
Fluorinated Organic Paramagnetic Building Blocks for Cross-Coupling Reactions


 , ,
, , 
Abstract
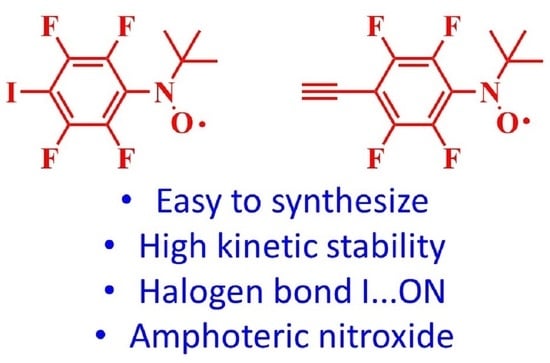
1. Introduction
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. Materials and Measurements
4.2. Synthetic Procedures
4.2.1. N-tert-Butyl-2,3,5,6-tetrafluoro-4-iodoaniline (1)
4.2.2. N-tert-Butyl-2,3,5,6-tetrafluoro-4-((trimethylsilyl)ethynyl)aniline (2)
4.2.3. N-tert-Butyl-4-ethynyl-2,3,5,6-tetrafluoroaniline (3)
4.2.4. N-tert-Butyl-N-oxyamino-2,3,5,6-tetrafluoro-4-iodobenzene (4)
4.2.5. N-tert-Butyl-N-oxyamino-2,3,5,6-tetrafluoro-4-ethynylbenzene (5)
4.2.6. trans-Bis(1,1,1,5,5,5-hexafluoropentane-2,4-dionato-κ2O,O′)bis{N-tert-butyl-N-oxyamino-2,3,5,6-tetrafluoro-4-iodobenzene-κO}copper(II) ([Cu(hfac)2(4)2])
4.3. Single-Crystal XRD Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hicks, R.G. Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds; John Wiley and Sons: Chichester, UK, 2010. [Google Scholar]
- Tretyakov, E.V.; Ovcharenko, V.I. The chemistry of nitroxide radicals in the molecular design of magnets. Russ. Chem. Rev. 2009, 78, 971–1012. [Google Scholar] [CrossRef]
- Tretyakov, E.; Fedyushin, P.; Panteleeva, E.; Gurskaya, L.; Rybalova, T.; Bogomyakov, A.; Zaytseva, E.; Kazantsev, M.; Shundrina, I.; Ovcharenko, V. Aromatic SNF-approach to fluorinated phenyl tert-butyl nitroxides. Molecules 2019, 24, 4493. [Google Scholar] [CrossRef] [PubMed]
- Fedyushin, P.; Rybalova, T.; Asanbaeva, N.; Bagryanskaya, E.; Dmitriev, A.; Gritsan, N.; Kazantsev, M.; Tretyakov, E. Synthesis of nitroxide diradical using a new approach. Molecules 2020, 25, 2701. [Google Scholar] [CrossRef] [PubMed]
- Slota, M.; Keerthi, A.; Myers, W.K.; Tretyakov, E.; Baumgarten, M.; Ardavan, A.; Sadeghi, H.; Lambert, C.J.; Narita, A.; Müllen, K.; et al. Magnetic edge states and coherent manipulation of graphene nanoribbons. Nature 2018, 557, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Stass, D.; Tretyakov, E. Estimation of absolute spin counts in nitronyl nitroxide-bearing graphene nanoribbons. Magnetochemistry 2019, 5, 32. [Google Scholar] [CrossRef]
- Morozov, V.; Tretyakov, E. Spin polarization in graphene nanoribbons functionalized with nitroxide. J. Mol. Model. 2019, 25, 58. [Google Scholar] [CrossRef] [PubMed]
- Ten, Y.A.; Troshkova, N.M.; Tretyakov, E.V. From spin-labelled fused polyaromatic compounds to magnetically active graphene nanostructures. Russ. Chem. Rev. 2020, 89, 693–712. [Google Scholar] [CrossRef]
- Ohshita, J.; Iida, T.; Ohta, N.; Komaguchi, K.; Shiotani, M.; Kunai, A. Synthesis of Phenylnitroxides Bridged by an sp3-Linkage. Org. Lett. 2002, 4, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-L.; Song, Y.; Du, C.-F. CCDC 244384, Experimental Crystal Structure Determination. 2014. Available online: https://www.ccdc.cam.ac.uk/structures/search?id=doi:10.5517/cc869c3&sid=DataCite (accessed on 21 October 2020).
