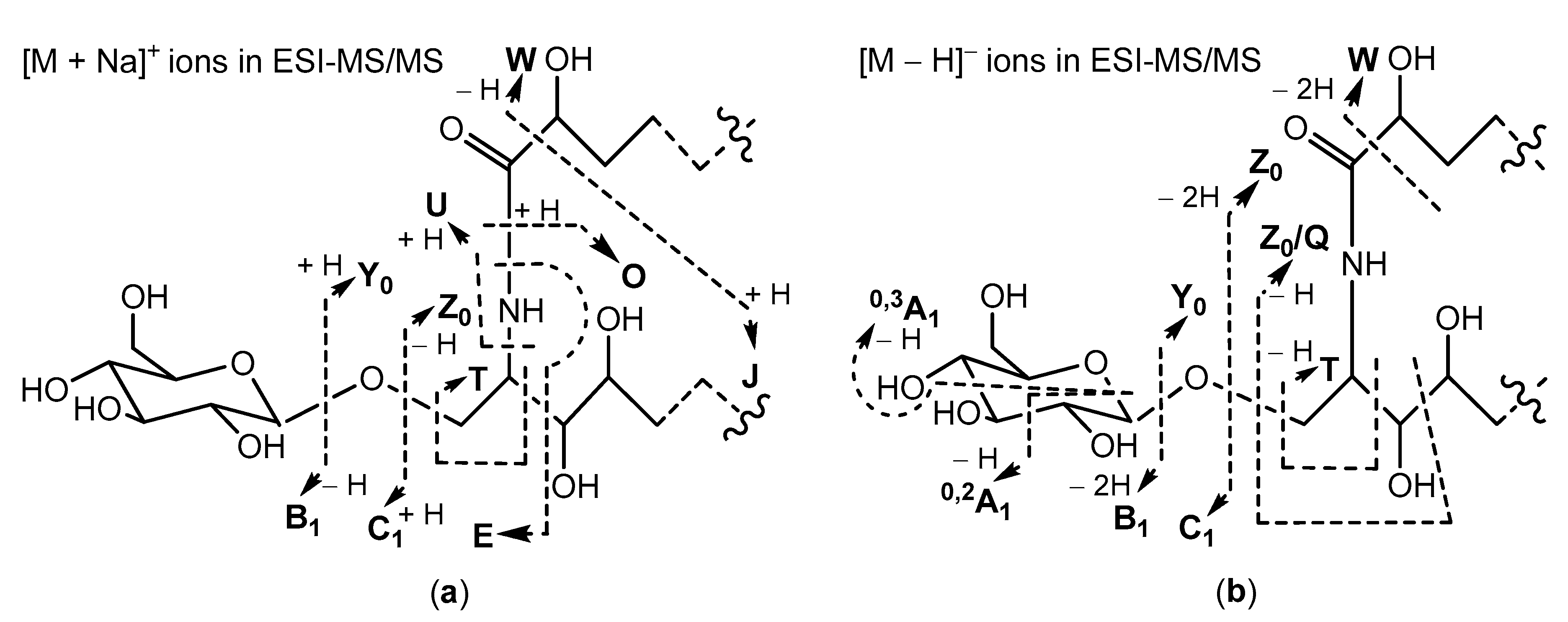
2.1. Positive and Negative Ion Mode ESI-MS/MS Analyses of Oxidized Cerebrosides
The molecular formulae, С
49H
93NO
12 for allylic hydroperoxides (
1a,
1b,
2a,
2b,
3c,
3d), С
49H
93NO
11 for allylic alcohols (
1a/,
1b/,
2a/,
2b/,
3c/,
3d/), and С
49H
91NO
11 for enones (
1a//,
1b//,
2a//,
2b//,
3c//,
3d//), were determined by HR-ESI-MS (high resolution ESI-MS) analyses in positive-((+)ESI-MS) and negative-((–)ESI-MS) ion modes. Complementary (+)ESI- and (–)ESI-MS/MS analyses of these glucosylceramides, containing 2-hydroxy acyl chains and phytosphingosine-type backbones, resulted in a series of fragment ions, as shown in
Scheme 1.
Many of the fragment ions, shown in
Scheme 1, have also been detected in our ESI-MS/MS studies of non-oxidized cerebrosides isolated from
Aulosaccus sp. Namely, (+)ESI-MS/MS spectra of sodium adducts from non-oxidized glycosphingolipids have been characterized by prominent peaks, corresponding to [M + Na]
+ (base peak),
Y0,
Z0,
O, and
C1 ions, and by small peaks, representing [M + Na − H
2O]
+,
E, and
B1 ions. However, the (+)ESI-MS/MS spectrum of the [M + Na]
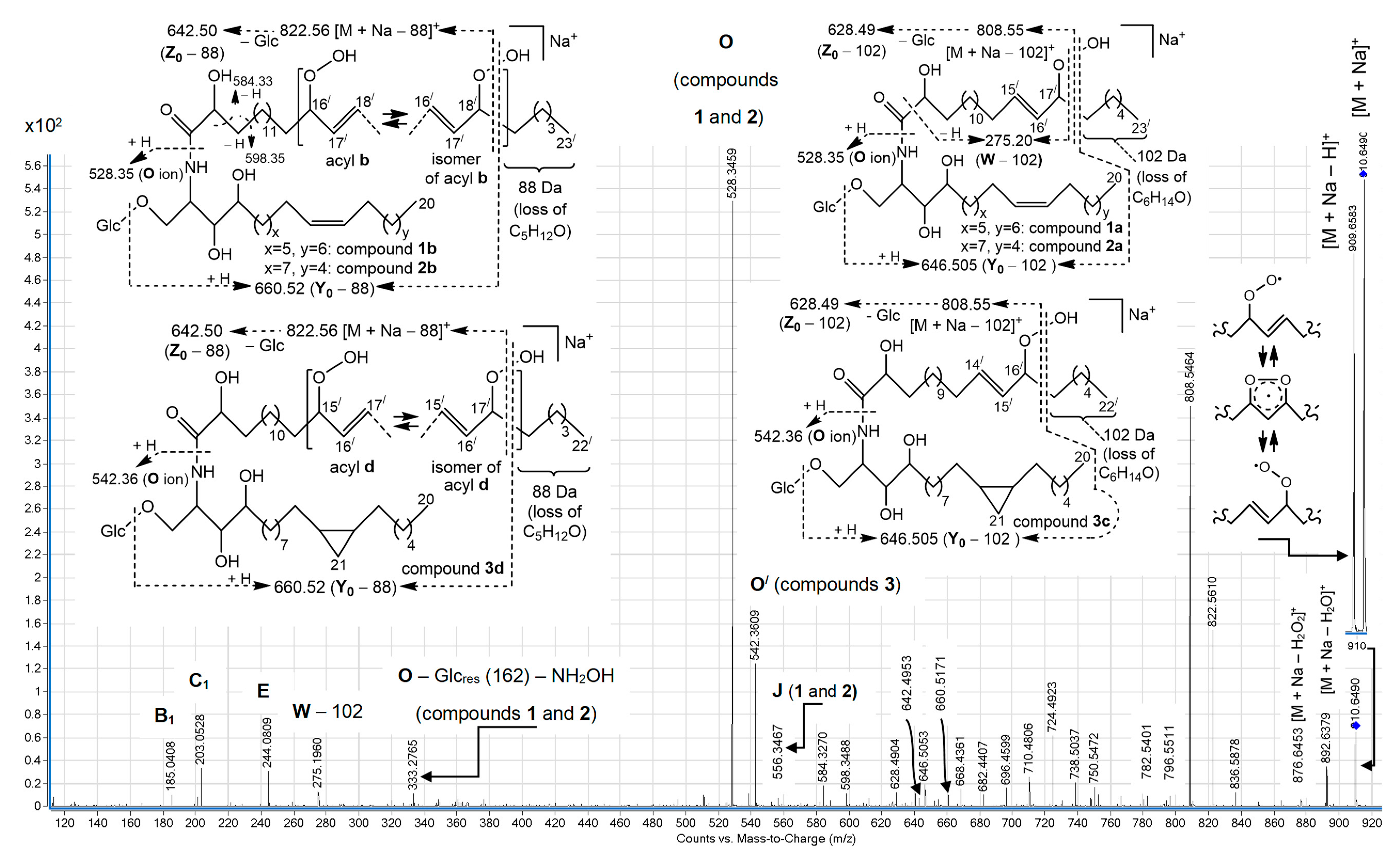
+ ion of isomeric hydroperoxy cerebrosides (
Figure 2) showed other relative abundances for a variety of these ions. In particular, the
O ions of
m/
z 528.35, representing isomeric monoglucosylated monounsaturated С
20 sphingoid base backbones
1 and
2, constituted the base peak of this spectrum. The spectrum also exhibited a homologous, less abundant
O/ ion (
m/
z 542.36), containing a monoglucosylated cyclopropane С
21 sphingoid base backbone
3. A relatively low intensity pseudo-molecular ion peak (
m/
z 910.65, [M + Na]
+) and very small peaks, corresponding to
Y0 (
m/
z 748.60) and
Z0 (
m/
z 730.58) ions (not shown), were observed. The presence of the [M + Na − Н]
+ peak, comparable with the pseudo-molecular ion peak of the hydroperoxides, was explained by hydrogen atom abstraction, followed by electronic delocalization in the resulting radical, which might yield rearranged products. At the same time, the (+)ESI-MS/MS spectrum revealed more abundant [M + Na − 102]
+ (
m/
z 808.55), [M + Na − 88]
+ (
m/
z 822.56), [
Y0 − 102] (
m/
z 646.505), [
Y0 − 88] (
m/
z 660.52), [
Z0 − 102] (
m/
z 628.49), and [
Z0 − 88] (
m/
z 642.50) ions. These fragments and fragment [
W − 102] (
m/
z 275.20), each with a terminal α,β-unsaturated aldehyde, were thought to arise from specific α-cleavages of lipid hydroperoxides (
Figure 2). Related fragment ions, formed by C
nH
2n+2O losses from [M + Na]
+ ions, were also found in the MS/MS studies of some monoenoic [
6] and polyenoic [
11,
12,
13] FA moieties or free FAs, in which an allylic hydroperoxy group was between double bond(s) and a terminal methyl group. In general, the [M + Na − C
nH
2n+2O]
+ ions were also more abundant in MS/MS spectra of those compounds compared to their [M + Na]
+ ions.
