
DFT Study on the Mechanism of Iron-Catalyzed Diazocarbonylation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
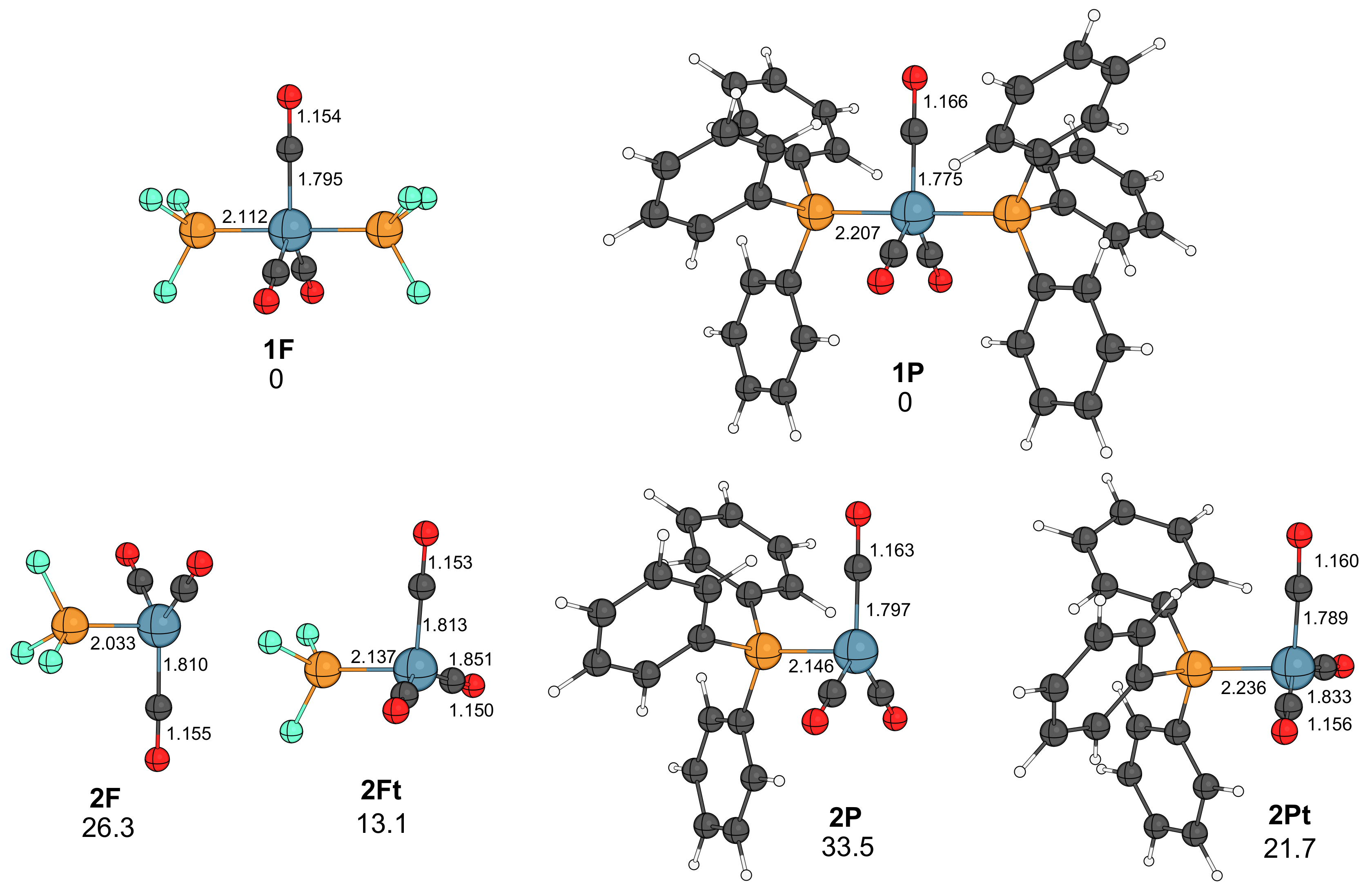
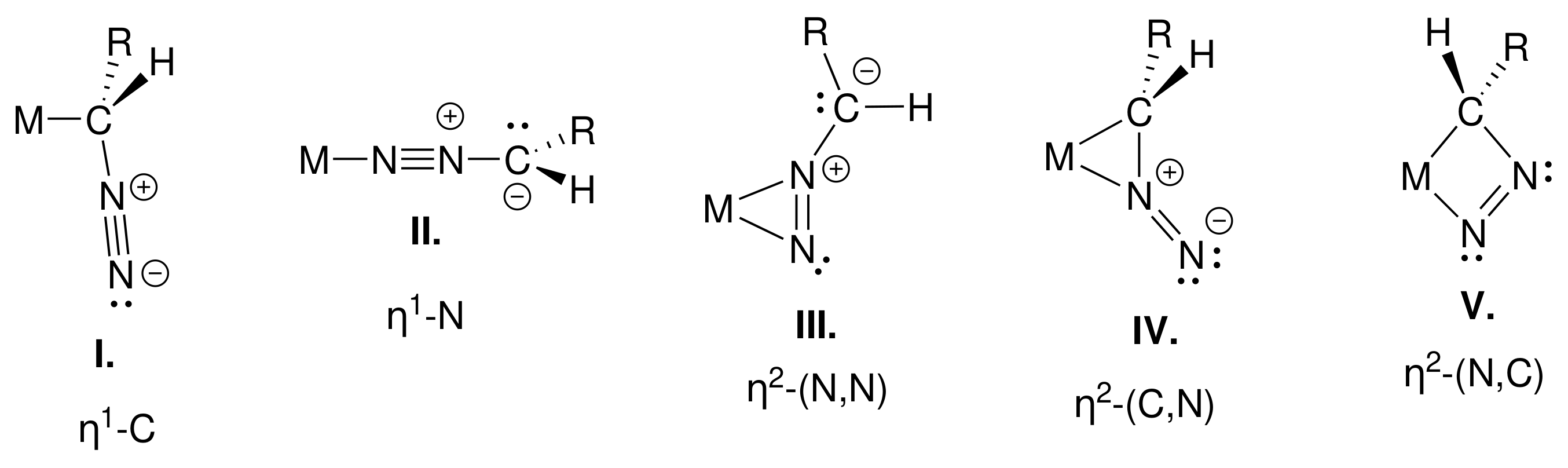
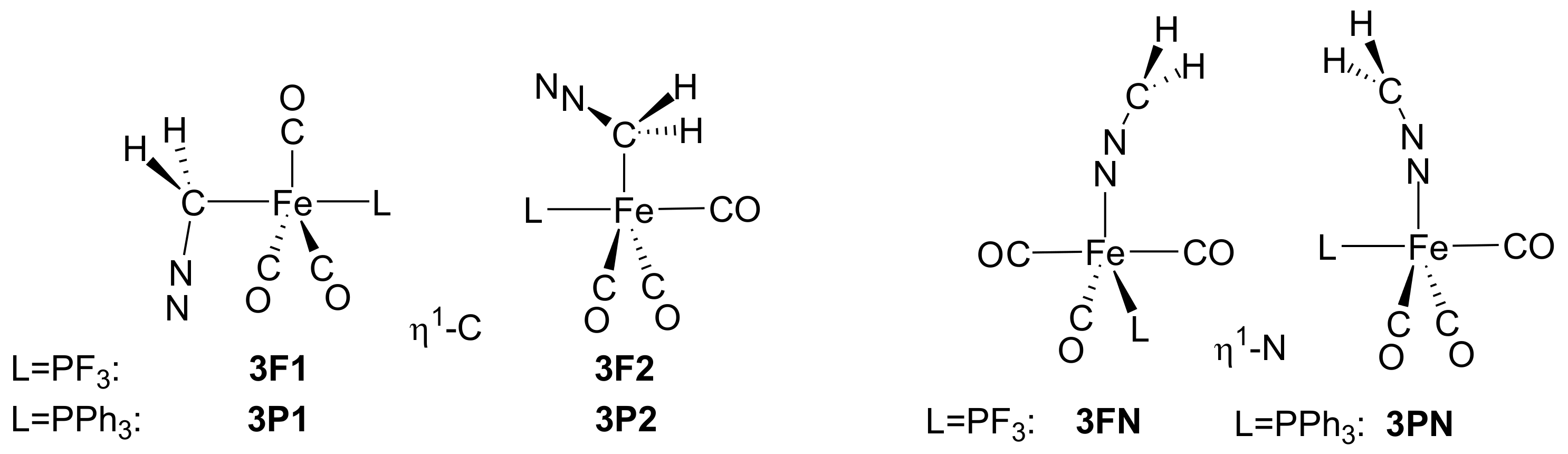