- Wagner, B.; Gompper, R.; Polborn, K. CSD Communication (Private Communication); CCDC 198532; Department of Chemistry, University of Munich: Munchen, Germany, 2002. [Google Scholar]
- Suzuki, S.; Nakamura, F.; Naota, T. A direct synthetic method for (nitronyl nitroxide)-substituted π-electronic compounds via a palladium-catalyzed cross-coupling reaction with a zinc complex. Mater. Chem. Front. 2018, 2, 591–596. [Google Scholar] [CrossRef]
- Espallargas, G.M.; Recuenco, A.; Romero, F.M.; Brammer, L.; Libri, S. One-dimensional organization of free radicals via halogen bonding. CrystEngComm 2012, 14, 6381–6383. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Caneschi, A.; Gatteschi, D.; Grand, A.; Laugier, J.; Pardi, L.; Rey, P. Moderate Ferromagnetic Exchange between Copper(II) and a Nitronyl Nitroxide in a Square-Pyramidal Adduct. MO Interpretation of the Mechanism of Exchange in Copper(II)–Nitroxide Complexes. Inorg. Chem. 1988, 27, 1031–1035. [Google Scholar]
- Iwahori, F.; Markosyan, A.S.; Inoue, K. Structures and Magnetic Properties of the Complexes Made up by Cu(hfac)2 and Bisnitroxide Radical Derivatives. Mol. Cryst. Liq. Cryst. 2002, 376, 449–454. [Google Scholar] [CrossRef]
- Ishimaru, Y.; Kitano, M.; Kumada, H.; Koga, N.; Iwamura, H. Regiospecificity in the Exchange Coupling of the Spins of Copper(II) Ion Coordinated with the Ring Nitrogen Atoms and N-tert-Butylaminoxyl Radical Attached as a Substituent on the Pyridine and N-Phenylimidazole Rings. Inorg. Chem. 1998, 37, 2273–2280. [Google Scholar] [CrossRef] [PubMed]
- Field, L.M.; Lahti, P.M.; Palacio, F.; Paduan-Filho, A. Manganese(II) and Copper(II) Hexafluoroacetylacetonate 1:1 Complexes with 5-(4-[N-tert-Butyl-N-aminoxyl]phenyl)pyrimidine: Regiochemical Parity Analysis for Exchange Behavior of Complexes between Radicals and Paramagnetic Cations. J. Am. Chem. Soc. 2003, 125, 10110–10118. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.A.; Kaplan, R.I. A study of bis(hexafluoroacetylacetonato)copper(II). Inorg. Chem. 1966, 5, 489–491. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin: Simulating cw ESR spectra. Biol. Magn. Reson. 2007, 27, 299–321. [Google Scholar]
- Sheldrick, G.M. SADABS, Program for Area Detector Adsorption Correction; Institute for Inorganic Chemistry, University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELX-97. Programs for Crystal Structure Analysis (Release 97-2); University of Göttingen: Göttingen, Germany, 1998. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 4 | [Cu(hfac)2(4)2] | [Cu(hfac)2(5)] |
|---|---|---|---|
| Empirical formula | C10H9F4INO | C30H20CuF20I2N2O6 | C22H12CuF16NO5 |
| Formula weight | 362.08 | 1201.82 | 737.87 |
| Crystal system | Orthorhombic | Triclinic | Triclinic |
| Space group | Pbca | P¯1 | P¯1 |
| Unit cell dimensions a, Å | 11.5462(4) | 10.0763(9) | 9.132(1) |
| b, Å | 12.1680(5) | 13.784(1) | 10.472(1) |
| c, Å | 17.8601(7) | 16.009(2) | 17.273 (2) |
| α, ° | 90 | 96.469(3) | 75.370(6) |