Ions, possibly formed by Hock cleavage [
14,
15], were insignificant in our (+)ESI-MS/MS analysis of allylic hydroperoxides. These fragments included
m/
z 796.55 (for
1b,
2b, and
3d) and 782.54 (for the allylic isomers of
1a,
2a, and
3c) ions, as illustrated in
Scheme S1 (Supplementary Materials) and
Figure 2 (allylic rearrangements for acyl chains
a and
c are not shown). In contrast, the relatively abundant ions, presumably formed by analogous cleavage from hemiacetal derivatives, were reported for MS/MS fragmentations of [M + Na]
+ precursors of some free polyenoic FAs in which an allylic hydroperoxy group was between the double bond system and C-1 [
12,
13].
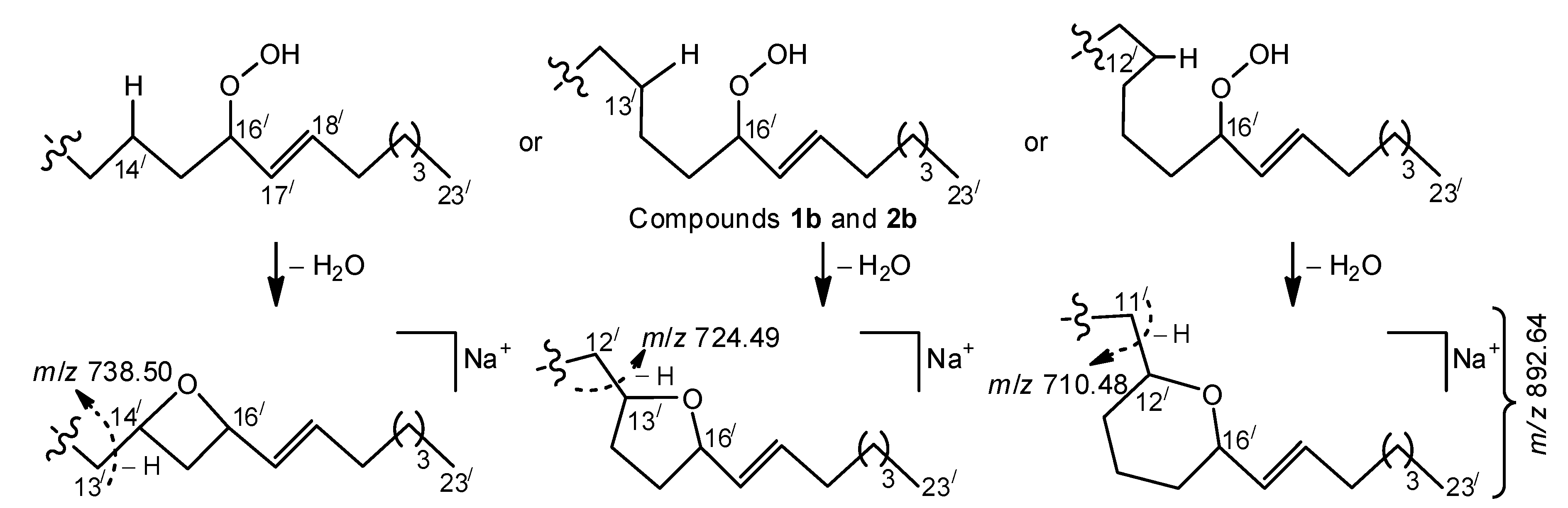
Favorable cleavage of the isomers, formed by allylic rearrangements of acyl chains
b and
d (
Figure 2), occurred, losing an 88 Da fragment. At the same time, compounds with parent acyl chains
b and
d underwent other favorable fragmentations, yielding a distinct group of homologous ions (from
m/
z 668.44 to 738.50), containing the most significant fragment of
m/
z 724.49. We suggest that the occurrence of these ions may be connected with homolysis of a weak RO-̸-OH bond, formation of an alkoxyl radical, and subsequent formation of a radical centered on a remote and non-activated carbon atom of a saturated hydrocarbon chain. This may lead to fast cyclization that, in turn, leads to MS fragmentations of the resulting cyclic ethers, shown in
Scheme 2. The proposed cyclization reaction is reminiscent of the formation of cyclic (mainly, five-membered) ethers from acyclic saturated monohydroxy alcohols, occurring via radical (primarily alkoxyl) intermediates under appropriate chemical and thermal conditions (for reviews, see References [
16,
17]). In this process, secondary aliphatic alcohols yielded 2,5-dialkyltetrahydrofurans [
18]. A possible mechanism for the formation of such products includes 1,5-transposition of the radical center from the oxygen atom of the alkoxyl radical to
δ-carbon atom, involving a 1,5-hydrogen transfer through a chair-like six-membered transition state. For a linear hydrocarbon chain with an initial alkoxyl radical, the 1,5-hydrogen atom transfer may be considered the most common reaction, even though intramolecular abstractions of the hydrogen atom from other positions (1,4-migrations, 1,6-migrations, and 1,7-migrations, etc.) may be observed [
17]. Similar processes may have occurred in our MS/MS experiment because alkyl hydroperoxides are known to form alkoxyl radicals by thermal or photolytic decomposition [
17,
19]. Apparently, the ability to undergo favorable cyclic transition states affected the fragmentation process, leading to the formation of major homologous ions, as illustrated in
Scheme 2.
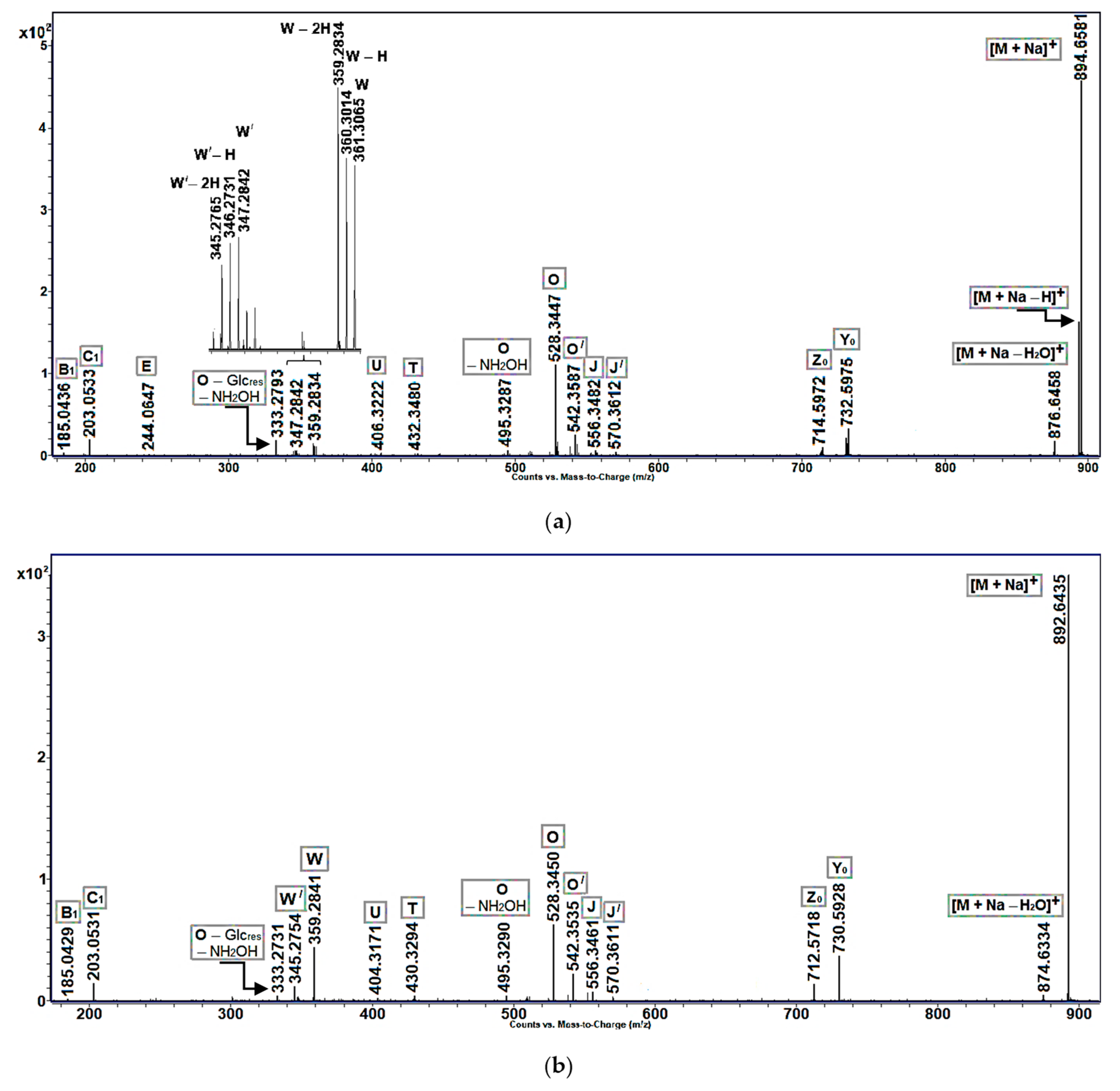
Ion intensity profiles, obtained for cerebrosides with allylic hydroxy or keto groups, were, in general, similar to those of non-oxidized cerebrosides found in
Aulosaccus sp. In particular, the MS/MS spectra of sodiated molecular ions of allylic alcohols (
Figure 3a:
m/
z 894.66 [M + Na]
+) and enones (
Figure 3b:
m/
z 892.64 [M + Na]
+) showed predominant peaks of pseudo-molecular ions, along with peaks of lower intensities, which represented [M + Na − H
2O]
+,
Y0,
Z0, and
O ions. However, unlike [M + Na]
+ ions of non-oxidized cerebrosides, the sodium adducts of allylic alcohols and enones fragmented to give discernible ions of
W-type and minor
U and
T ions. Additionally, the acyl-containing ions of allylic alcohols had a tendency to lose one, or even two, hydrogen atoms. In this case, a trend was observed toward the increased loss of hydrogen atoms with decreasing ion masses. For example, a significant difference was noted between the relative intensities of [M + Na]
+ and [M + Na − H]
+ peaks ([M + Na − 2H]
+ ions were not even detected), but the intensities of
W, [
W − H], and [
W − 2H] peaks were comparable (
Figure 3a).