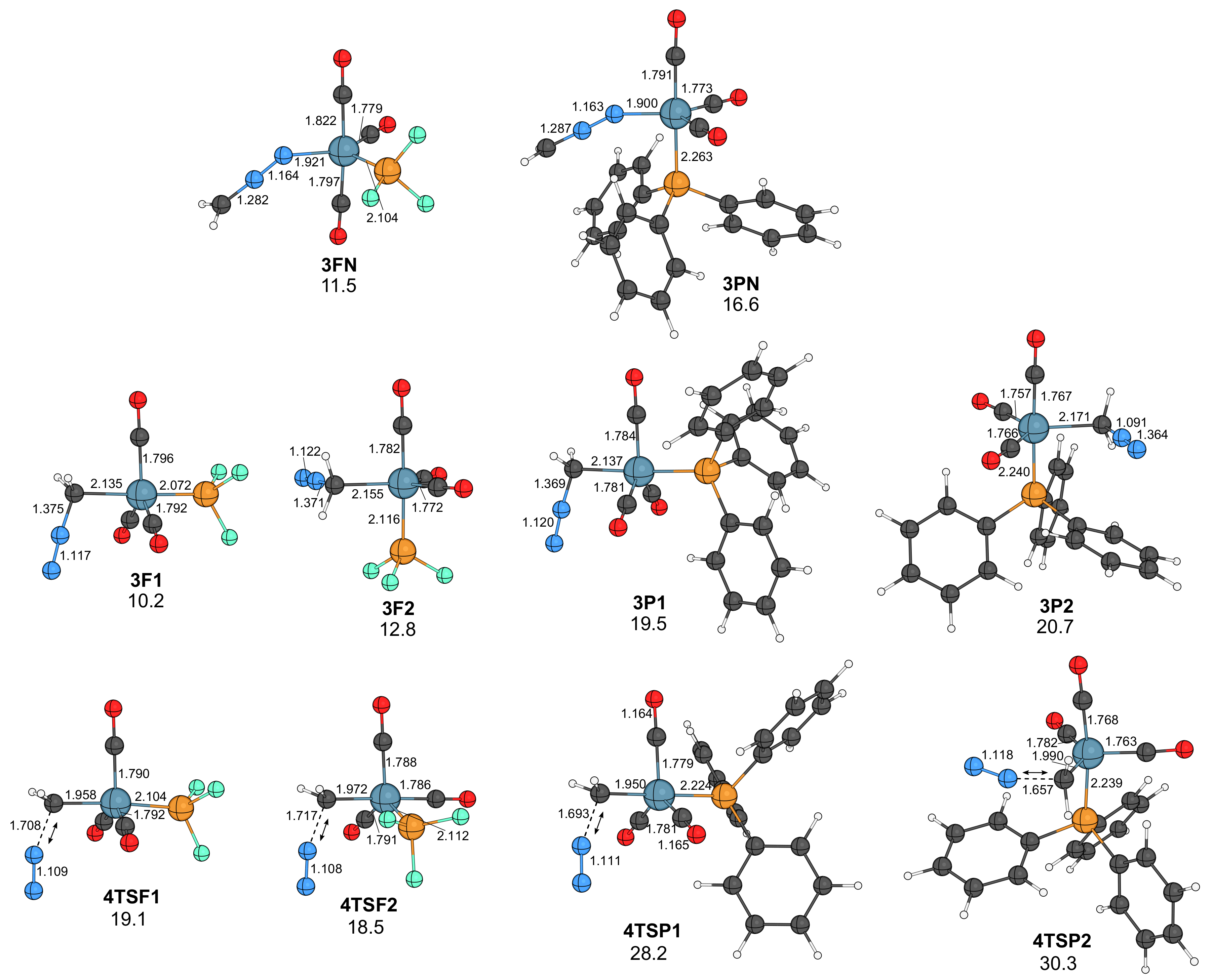
2.1. Reaction of Diazomethane with Phosphine Substituted Iron Carbonyls
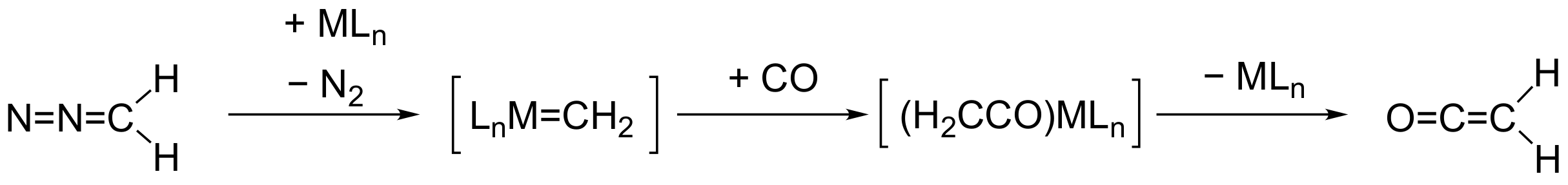
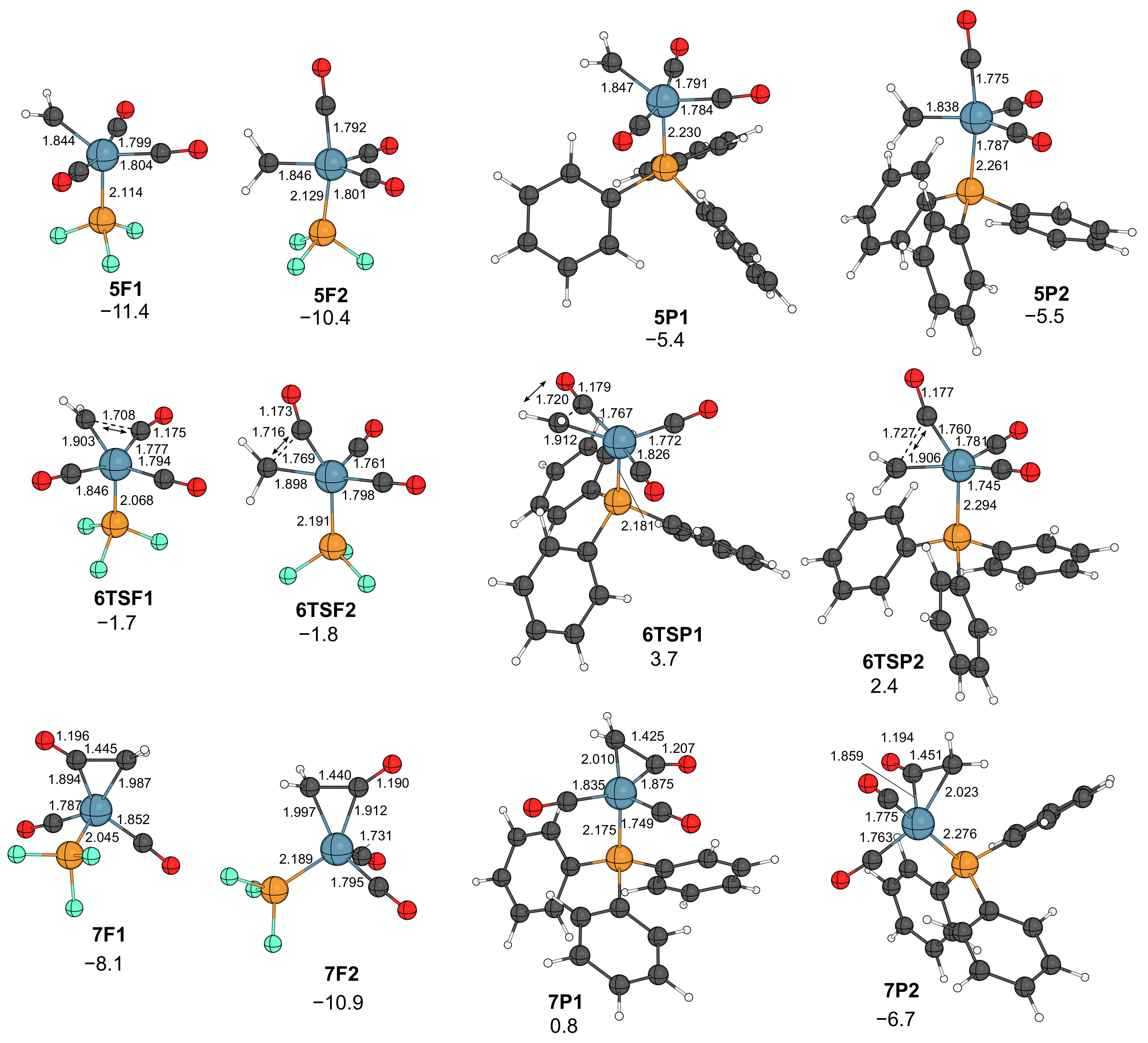
