Targeted c-Met with Antitumor Activity. Molecules. 2020; 25(3):451. https://doi.org/10.3390/molecules25030451
Chicago/Turabian StyleXia, Chunlei, Ying Wang, Chen Liu, Liwen Wang, Xinmei Gao, Dongping Li, Weiyan Qi, Roujin An, and Hanmei Xu. 2020. "Novel Peptide CM 7 Targeted c-Met with Antitumor Activity" Molecules 25, no. 3: 451. https://doi.org/10.3390/molecules25030451
APA StyleXia, C., Wang, Y., Liu, C., Wang, L., Gao, X., Li, D., Qi, W., An, R., & Xu, H. (2020). Novel Peptide CM 7 Targeted c-Met with Antitumor Activity. Molecules, 25(3), 451. https://doi.org/10.3390/molecules25030451