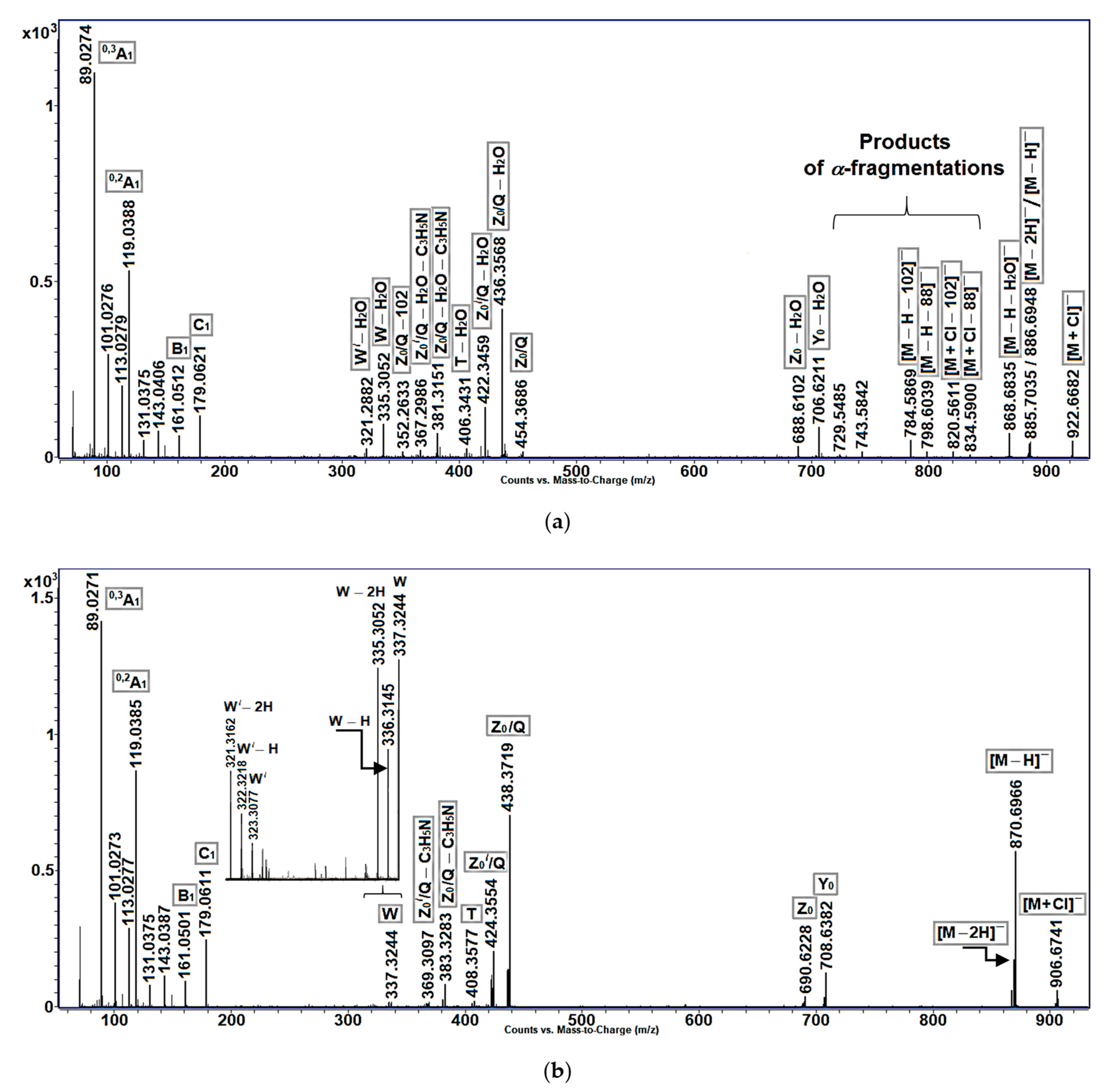
In (−)ESI-MS/MS experiments with [M − H]
− and [M + Cl]
− ions (
Figure 4a–c), allylic hydroperoxides again produced more fragments than allylic alcohols and enones. The main feature of (−)ESI-MS/MS fragmentation of [M − H]
−,
Y0,
Z0,
Z0/
Q,
T,
W, and other precursor ions, containing an allylic hydroperoxy group, was the loss of water to produce fragments with enone functionality in acyl chains, as described for [M − H]
− ions of hydroperoxy-eicosatetraenoic acids [
20]. In particular, the homologous ions of
m/
z 436.4 ([
Z0/
Q − H
2O]) and 422.4 ([
Z0//
Q − H
2O]), containing acyl C
23 and С
22 chains, respectively (
Figure 4a), were also observed in (−)ESI-MS/MS spectrum of enones (
Figure 4c). Then, relatively low-intensity pseudo-molecular ion peaks ([M + Cl]
− and [M − H]
−) and very small peaks, corresponding to
Y0 and
Z0 ions (not shown), were seen in the (−)ESI-MS/MS spectrum of allylic hydroperoxides. A discernible [M − 2H] peak was comparable with a pseudo-molecular [M − H] peak. This spectrum also exhibited [M + Cl − 102]
−, [M + Cl − 88]
−, [M − H − 102]
−, [M − H − 88]
−, and [
Z0/Q − 102] ions, interpreted as α-cleavage ions with a terminal α,β-unsaturated aldehyde. Additionally, the two minor ions of
m/
z 743.6 and 729.55 could be formed by α-cleavages of compounds in which an allylic hydroperoxy group was between a double bond and C-1
/. In particular, the
m/
z 743.6 ions could be fragments of isomeric compounds
1b,
2b (C-15
/−C-16
/ bond fission), and
3d (C-14
/−C-15
/ bond fission), while the less abundant
m/
z 729.55 ions could be fragments of the allylic isomers of compounds
1a,
2a (C-14
/−C-15
/ bond fission), and
3c (C-13
/−C-14
/ bond fission).
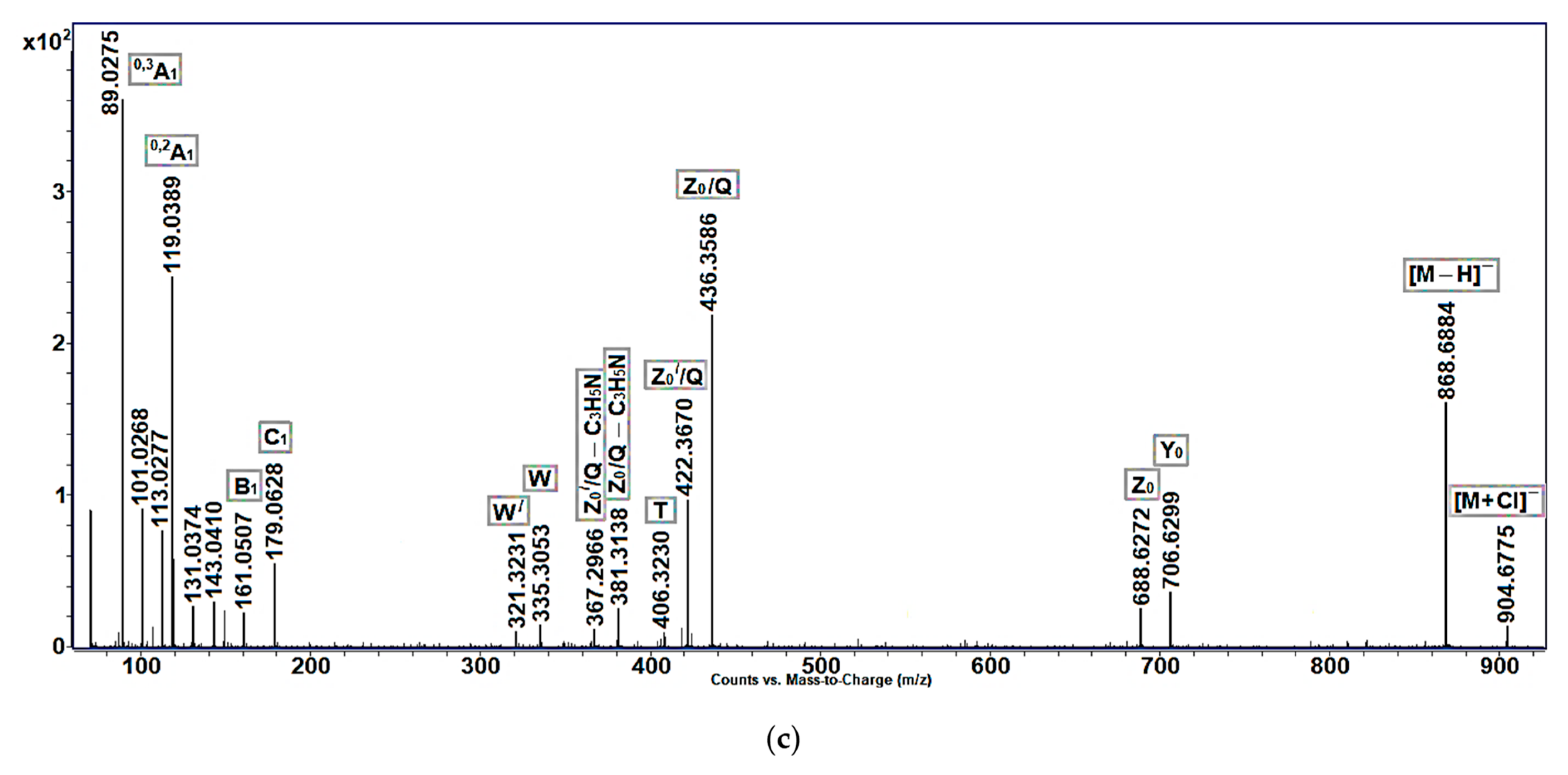
Like the (−)ESI-MS/MS spectra of the non-oxidized cerebrosides of
Aulosaccus sp., those of allylic alcohols (
Figure 4b) and enones (
Figure 4c) exhibited significant peaks corresponding to [M − H]
− and
Z0/
Q ions, with lower intensity peaks representing [M + Cl]
−,
Y0,
Z0, [
Z0/
Q − C
3H
5N], and
W ions. The negatively charged acyl-containing ions of allylic alcohols (
Figure 4b), like their positively charged acyl-containing counterparts (
Figure 3a), tended to lose hydrogen atoms in the MS/MS experiment.