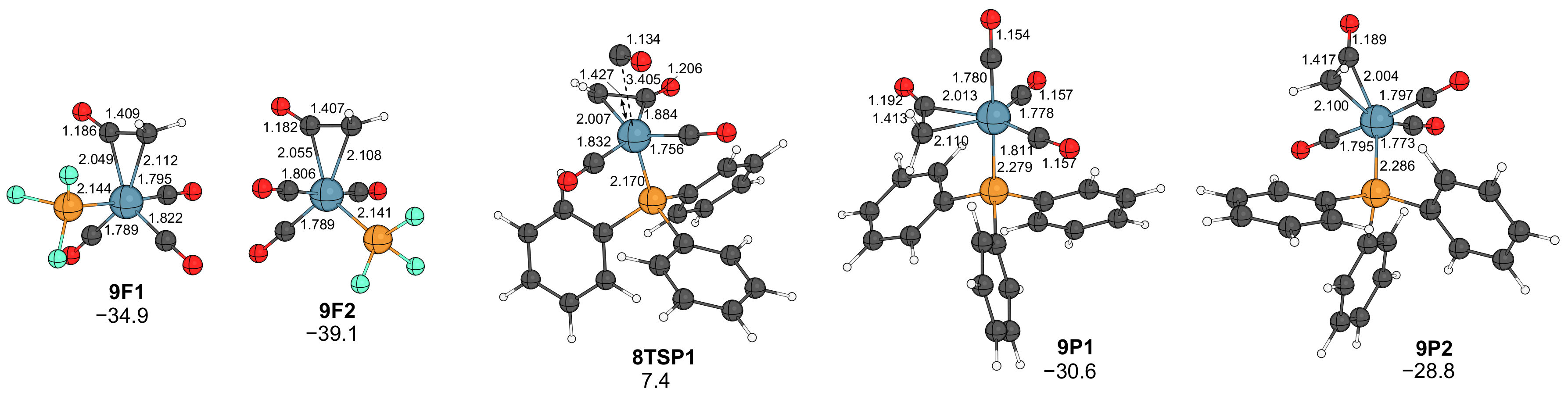
2.2. Formation of Ketene Complexes
2.3. Overall Reaction Mechanism
3. Computational Details
4. Conclusions
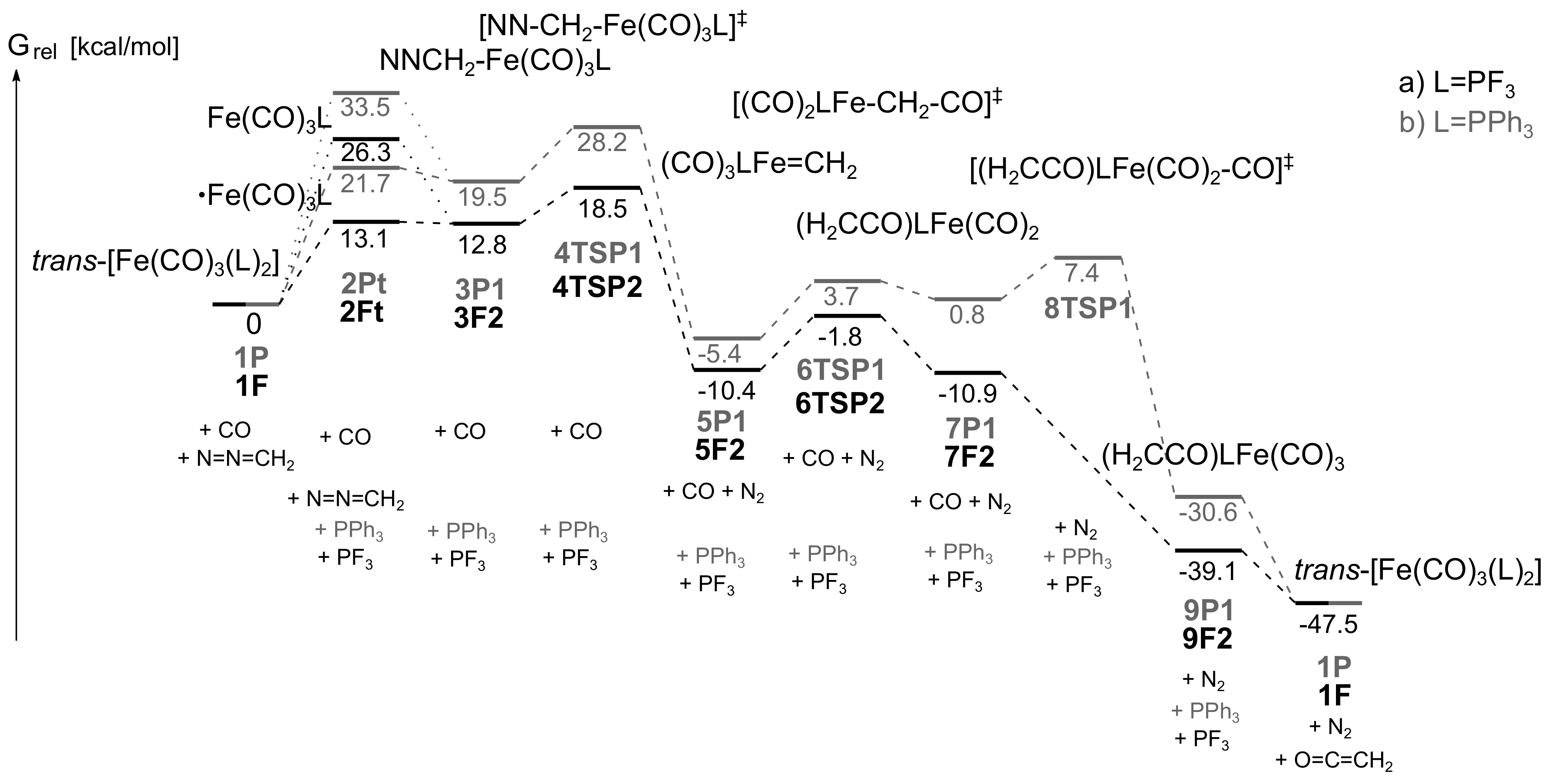
- In the presence of phosphine, the course of the reaction resembles that obtained for the nickel–carbonyl catalysts; that is, the –diazoalkane complexes lose N2, and the resulting carbenoids undergo carbene–carbonyl coupling affording coordinatively unsaturated ketene complexes.