| β, ° | 90 | 102.396(4) | 83.253(6) |
| γ, ° | 90 | 104.941(3) | 65.505(5) |
| Volume, Å3 | 2509.2(2) | 2064.7(3) | 1454.3(3) |
| Z | 8 | 2 | 2 |
| F(000) | 1384 | 1154 | 728 |
| Density (calcd.), Mg·m−3 | 1.917 | 1.933 | 1.685 |
| Abs. coefficient, mm−1 | 2.59 | 2.16 | 0.89 |
| Crystal size, mm | 0.90 × 0.30 × 0.10 | 0.30 × 0.15 × 0.01 | 0.25 × 0.24 × 0.14 |
| Tmin, Tmax | 0.544, 0.862 | 0.722, 0.862 | 0.825, 0.928 |
| Reflections collected | 28215 | 34134 | 28365 |
| Independent reflections [Rint] | 3516 | 7322 | 5688 |
| Reflections observed (I > 2σ(I)) | 2741 | 3850 | 2856 |
| Rint | 0.048 | 0.089 | 0.074 |
| θ range for data collection (°) | 2.3–30.4 | 1.6–25.2 | 2.194–26.088 |
| Range of h, | −13→16 | −12→11 | −11→11 |
| k, | −16→16 | −16→16 | −12→12 |
| l | −23→24 | −19→18 | −21→21 |
| Completeness to θ 50° (%) | 100 | 99.1 | 99.7 |
| Data/restraints/parameters | 3516/0/157 | 7322/0/553 | 5688/6/503 |
| Final R [F2 > 2 σ (F2)] | 0.040 | 0.062 | 0.087 |
| Final wR(F2) (all data) | 0.115 | 0.173 | 0.324 |
| Goodness-of-fit on F2 | 1.03 | 1.01 | 1.05 |
| Largest diff. peak/hole, e·Å−3 | 1.44, −1.12 | 0.71, −0.64 | 0.64, −0.56 |
| Compound | 4 | [Cu(hfac)2(4)2] | [Cu(hfac)2(4)2]′ |
|---|---|---|---|
| Bond lengths, Å | |||
| Cu1–O1 | - | 2.401(8) | 2.380(7) |
| Cu1–O2 | - | 1.933(6) | 1.954(6) |
| Cu1–O3 | - | 1.955(6) | 1.954(5) |
| C4–I1 | 2.079(4) | 2.062(9) | 2.060(8) |
| N1–O1 | 1.278(4) | 1.29(1) | 1.283(9) |
| N1–C1 | 1.428(4) | 1.43(1) | 1.41(1) |
| N1–C7 | 1.493(5) | 1.47(1) | 1.50(1) |
| Bond angles, ° | |||
| O2–Cu1–O1 | - | 95.7(2) | 98.6(2) |
| O3–Cu1–O1 | - | 91.5(3) | 91.2(2) |
| O2–Cu1–O3 | - | 92.3(2) | 92.3(2) |
| O1–N1–C1 | 115.7(3) | 115.8(7) | 115.9(6) |
| O1–N1–C7 | 118.0(3) | 117.2(7) | 116.8(6) |
| C1–N1–C7 | 125.3(3) | 125.7(7) | 126.4(6) |
Sample Availability: Samples of all described compounds are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Politanskaya, L.V.; Fedyushin, P.A.; Rybalova, T.V.; Bogomyakov, A.S.; Asanbaeva, N.B.; Tretyakov, E.V. Fluorinated Organic Paramagnetic Building Blocks for Cross-Coupling Reactions. Molecules 2020, 25, 5427. https://doi.org/10.3390/molecules25225427
Politanskaya LV, Fedyushin PA, Rybalova TV, Bogomyakov AS, Asanbaeva NB, Tretyakov EV. Fluorinated Organic Paramagnetic Building Blocks for Cross-Coupling Reactions. Molecules. 2020; 25(22):5427. https://doi.org/10.3390/molecules25225427
Chicago/Turabian StylePolitanskaya, Larisa V., Pavel A. Fedyushin, Tatyana V. Rybalova, Artem S. Bogomyakov, Nargiz B. Asanbaeva, and Evgeny V. Tretyakov. 2020. "Fluorinated Organic Paramagnetic Building Blocks for Cross-Coupling Reactions" Molecules 25, no. 22: 5427. https://doi.org/10.3390/molecules25225427
APA StylePolitanskaya, L. V., Fedyushin, P. A., Rybalova, T. V., Bogomyakov, A. S., Asanbaeva, N. B., & Tretyakov, E. V. (2020). Fluorinated Organic Paramagnetic Building Blocks for Cross-Coupling Reactions. Molecules, 25(22), 5427. https://doi.org/10.3390/molecules25225427