2.2. NMR Characterization of Oxidized Cerebrosides
The
1Н-NMR and
13С-NMR spectra (CD
3OD) of oxidized cerebrosides (
Table 1,
Figures S2 and S3), as well as the corresponding spectra of the non-oxidized cerebrosides of
Aulosaccus sp. [
8], showed signals of β-glucopyranosyl-(1→1)-ceramides that had monoenoic or cyclopropane-containing phytosphingosine-type backbones,
N-acylated with 2-hydroxy FAs. In particular, H-2 (
δН 4.25, m) of the sphingoid base moieties displayed a characteristic cross signal with
N-acyl C-1
/ (
δC 177.7), as demonstrated by an НMBC experiment with sphingolipids
1–
3.
1Н,
1Н-COSY diagram indicated that several protons, starting from −O−CH
2− (
δН 3.80 and 4.045, dd, CH
2-1) and ending with alkyl −CH
2− (
δН 1.31 and 1.55, m, CH
2-6), formed a linear spin system of phytosphingosine-type moieties of
1–
3. Another spin system consisted of CH-2
/ (
δН 4.01, dd), CH
2-3
/ (
δН 1.60 and 1.74, m), and CH
2-4
/ (
δН 1.42, m) protons of 2-hydroxy acyl chains. The signals of a β-glucopyranoside moiety were sequentially assigned by
1Н,
1Н-COSY, HSQC (heteronuclear single-quantum correlation spectroscopy), and НМВС experiments, starting from the signal of anomeric CH-1
// (
δН 4.28, d,
J = 7.8 Hz;
δC 105.3). Accordingly, cross signals C
Н2-1/
С-1
// and C
Н-1
///
С-1 were observed in the НМВС diagram of glycosides
1–
3. Then, the
1Н- and
13С-NMR spectra of these sphingolipids showed signals of long hydrocarbon chains (−(CH
2)
n−,
δН 1.22–1.42, m,
δС 30.6–32.0) and terminal methyl groups (
δН 0.89–0.90, several overlapping triplets,
δС 15.0–15.05, broad signal). The
δ values of allylic CH
2 (
δC 28.7–28.85,
δН 2.02, m) and olefinic CH (
δН 5.34, m) were used to characterize
cis-double bonds of backbones
1 and
2. These data were in good agreement with
1Н-NMR and
13С-NMR (CD
3OD) data on some cerebrosides [
21] and FA standards [
22] with isolated
cis-double bonds (
cis-isomers:
δC 28.7–28.9,
δН 2.02–2.03 m, allylic CH
2, and
δH 5.335–5.34 m, olefinic CH,
trans-isomers:
δC 34.15–34.2,
δН 1.97–1.975 m, allylic CH
2, and
δН 5.37–5.38 m, olefinic CH). A
cis-cyclopropane ring in backbone
3 caused three upfield shifted signals at
δН −0.33 (dt,
J = 4.1, 5.3 Hz, H-21a), 0.58 (ddd,
J = 4.1, 8.3, 8.3 Hz, H-21b), and 0.67 (m, H-13, H-14).
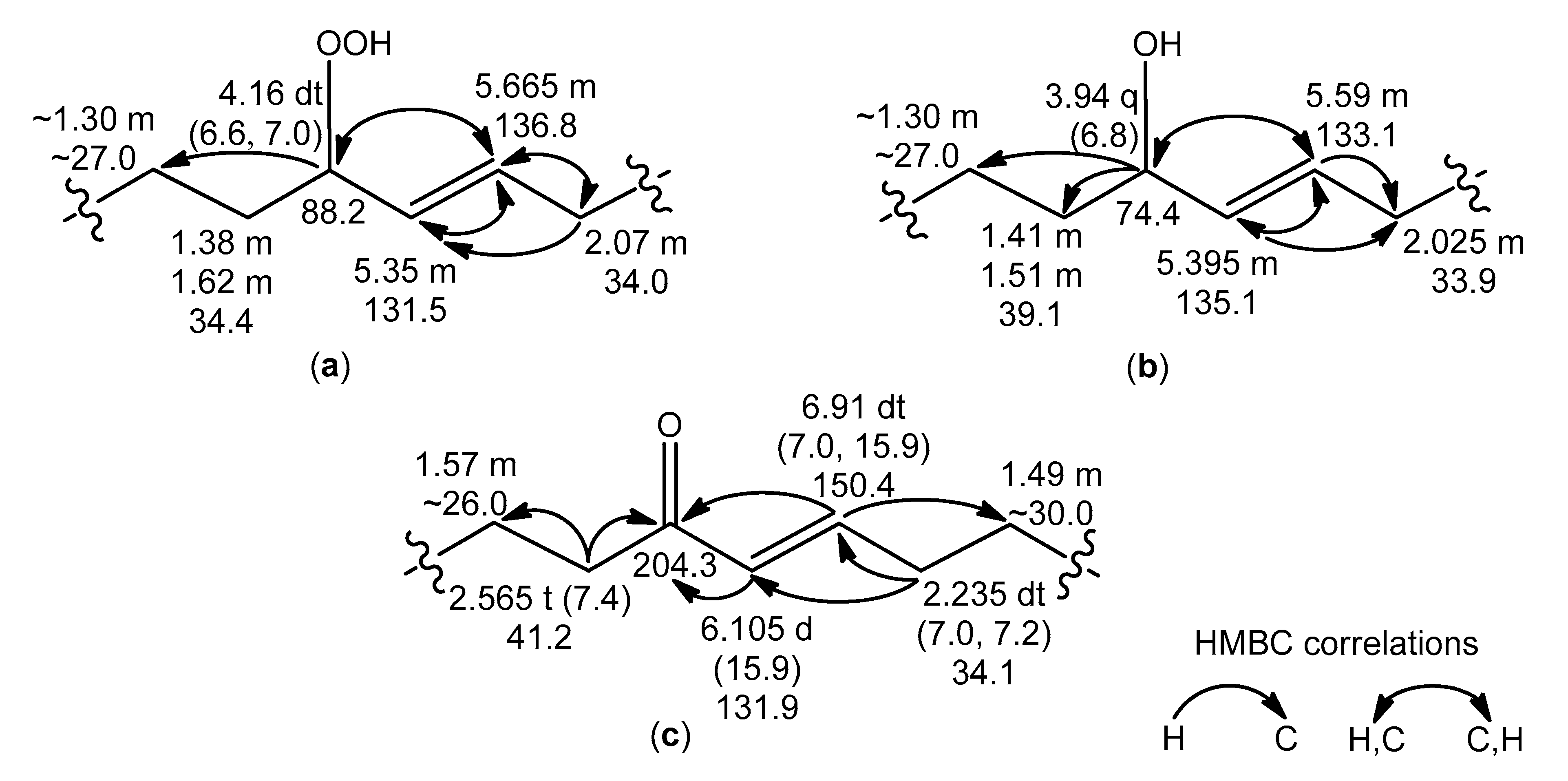
Apart from the previously mentioned NMR resonances, the signals of
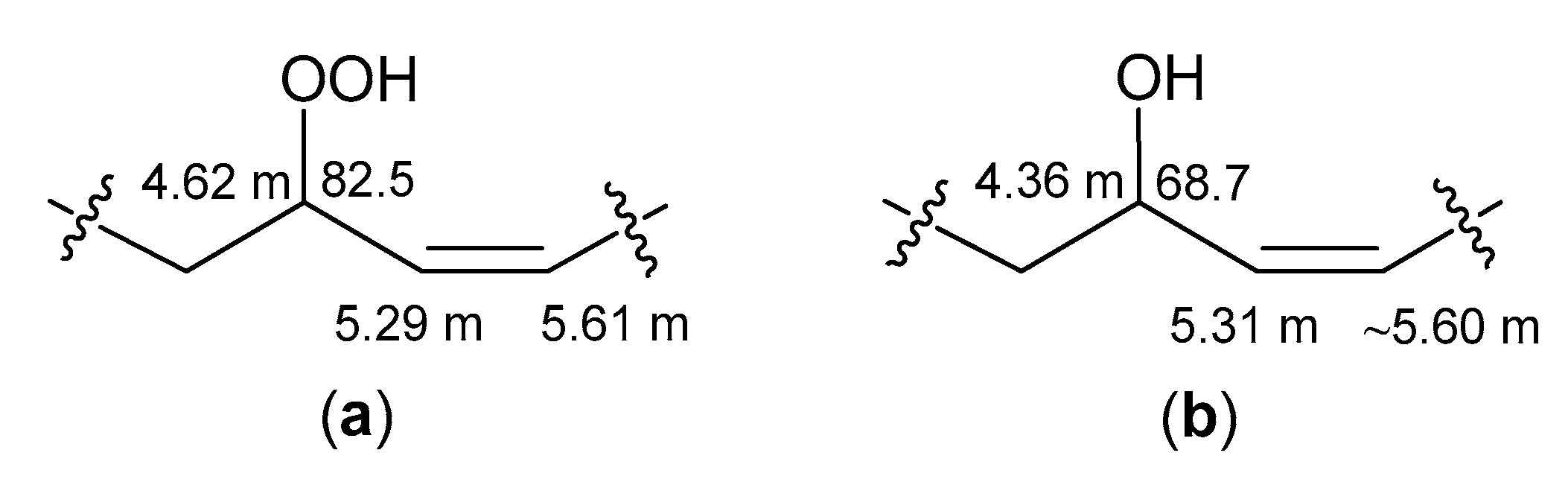
trans-monoenoic acyl moieties with an allylic hydroperoxy, hydroxy, or keto group (
Figure 5a–c) were observed in the NMR spectra of the RP-HPLC fraction, containing oxidized cerebrosides. The
δH values (CD
3OD) of the signals for –
HC(OOH)– (4.16, dt), –
HC(OH)– (3.94, q), and −CH=C
H–CO– (6.105, d) were close to
δH values (CDCl
3) of signals of corresponding protons in the
trans-monoenoic allylic hydroperoxides (4.2, q), allylic alcohols (4.0, q), and enones (6.1, d), respectively, prepared from oleic acid [
23]. Deviations of
δH values (this study) from those in other experiments [
23] arose from solvent effects. According to
1Н,
1Н-COSY correlations, a spin system, assigned to acyl chains
a–
d (
Figure 5a), included protons of allylic hydroperoxide, especially two olefinic CH (
δН 5.35 and 5.665, m) and allylic CH (
δН 4.16, dt,
J = 6.6, 7.0 Hz), bearing −OOH. Allylic CH (
δН 3.94, q,
J = 6.8 Hz), bearing –OH, and two olefinic CH (
δН 5.395 and 5.59, m) groups, belonging to allylic alcohol, were part of a spin system within acyl chains
a/–
d/ (
Figure 5b). In experiments, involving selective irradiation of allylic protons, the coupling constants
J = 15.6 Hz and
J = 15.4 Hz for the
trans-olefinic protons of the allylic hydroperoxides and allylic alcohols, respectively, were detected. The characteristic NMR resonances of two
trans-alkenyl CH (
δН 6.105, d,
J = 15.9 Hz, and 6.91, dt,
J = 7.0, 15.9 Hz), conjugated C=O (
δС 204.3), and α-CH
2 (
δН 2.565, t,
J = 7.4 Hz) groups were used for determining substructures of enones in acyl chains
a//–
d// (
Figure 5c). The structures of the allylic hydroperoxides, allylic alcohols, and enones were confirmed by HMBC correlations, as depicted in
Figure 5.