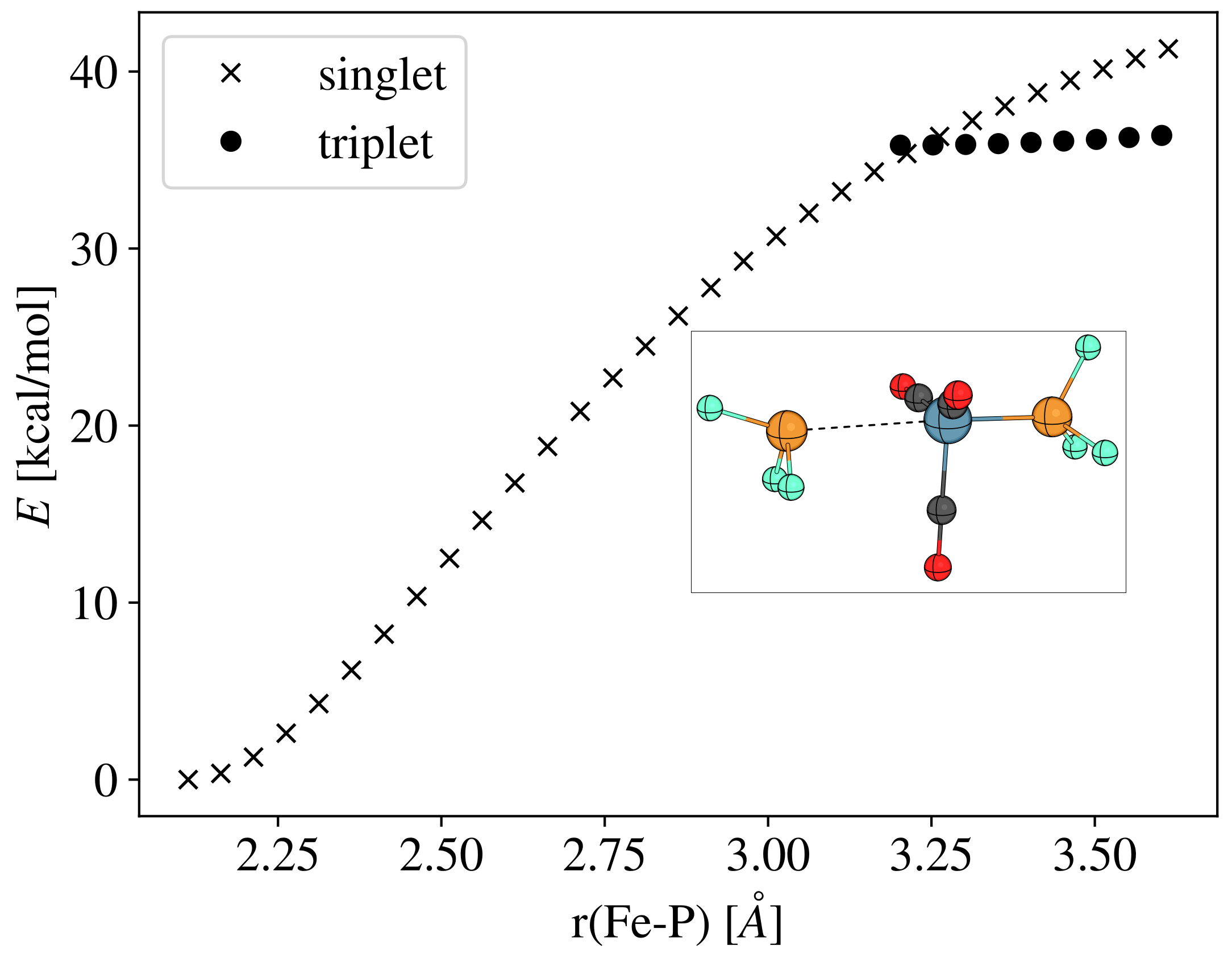
- The active catalysts for the iron-catalyzed diazocarbonylation are predicted to be the triplet state Fe(CO)3(P) complexes, which are formed via a spin change from the corresponding singlet state Fe(CO)3(P)2 species. The coordination of the diazoalkane proceeds through another spin change.
- As for the other metals, the rate-limiting step is the combination of catalyst formation, diazo coordination and the exergonic N2 extrusion. The carbene–carbonyl coupling is slightly exergonic for PF3 and endergonic for PPh3. The CO uptake, leading to the coordinatively saturated ketene complexes, is exergonic, as well as the dissociation of the ketene ligand from the iron center.
- Electron-withdrawing P-donor ligands, such as phosphorus trifluoride, are predicted to increase the reaction rate, in comparison to that obtained with triphenylphosphine.
- Diazomethane can follow -C or -N coordination; both types of adducts are close to each other in terms of relative free energy.