Some very weak signals in the
1Н-NMR,
1Н,
1Н-COSY, and HSQC spectra of oxidized cerebrosides found in the present study were attributed to
cis-double bonds of allylic hydroperoxides and allylic alcohols (
Appendix A,
Figure A1). The complete structures of these compounds could not be elucidated due to their trace amounts.
2.3. Analyses of FAs, Sphingoid Bases, and Sugar Obtained from Oxidized Cerebrosides
The RP-HPLC oxidized cerebroside fraction was divided into two parts (parts 1 and 2), which were treated using different chemical procedures before hydrolysis. Then, we applied MeCN/HCl hydrolysis [
24] for chemical degradation of cerebrosides. In our experience [
8], this procedure causes less disruption of spingoid bases than methanolysis (MeOН/HCl), which is most widely used in studies of complex lipids.
Analysis of FAs from Part 1. Part 1 of the oxidized cerebrosides was subjected to hydrogenation (with Adams’ catalyst) to fix the positions of allylic oxygen-containing groups before hydrolysis. Upon hydrolysis, liberated FAs were acetylated and methylated. The
1Н-NMR spectrum (CDCl
3) of FA derivatives showed proton signals of mid-chain substructures, including
−H2C−CO−C
H2− at
δН 2.38 (t,
J = 7.4 Hz) and −С
Н(OAс)– at
δH 4.855 (m). GC-MS analysis (electron impact ionization) of these derivatives revealed methyl esters of 2-acetyloxy C
23 and C
22 acids, containing an isolated keto or acetyloxy group or no additional oxygenated group. Similar products, namely keto, hydroxy, and non-oxygenated acid derivatives, have been previously reported for the hydrogenation (in EtOH over Adams’ catalyst) of allylic 9-hydroperoxides and 10-hydroperoxides, obtained from methyl oleate [
25].
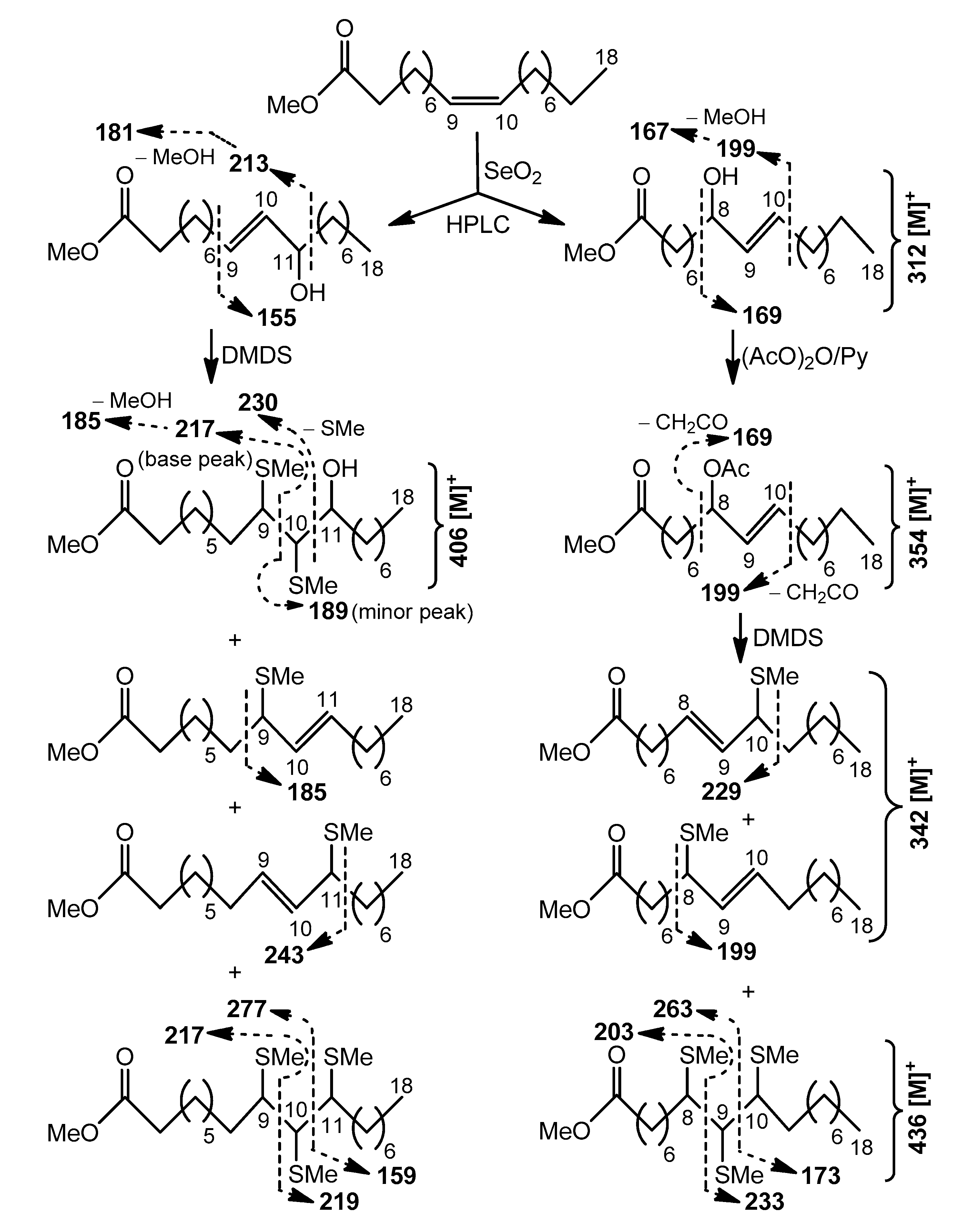
In the present report, mass spectra exhibited base peaks at
m/
z 339 and 325 ([M − MeOCO − CH
2CO]
+) for methyl esters of 2-acetyloxy keto C
23 (440 [M]
+) and C
22 (426 [M]
+) acids, respectively (
Figures S4–S7). Ions produced by cleavage β to a keto group and ions formed from the methyl end of the molecules by cleavage α to the keto group were prominent in the mass spectra, as described for MS fragmentations of several methyl esters of oxo (keto) FAs [
26]. In particular, mass spectra of methyl esters of C
23 and C
22 acids displayed homologous pairs of
m/
z 127/142 and 113/128 fragments, containing methyl ends of the molecules with keto groups on the (
n–8) or (
n–7) carbon, respectively. These keto group positions were confirmed by a number of ions containing the polar end of the FA esters that also produced daughter ions due to loss of AcOH, CH
2CO, or MeOH (
Figures S4–S7).
Hydrogenation of part 1, followed by hydrolysis, acetylation, and methylation, also yielded the methyl esters of 2-acetyloxy C
23 and C
22 acids, containing an additional isolated acetyloxy group. Expectedly, [M]
+ peaks were absent in the mass spectra of the methyl esters of these diacetylated acids, but [M − CH
3CO]
+ and [M − AcOH]
+ ions were observed in high mass regions (
Figures S8–S11). Additionally, these compounds, as derivatives of 2-acetyloxy FAs, fragmented to give abundant [M − MeOCO − AcOH]
+ ions of
m/
z 365 and 351 for methyl esters of C
23 and C
22 acids, respectively. Isomers, containing an isolated acetyloxy group in different positions, were discerned based on the presence of diagnostic α-cleavage ions and more abundant product ions, formed by elimination of CH
2CO or AcOH from α-fragments, as described for acetates of secondary alcohols [
27]. In particular, the α-fragments included the ions of
m/
z 385 and 399 (
Figures S8 and S9) for the derivatives of 2,16-, and 2,17-diacetyloxy C
23 acids, respectively, and the ions of
m/
z 371 and 385 (
Figures S10 and S11) for the 2,15- and 2,16-diacetyloxy C
22 acid derivatives, respectively. Moreover, some homologous ions, which arose from C-1–⁄–C-2 bond fission and cleavage α to an isolated acetyloxy group, were specific for the different positions of this group in acyl chains. For example, the abundant ions of
m/
z 283 and 297 (
Figures S8 and S9), which were detected in the mass spectra of the methyl esters of isomeric 2-acetyloxy C
23 acids, confirmed the presence of second acetyloxy groups on C-16 and C-17, respectively. Similarly,
m/
z 269 and 283 ions in the mass spectra of the methyl esters of 2-acetyloxy C
22 acids (
Figures S10 and S11) indicated a second acetoxy group on C-15 and C-16, respectively. Additionally, α-fragmentation of isomers with an acetyloxy group in the (
n–8) or (
n–7) positions and subsequent loss of AcOH gave rise to
m/
z 111 and 97 ions, respectively, containing the methyl end of acyl chains.
As a result of the FA analyses, hydrogenated minor derivatives of allylically oxygenated FAs were also detected. While the major derivatives formed from fatty acyl groups containing hydroperoxy/hydroxy/keto groups in the (
n–8) or (
n–7) positions, the minor derivatives were rearranged products of these FA moieties (
Appendix B,
Figures S12–S20).
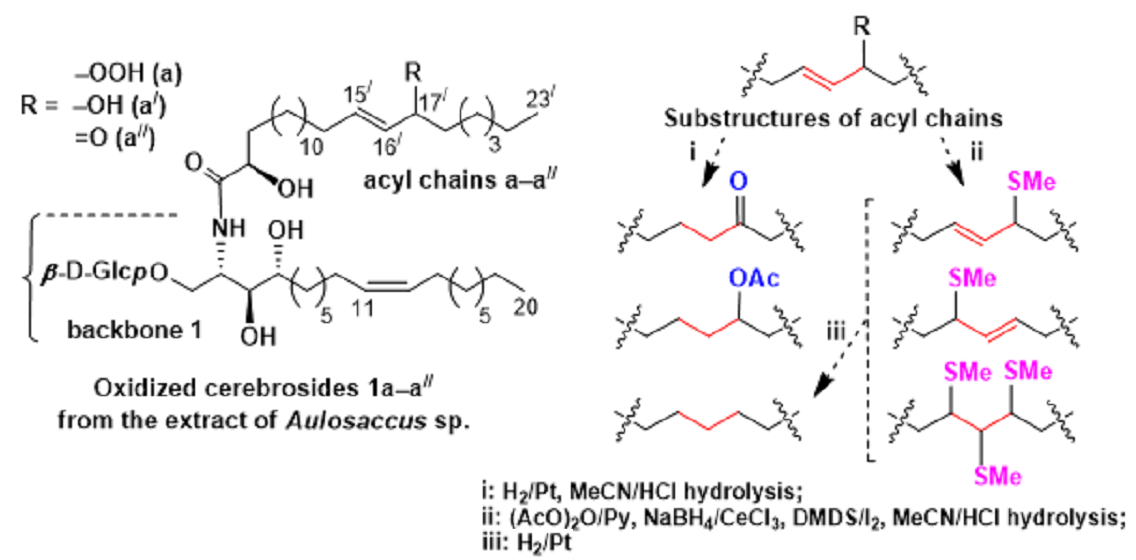
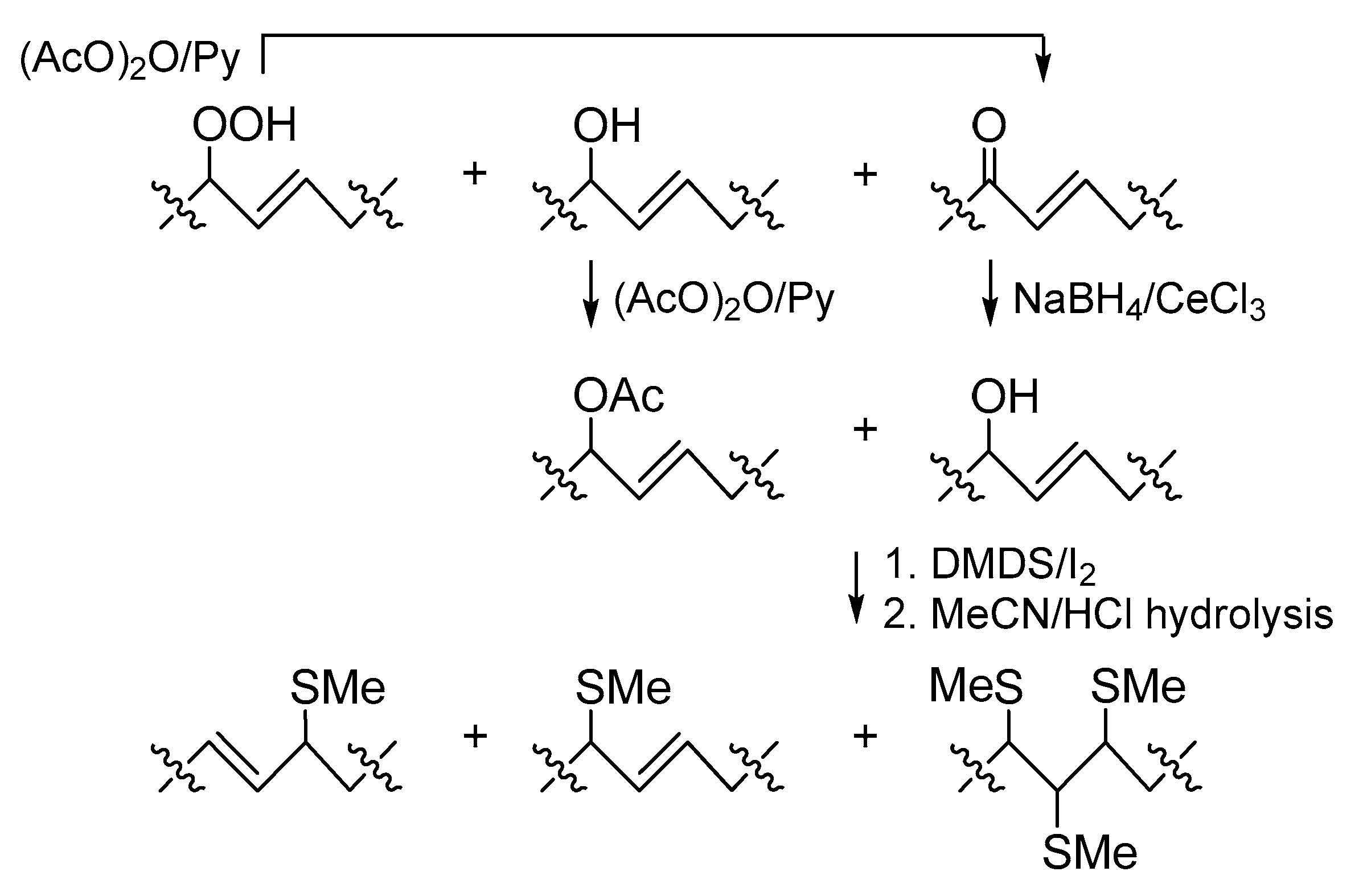
Analysis of FAs from Part 2. In this investigation, we tried to use S-methyl groups as markers for oxidized cerebroside double bonds. However, enones with polarized double bonds that did not react with DMDS under mild conditions, and labile allylic hydroperoxides were not suitable for this purpose. Therefore, enones and allylic hydroperoxides were converted into allylic alcohols that were expected to add to DMDS.
The oxidized cerebrosides from part 2 were acetylated to increase solubility of these relatively polar compounds in DMDS and other low-polarity or non-polar organic solvents. In this process, allylic hydroperoxides were transformed into enones (
Scheme 3), as reported by Porter and Wujek [
23]. The mixture obtained after acetylation was treated with NaBH
4/CeCl
3 [
28] to convert enones into allylic alcohols. The resulting derivatives, containing an allylic hydroxy or acetyloxy group, were treated with DMDS, and the products of this reaction were hydrolyzed in MeCN/HCl. Liberated 2-hydroxy FAs were acetylated, methylated, and analyzed by a GC-MS method that revealed major mono(methylthio) and minor tris(methylthio) derivatives (
Scheme 3 and
Scheme 4). The mono(methylthio) compounds, containing an allylic methylthio group, were characterized by
1H-NMR resonances (CDCl
3) of two
trans-olefinic CH (
δН 5.175 dd,
J = 9.0, 15.2 Hz, and 5.40 dt,
J = 6.9, 15.2 Hz) and one CH (
δН 2.99 m), bearing a –SMe group (
δH 1.97 s). The presence of the mid-chain allylic substructure was confirmed by a TOCSY (total correlation spectroscopy) experiment.
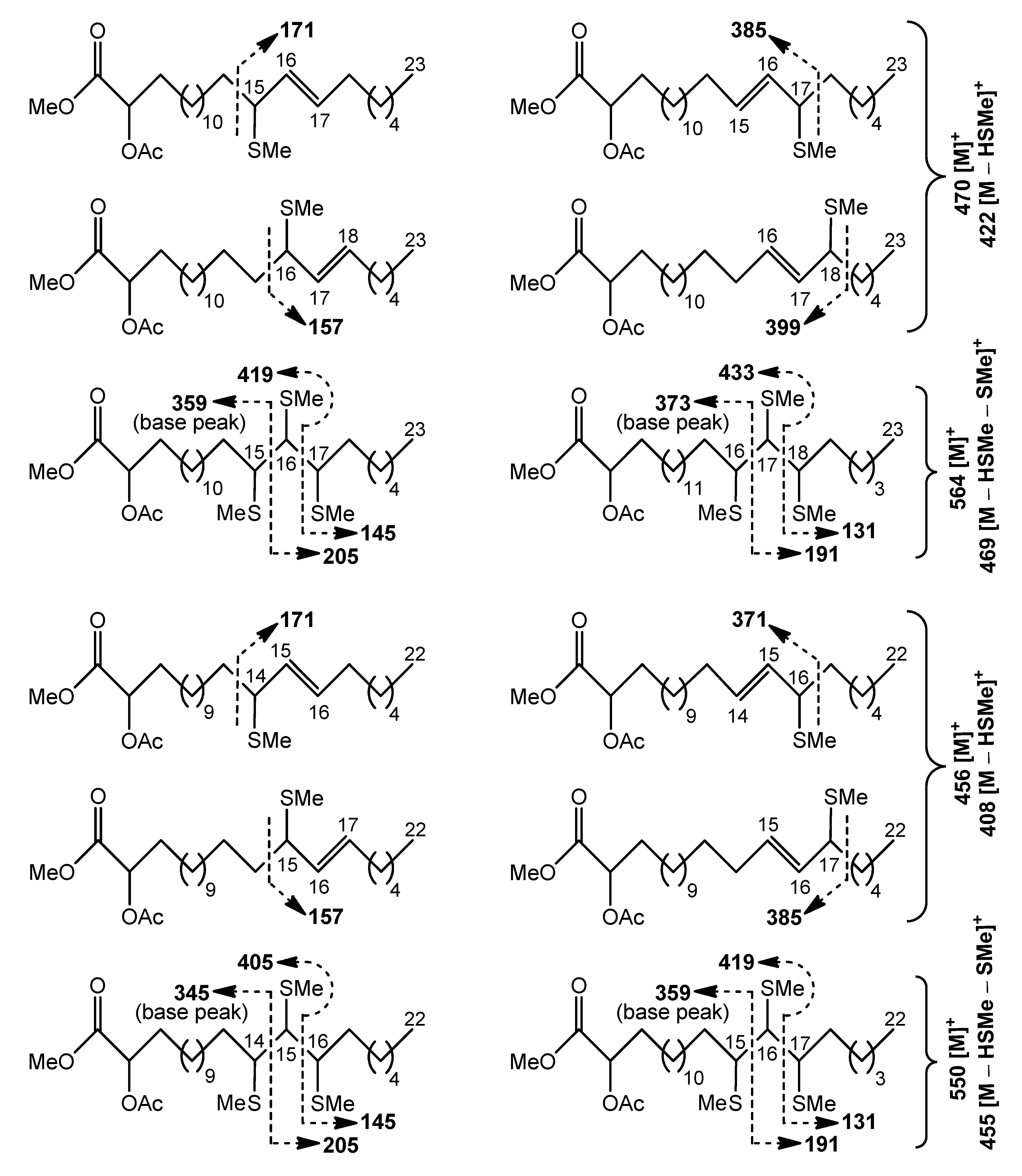
Cleavage patterns for methyl esters of 2-acetyloxy С
23 (470 [M]
+) and С
22 (456 [M]
+) acids, containing an allylic methylthio group, are depicted in
Scheme 4. The positional isomers of these allylic thioethers were only partially GC-separated. The mass spectra of isomeric allylic thioethers (
Figures S21–S28) were characterized by diagnostic peaks, corresponding to ions produced by α-cleavage. The fragments, formed by cleavage α to the carbon carrying an allylic methylthio group on the side remote from the carboxyl group included
m/
z 385 and 399 ions for the methyl esters of 2-acetyloxy C
23 acids with –SMe group in the (
n–7) and (
n–6) positions, respectively (
Figures S22 and S24). The peaks at
m/
z 371 and 385, observed in the mass spectra of methyl esters of homologous 2-acetyloxy C
22 acids, represented fragments of the same origin (
Figures S26 and S28). α-fragmentation of other mono(methylthio) derivatives gave rise to the ions at
m/
z 171 and 157, containing the methyl end of isomers with an allylic –SMe group in the (
n–9) and (
n–8) positions, respectively (
Figures S21, S23, S25, and S27). Relative abundances of α-fragments in average mass spectrum were used to quantify isomer distribution, and approximately equal amounts of the four isomeric allylic thioethers were found. This finding may reflect the fact that these
S-methyl derivatives were possibly products of allylic rearrangements, occurring prior to GC-MS analysis.