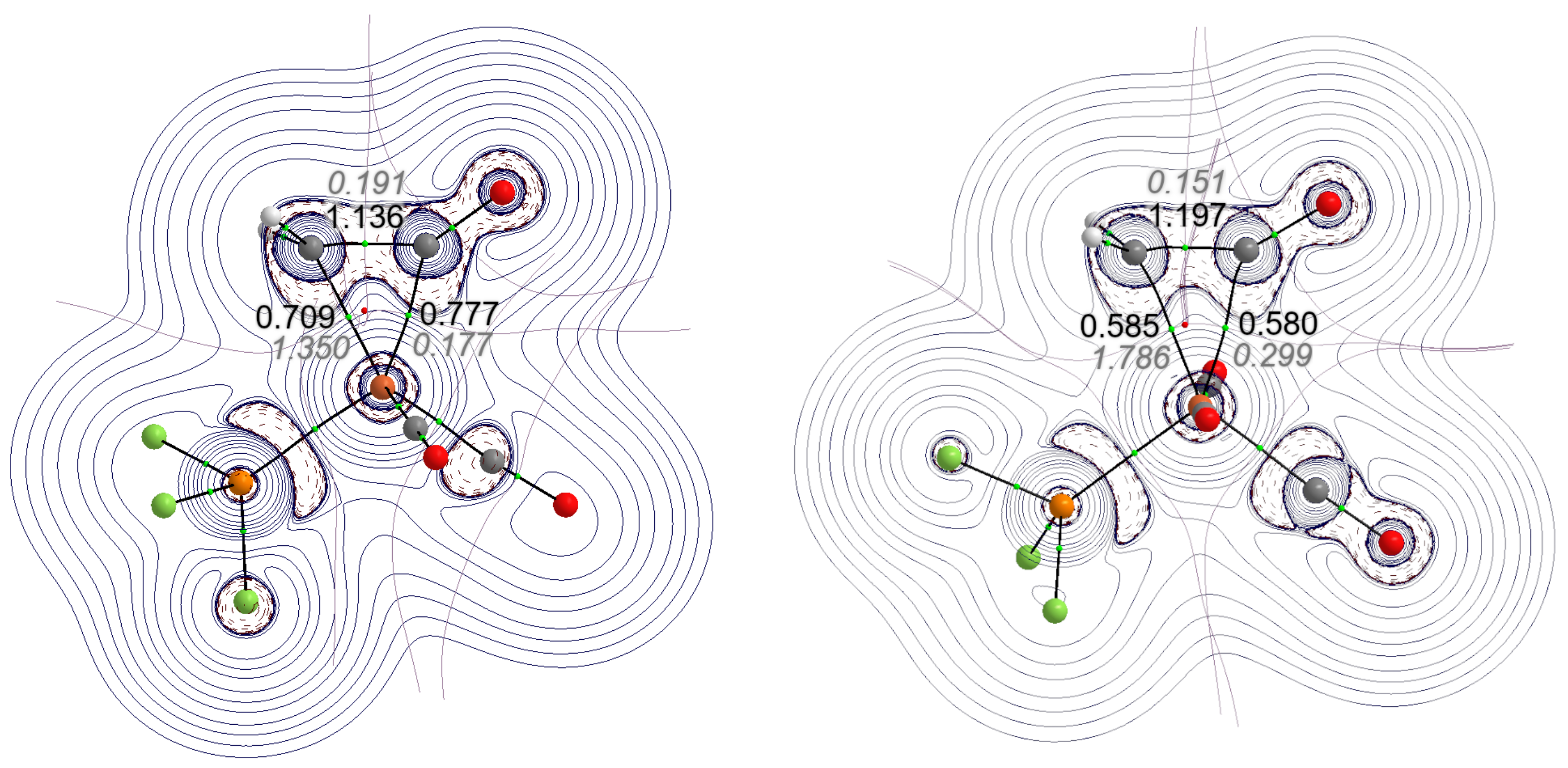
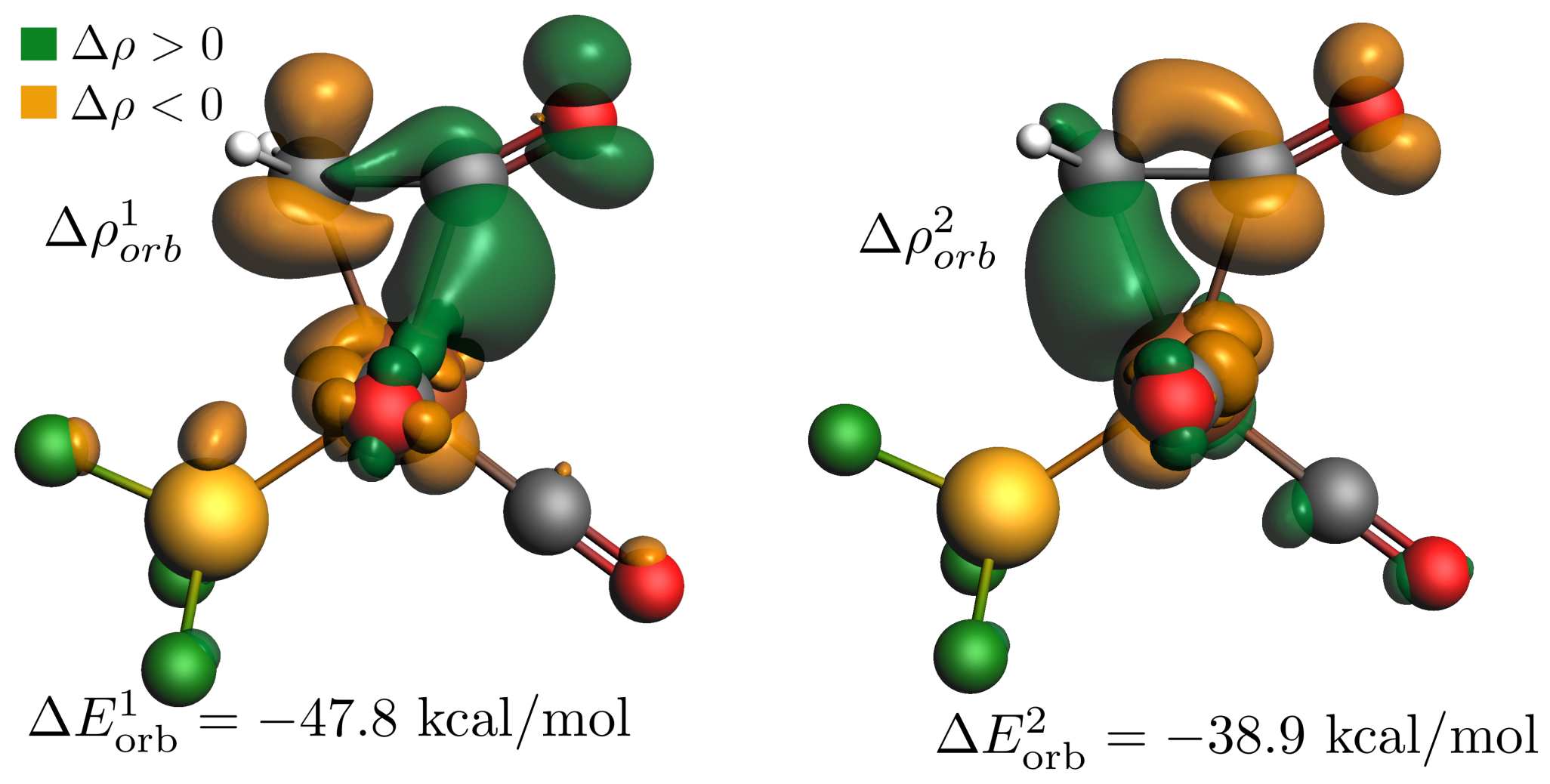
- According to EDA–NOCV calculations, the charge flow cannot be separated clearly between the ketene and the metal-containing fragment. The more localized NBO approach shows, however, that the main source of the -donor interaction is the lone pair of the terminal carbon, mainly based on the 2 natural atomic orbital, whereas the back-donation is based mainly on the lone pair of iron, which is an out-of-phase hybrid of the 3 and the 3 natural atomic orbitals, interacting with the orbital of bound ketene.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tidwell, T.T. Ketenes; John Wiley & Sons: Hoboken, NJ, USA, 1995. [Google Scholar]
- Tidwell, T.T. Ketene chemistry: The second golden age. Acc. Chem. Res. 1990, 23, 273–279. [Google Scholar] [CrossRef]
- Tidwell, T.T. The first century of ketenes (1905–2005): The birth of a versatile family of reactive intermediates. Angew. Chem. Int. Ed. 2005, 44, 5778–5785. [Google Scholar] [CrossRef] [PubMed]
- Tidwell, T.T. Ketene chemistry after 100 years: Ready for a new century. Eur. J. Org. Chem. 2006, 563–576. [Google Scholar] [CrossRef]
- Allen, A.D.; Tidwell, T.T. New Directions in Ketene Chemistry: The Land of Opportunity. Eur. J. Org. Chem. 2012, 2012, 1081–1096. [Google Scholar] [CrossRef]
- Tidwell, T.T. Ketenes II, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Tidwell, T.T. Hugo (Ugo) Schiff, Schiff Bases, and a Century of β-Lactam Synthesis. Angew. Chem. Int. Ed. 2008, 47, 1016–1020. [Google Scholar] [CrossRef]
- Allen, A.D.; Tidwell, T.T. Ketenes and other cumulenes as reactive intermediates. Chem. Rev. 2013, 113, 7287–7342. [Google Scholar] [CrossRef]
- Fördős, E.; Tuba, R.; Párkányi, L.; Kégl, T.; Ungváry, F. Application of the Octacarbonyldicobalt-Catalyzed Carbonylation of Ethyl Diazoacetate for the One-Pot Synthesis of N-tert-Butyl-trans-α-ethoxycarbonyl-β-phenyl-β-lactam. Eur. J. Org. Chem. 2009, 2009, 1994–2002. [Google Scholar] [CrossRef]
- Balogh, J.; Kégl, T.; Ungváry, F.; Skoda-Földes, R. Co2(CO)8-induced domino reactions of ethyl diazoacetate, carbon monoxide and ferrocenylimines leading to 2-(1-ferrocenyl-methylidene)-malonic acid derivatives. Tetrahedron Lett. 2009, 50, 4727–4730. [Google Scholar] [CrossRef]
- Balogh, J.; Kégl, T.; Párkányi, L.; Kollár, L.; Ungváry, F.; Skoda-Földes, R. Synthesis of (E)-2-(1-ferrocenylmethylidene) malonic acid derivatives by a cobalt-catalyzed domino reaction of ethyl diazoacetate, carbon monoxide and ferrocenylimines. J. Organomet. Chem. 2011, 696, 1394–1403. [Google Scholar] [CrossRef]
- Rüchardt, C.; Schrauzer, G.N. Über die Carbonylierung von Carbenen und die katalytische Zersetzung von Diazoalkanen mit Nickelcarbonyl. Chem. Ber. 1960, 93, 1840–1848. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Bikzhanova, G.A.; Collins, L.S.; Concolino, T.; Lam, K.C.; Rheingold, A.L. Controlled, reversible conversion of a ketene ligand to carbene and CO ligands on a single metal center. J. Am. Chem. Soc. 2000, 122, 5222–5223. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Collins, L.S.B.; Wolpert, M.; Bikzhanova, G.A.; Lo, H.C.; Combs, D.; Hubbard, J.L. First Direct Structural Comparison of Complexes of the Same Metal Fragment to Ketenes in Both C,C- and C,O-Bonding Modes. J. Am. Chem. Soc. 2001, 123, 8260–8270. [Google Scholar] [CrossRef] [PubMed]
- Tuba, R.; Ungváry, F. Octacarbonyl dicobalt-catalyzed selective transformation of ethyl diazoacetate into organic products containing the ethoxycarbonyl carbene building block. J. Mol. Catal. A Chem. 2003, 203, 59–67. [Google Scholar] [CrossRef]
- Ungvári, N.; Kégl, T.; Ungváry, F. Octacarbonyl dicobalt-catalyzed selective carbonylation of (trimethylsilyl)diazomethane to obtain (trimethylsilyl)ketene. J. Mol. Catal. A Chem. 2004, 219, 7–11. [Google Scholar] [CrossRef]
- Fördős, E.; Ungvári, N.; Kégl, T.; Ungváry, F. Reactions of 13CO with Ethoxycarbonylcarbene-Bridged Dicobalt Carbonyl Complexes: [μ2-Ethoxycarbonyl (methylene)-μ2-(carbonyl) bis (tricarbonylcobalt)(Co–Co)] and [Di-μ2-ethoxycarbonyl (methylene) bis (tricarbonylcobalt)(Co–Co)]. Eur. J. Inorg. Chem. 2006, 2006, 1875–1880. [Google Scholar] [CrossRef]
- Kégl, T.; Ungváry, F. Internal carbon monoxide exchange and CO dissociation in cobalt carbonyl carbene complexes. A density functional study. J. Organomet. Chem. 2007, 692, 1825–1833. [Google Scholar] [CrossRef]
- Kégl, T.; Ungváry, F. The cobalt-catalyzed ketene formation from diazoalkanes. Lett. Org. Chem. 2010, 7, 634–644. [Google Scholar] [CrossRef]
- Ungvári, N.; Fördős, E.; Kégl, T.; Ungváry, F. Mechanism of the cobalt-catalyzed carbonylation of ethyl diazoacetate. Inorg. Chim. Acta 2010, 363, 2016–2028. [Google Scholar] [CrossRef]
- Ungvári, N.; Fördős, E.; Balogh, J.; Kégl, T.; Párkányi, L.; Ungváry, F. Triphenylphosphane-modified cobalt catalysts for the selective carbonylation of ethyl diazoacetate. Organometallics 2010, 29, 3837–3851. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Y.; Ling, L.; Li, Y.; Dong, Y.; Gong, M.; Zhao, X.; Zhang, Y.; Wang, J. Pd-catalyzed carbonylation of diazo compounds at atmospheric pressure: A catalytic approach to ketenes. J. Am. Chem. Soc. 2011, 133, 4330–4341. [Google Scholar] [CrossRef]
- Barcs, B.; Kollár, L.; Kégl, T. Density Functional Study on the Mechanism of Nickel-Mediated Diazo Carbonylation. Organometallics 2012, 31, 8082–8097. [Google Scholar] [CrossRef]