The minor tris(methylthio) derivatives of the methyl esters of 2-acetyloxy C
23 (564 [M]
+) or C
22 (550 [M]
+) acids produced more diagnostic fragments than the previously mentioned major mono(methylthio) derivatives. Expectedly, the cleavages of minor
S-methylated compounds occurred between methylthio-carrying carbons to yield substantial fragment ions, as illustrated in
Scheme 4 and
Figures S29–S32. A cluster of four GC peaks for isomeric tris(methylthio) derivatives was observed. According to fragmentation patterns, there were two peaks representing positional isomers and two peaks that represented stereoisomers of these regioisomers on the chromatogram.
The results of the transformations of allylic alcohols and their acetates into methylthio derivatives (
Scheme 3) were confirmed by experiments with model compounds, methyl esters of 11-hydroxy and 8-acetyloxy elaidic acids (prepared from methyl oleate,
Appendix C,
Scheme A1,
Figures S33–S37). Under the conditions used here, the allylic alcohol acetates reacted with DMDS to give major allylic thioethers and minor tris(methylthio) derivatives. Allylic alcohols reacted with DMDS to give major DMDS adducts and minor allylic thioethers and tris(methylthio) derivatives. However, in contrast to the DMDS adducts of monoenes with an isolated double bond, the bis(methilthio) derivatives of allylic alcohols were destroyed during MeCN/HCl hydrolysis.
Previously, the synthesis of allylic thioethers from allylic alcohols and thiols was reported by Zhang et al. ([
29]: iodine-catalyzed process) and Tabarelli et al. ([
30]: catalyst-free approach). Regio-isomeric mixtures of allylic thioethers were produced when the allylic alcohol contained two different substituents. To explain the presence of regio-isomer products of 1,3-isomerization, the allylic cation, formed by water loss from the allylic alcohol, was proposed to be an intermediate in the reaction pathway [
30]. The existence of a similar mechanism explains the formation of regio-isomeric allylic thioethers and their tris(methilthio) derivatives in the iodine-catalyzed reaction of allylic alcohols and their acetates with DMDS reported here. Additionally, allylic thioethers, which could undergo 1,3-isomerization under acidic conditions, were possibly formed from the bis-DMDS adducts of allylic alcohols during MeCN/HCl hydrolysis. As a result of these rearrangements, the FA methylthio derivatives, obtained from oxidized cerebrosides, gave characteristic mass spectra, permitting locations of three-carbon allylically oxygenated substructures, rather than double bonds in the starting acyl chains.
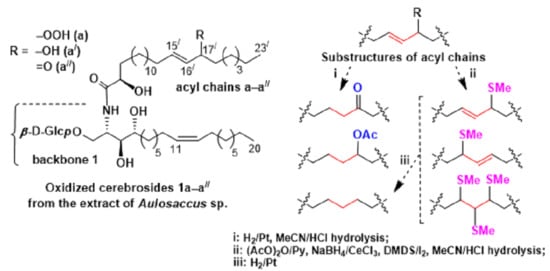
We clarified the acyl structures (
Figure 1) using a logical approach. The GC-MS analyses of the methyl esters of 2-acetyloxy methylthio FAs (
Scheme 4) revealed that the three-carbon allylically functionalized substructures of oxidized cerebrosides included С-15
/–С-16
/–С-17
/ and С-16
/–С-17
/–С-18
/ fragments for 2-hydroxy С
23 acyl chains and С-14
/–С-15
/–С-16
/ and С-15
/–С-16
/–С-17
/ fragments for 2-hydroxy С
22 acyl chains. According to the GC-MS analyses of the hydrogenated FA derivatives, the amide-linked FAs of oxidized cerebrosides contained hydroperoxy, hydroxy, or keto groups in the (
n–8) or (
n–7) positions, more specifically in the 16
/ and 17
/ positions of 2-hydroxy С
23 acyl chains and the 15
/ and 16
/ positions of 2-hydroxy С
22 acyl chains. A priori, an allylic hydroperoxy, hydroxy, or keto group should be located in the terminal points of the previously mentioned three-carbon substructures. Consequently, the С-15
/–С-16
/–С-17
/ fragments of the С
23 acyl chains (
a–
a//) contained such groups in position С-17
/ and the double bond between С-15
/ and C-16
/, while the С-16
/–С-17
/–С-18
/ fragments of the other C
23 acyl chains (
b–
b//) had 16
/-hydroperoxy/hydroxy/oxo groups and 17
/,18
/-double bonds. Similarly, the С-14
/–С-15
/–С-16
/ fragments of the С
22 acyl chains (
c–
c//) contained 16
/-hydroperoxy/hydroxy/oxo groups and 14
/,15
/-double bonds, while the С-15
/–С-16
/–С-17
/ fragments of the other C
22 acyl chains (
d–
d//) had 15
/-hydroperoxy/hydroxy/oxo groups and double bonds between C-16
/ and C-17
/.
The mono(methylthio) and tris(methylthio) derivatives of allylically oxygenated FAs were also used to explain other structural peculiarities of the starting material. Upon hydrogenation, all the methylthio derivatives lost
S-methyl groups, giving saturated hydrocarbon chains. In particular, the transformations, shown in
Scheme 3, with subsequent hydrodesulfurization allowed us to convert –CH=CH–CH(OOH/OH)– and –CH=CH–CO– substructures to –СН
2–СН
2–СН
2– chains without reducing other oxygen-containing groups in the molecules. As a result, the unbranched structures of the parent amide-linked FAs were clarified using retention times of the 2-acetyloxy tricosanoic and docosanoic acid methyl esters, obtained from the methylthio derivatives. Then, these methyl esters were converted into (2
S)-oct-2-yl esters of 2-hydroxy acids. The resulting (2
S)-oct-2-yl esters of 2-hydroxy tricosanoic and docosanoic acids coeluted in GC analyses with the reference (2
S)-oct-2-yl ester of (2
R)-2-hydroxy tricosanoic or docosanoic acids, respectively, indicating (2
R)-configurations of 2-hydroxy acids liberated from oxidized cerebrosides.
Thus, we used two complementary approaches for determining oxidized acyl chain structures in glycosphingolipids. Analysis of the FA derivatives from part 1 indicated the allylic oxygen-containing group positions, but hydrogenation resulted in the loss of information regarding the positions of double bonds in the starting material (approach 1). In the analysis of part 2 (approach 2), the data on the locations of three-carbon allylically oxygenated substructures in the FA derivatives and the information on their straight-chain structures and (2R)-configurations were obtained. Although there was no direct information about the position of the double bond in the FA esters, the combined data of approaches 1 and 2 allowed for determination of the double bond and allylic hydroperoxy, hydroxy, or keto group locations in each acyl chain.
Analyses of Sphingoid Bases and Sugar from Parts 1 and 2. The sphingoid bases, liberated by hydrolysis of oxidized cerebrosides, were obtained as acetylated derivatives. The
1Н-NМR data (
δН values of CH
2-1–CH-4; CDCl
3) and optical rotation value ([α]
25D = + 27.9, CHCl
3) of the hydrogenated sphingoid base acetates, isolated from the hydrolysate of part 1, indicated their (2
S,3
S,4
R)-configuration [
31]. The
1Н-NМR spectrum of these compounds also showed signals of the terminal methyl groups of dominant normal-chain (
δН 0.88, t,
J = 6.9 Hz) and minor cyclopropane-containing (
δН 0.89, t,
J = 6.9 Hz) constituents. Under our conditions of hydrogenation, ring opening was not the dominant process for the minor constituent, so the signals of
cis-cyclopropane protons at
δН −0.33 (dt,
J = 4.2, 5.5 Hz), 0.56 (ddd,
J = 4.2, 8.3, 8.3 Hz), and 0.645 (m) [
32] were observed. The hydrolysis of derivatized oxidized cerebrosides from part 2 with subsequent acetylation of products gave three sphingoid base derivatives. Two spingoid base derivatives were the DMDS adducts of acetylated isomeric monoenoic С
20 compounds. The mass spectrum of the DMDS adduct of major acetylated C
20 monoene exhibited significant peaks at
m/
z 432 [M − H
3CSC
9H
18]
+, 372 [M − H
3CSC
9H
18 − AcOH]
+ (base peak), and 173 [H
3CSC
9H
18]
+, indicating 11,12-double bond in backbone
1. Key peaks in the mass spectrum of the DMDS adduct of isomeric minor acetylated C
20 monoene, which have a longer retention time in GC-MS, were observed at
m/
z 460 [M − H
3CSC
7H
14]
+, 400 [M − H
3CSC
7H
14 − AcOH]
+ (base peak), and 145 [H
3CSC
7H
14]
+, indicating Δ13 unsaturation in backbone
2. A third acetylated sphingoid base, derived from cyclopropane-containing backbone
3, did not give a DMDS adduct. In the mass spectrum of this compound, an [M − AcOH]
+ ion fragmented to give discernible peaks at
m/
z 380 [M − AcOH − (CH
2)
6СH
3]
+ (α cleavage to a cyclopropane ring, at the C-14–C-15 position) and 394 [M − AcOH − (CH
2)
5СH
3]
+ (β-cleavage, at the C-15–C-16 position) that was characteristic of the peracetate of the С
21 sphingoid base containing a ring between C-13 and C-14.