- Harvey, J.N.; Aschi, M. Modelling spin-forbidden reactions: Recombination of carbon monoxide with iron tetracarbonyl. Faraday Discuss. 2003, 124, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.R.; Kepp, K.P. Spin propensities of octahedral complexes from density functional theory. J. Phys. Chem. A 2015, 119, 4041–4050. [Google Scholar] [CrossRef] [PubMed]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef]
- Michalak, A.; Mitoraj, M.; Ziegler, T. Bond orbitals from chemical valence theory. J. Phys. Chem. A 2008, 112, 1933–1939. [Google Scholar] [CrossRef]
- Mitoraj, M.; Michalak, A. Donor-Acceptor Properties of Ligands from the Natural Orbitals for Chemical Valence. Organometallics 2007, 26, 6576–6580. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A. σ-donor and π-acceptor properties of phosphorus ligands: An insight from the natural orbitals for chemical valence. Inorg. Chem. 2010, 49, 578–582. [Google Scholar] [CrossRef]
- Cohen, R.; Rybtchinski, B.; Gandelman, M.; Rozenberg, H.; Martin, J.M.; Milstein, D. Metallacarbenes from diazoalkanes: An experimental and computational study of the reaction mechanism. J. Am. Chem. Soc. 2003, 125, 6532–6546. [Google Scholar] [CrossRef]
- Fortman, G.C.; Kégl, T.; Li, Q.S.; Zhang, X.; Schaefer, H.F.; Xie, Y.; King, R.B.; Telser, J.; Hoff, C.D. Spectroscopic Detection and Theoretical Confirmation of the Role of Cr2(CO)5(C5R5)2 and ·Cr(CO)2(ketene)(C5R5) as Intermediates in Carbonylation of NNCHSiMe3 to OCCHSiMe3 by ·Cr(CO)3(C5R5) (R= H, CH3). J. Am. Chem. Soc. 2007, 129, 14388–14400. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol, Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 7.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A. AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA. 2019. Available online: aim.tkgristmill.com (accessed on 19 October 2019).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- ADF2019. SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. 2019. Available online: http://www.scm.com (accessed on 25 May 2019).


Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kégl, T.R.; Kollár, L.; Kégl, T. DFT Study on the Mechanism of Iron-Catalyzed Diazocarbonylation. Molecules 2020, 25, 5860. https://doi.org/10.3390/molecules25245860
Kégl TR, Kollár L, Kégl T. DFT Study on the Mechanism of Iron-Catalyzed Diazocarbonylation. Molecules. 2020; 25(24):5860. https://doi.org/10.3390/molecules25245860
Chicago/Turabian StyleKégl, Tímea R., László Kollár, and Tamás Kégl. 2020. "DFT Study on the Mechanism of Iron-Catalyzed Diazocarbonylation" Molecules 25, no. 24: 5860. https://doi.org/10.3390/molecules25245860
APA StyleKégl, T. R., Kollár, L., & Kégl, T. (2020). DFT Study on the Mechanism of Iron-Catalyzed Diazocarbonylation. Molecules, 25(24), 5860. https://doi.org/10.3390/molecules25245860