Methylthiolation of
cis-monoenes and
trans-monoenes with DMDS, as an anti-addition, leads to the threo-adducts and erythro-adducts, respectively [
33,
34]. The threo-diastereomers and erythro-diastereomers can be easily distinguished by NMR shifts of protons and carbons in and close to the 1,2-bis(alkylthio) moiety [
35]. In addition to the data presented in the NMR study of Knothe and Steidley [
35], we used
δН values of the DMDS adducts of two standards, methyl palmitoleate (
Figure 6a) and its
trans-isomer (
Figure 6b), to confirm the configurations of double bonds in the monoenoic sphingoid base moieties of the starting oxidized cerebrosides. The
δH values for CH
2, α to the –CH(SMe)–CH(SMe)– moiety, were obtained through correlations in
1Н,
1Н-COSY diagrams. The
1Н-NМR and
1Н,
1Н-COSY spectra (CDCl
3) of the DMDS derivatives of acetylated sphingoid bases, derived from backbones
1 and
2, showed superimposed resonances of two vicinal CH groups (
δН 2.685, m), linked to –SMe (
δH 2.10, s), and two α-CH
2 groups (
δН 1.84, m; 1.32, m), characteristic of threo-diastereomers. Consequently,
cis-monoenes (
1 and
2) were precursors of these compounds.
GC analyses of peracetylated (2
R)- and (2
S)-oct-2-yl glucosides showed a D-configuration of glucose, released from parts 1 and 2 [
36].
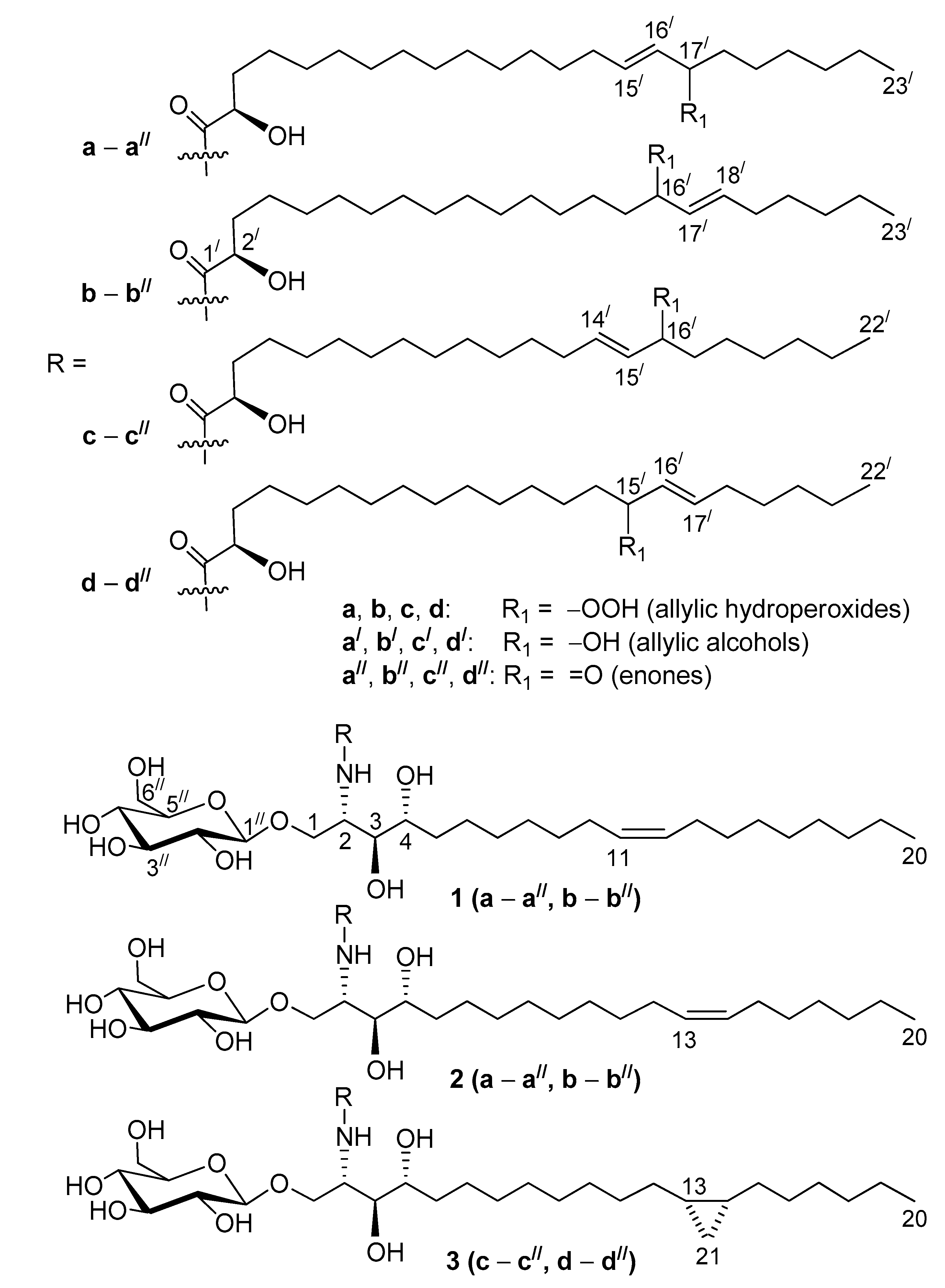
2.4. Oxidized Cerebrosides from the Extract of Aulosaccus sp.: Structures and Possible Origins
As a result of our study, structures of 18 previously unknown compounds, found in the complex mixture of the oxidized cerebrosides from the extract of Aulosaccus sp., were elucidated. These β-D-glucopyranosyl-(1→1)-ceramides (1a–a//, 1b–b//, 2a–a//, 2b–b//, 3c–c//, 3d–d//) were shown to contain phytosphingosine-type backbones, (2S,3S,4R,11Z)-2-aminoeicos-11-ene-1,3,4-triol (in 1), (2S,3S,4R,13Z)-2-aminoeicos-13-ene-1,3,4-triol (in 2), and (13S*,14R*)-2-amino-13,14-methylene-eicosane-1,3,4-triol (in 3). These backbones were N-acylated with straight-chain monoenoic (2R)-2-hydroxy acids that had allylic hydroperoxy/hydroxy/keto groups on C-17/ in the 15/E-23:1 chain (a–a//), C-16/ in the 17/E-23:1 (b–b//) and 14/E-22:1 (c–c//) chains, and C-15/ in the 16/E-22:1 chain (d–d//). Cerebrosides, having backbones 1, 2, and 3, comprised, respectively, 60%, 20%, and 20% of the mixture. The percentages were calculated from the integration of signals of a cyclopropane ring in the 1H-NMR spectra of this mixture (backbone 3) and from relative intensities of GC peaks, represented by DMDS derivatives of two acetylated isomeric monoenoic sphingoid bases (backbones 1 and 2, Δ11:Δ13 ≈ 3:1). The GC-MS analysis of the hydrogenation products of amide-linked FAs indicated that the a:b, c:d, a/:b/, c/:d/, a//:b//, and c//:d// isomer ratios were approximately 1:1.Therefore, the employed complementary instrumental and chemical methods clarified structures of oxidized cerebrosides in a complex mixture, without requiring isolation or complete separation.
Previously, the possible precursors of the oxidized cerebrosides were found in the major RP-HPLC fraction of glycosphingolipids, isolated from the extract of
Aulosaccus sp. These potential precursors were β-
d-glucopyranosyl-(1→1)-ceramides that contained backbones
1 (60% in the fraction) and
2 (20%),
N-acylated with (2
R,16
Z)-2-hydroxytricos-16-enoic acid, and backbone
3 (20%),
N-acylated with (2
R,15
Z)-2-hydroxydocos-15-enoic acid [
8]. Perhaps, peroxidation of the amide-linked FAs occurred symmetrically about the
cis-(
n–7) double bond, so a hydroperoxy, hydroxy, or keto group, found in the major
trans-monoenoic peroxidation products, was located at each of the carbon atoms, which originally formed the double bond (namely, in the (
n–8) and (
n–7) positions, ≈1:1). In particular, structures
a–
a// and
b–
b// could be formed from С
23Δ
16Z acyl chain while structures
c–
c// and
d–
d// could be formed from the С
22Δ
15Z acyl chain.
According to the product composition, photo-oxidation and autooxidation [
1] are possible mechanisms involved in the formation of the oxidized cerebrosides from the extract of
Aulosaccus sp. However, we would like to point to another possible origin of the oxidized cerebrosides in
Aulosaccus sp., taking into account the relationship between these oxidation products and other compounds isolated from the same sponge sample. In particular, some bacterial branched-chain, cyclopropane-containing FAs, and their monoenoic precursors were present in significant amounts in
Aulosaccus sp. [
32], and an overwhelming number of the sterols (stanols, Δ
5-, Δ
7-, and Δ
8(14)-sterols) of this sponge were oxidized to the corresponding 3-ketosteroids [
37]. The occurrence of these FAs and steroids in
Aulosaccus sp. suggested this sponge was associated with actinobacteria, known as sponge-specific microorganisms [
38] and sterol degraders [
39]. Cholesterol oxidase, produced by a variety of actinobacteria [
40], could catalyze the transformations of the previously mentioned sterols into 3-ketosteroids [
41] with the generation of H
2O
2. We suggest that H
2O
2 production in the enzymatic oxidation of
Aulosaccus sp. sterols led to oxidative transformations of a certain part of cerebrosides, located in the membranes of eukaryotic cells together with sterols.


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}