
Selective Interactions of O-Methylated Flavonoid Natural Products with Human Monoamine Oxidase-A and -B



 ,
,  ,
, 
Abstract
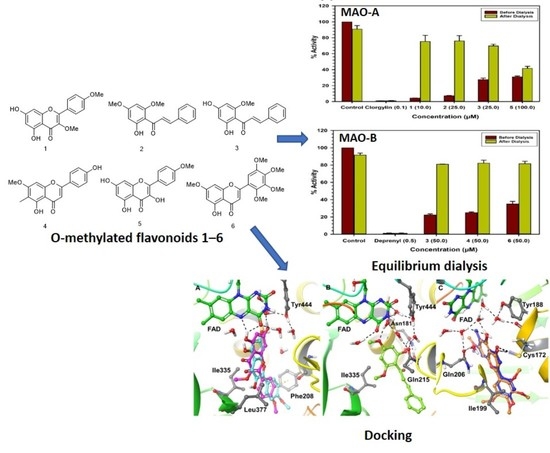
1. Introduction
2. Results
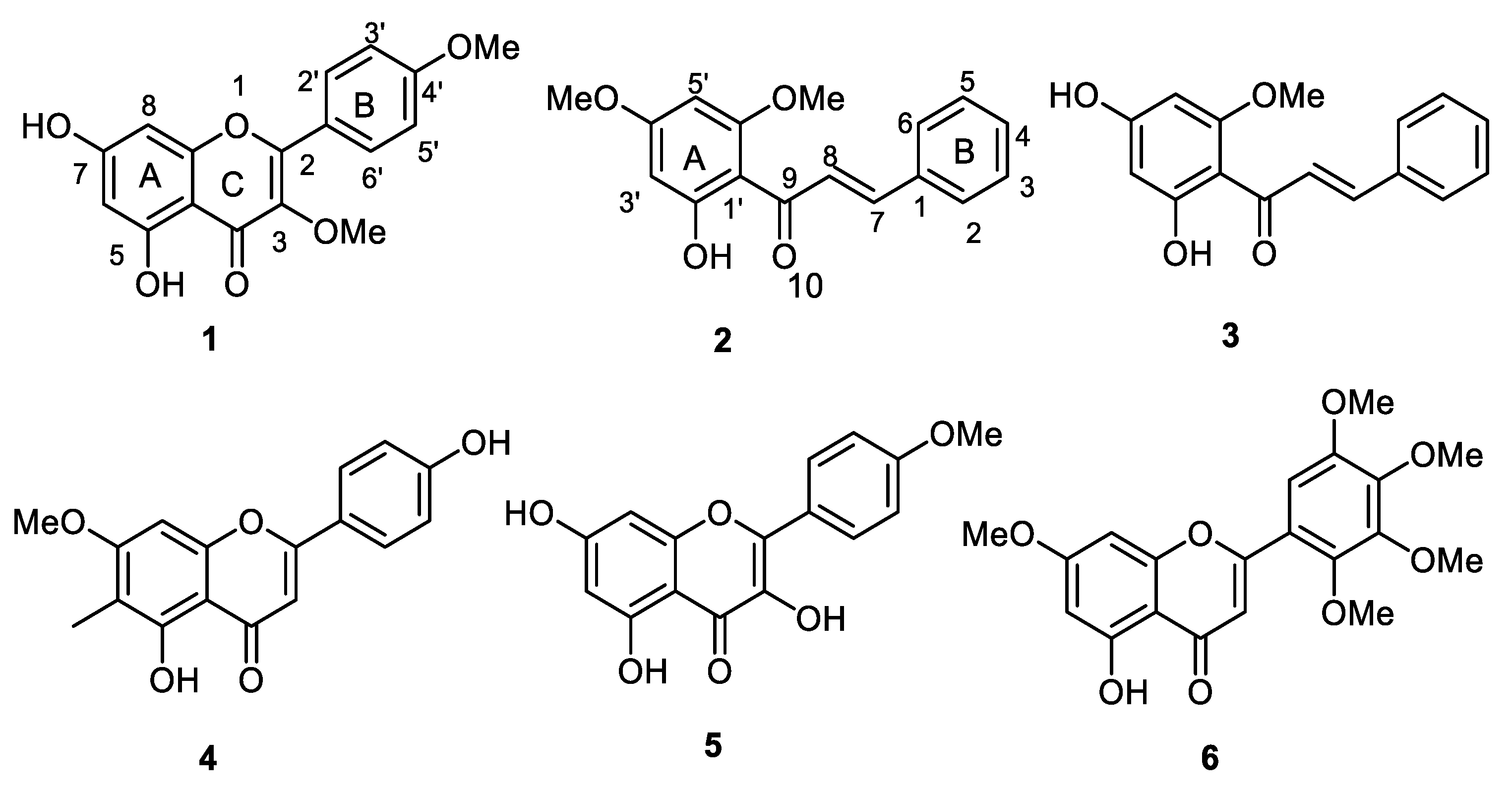
2.1. Isolation, Purification, and Characterization of O-Methylated Flavonoids
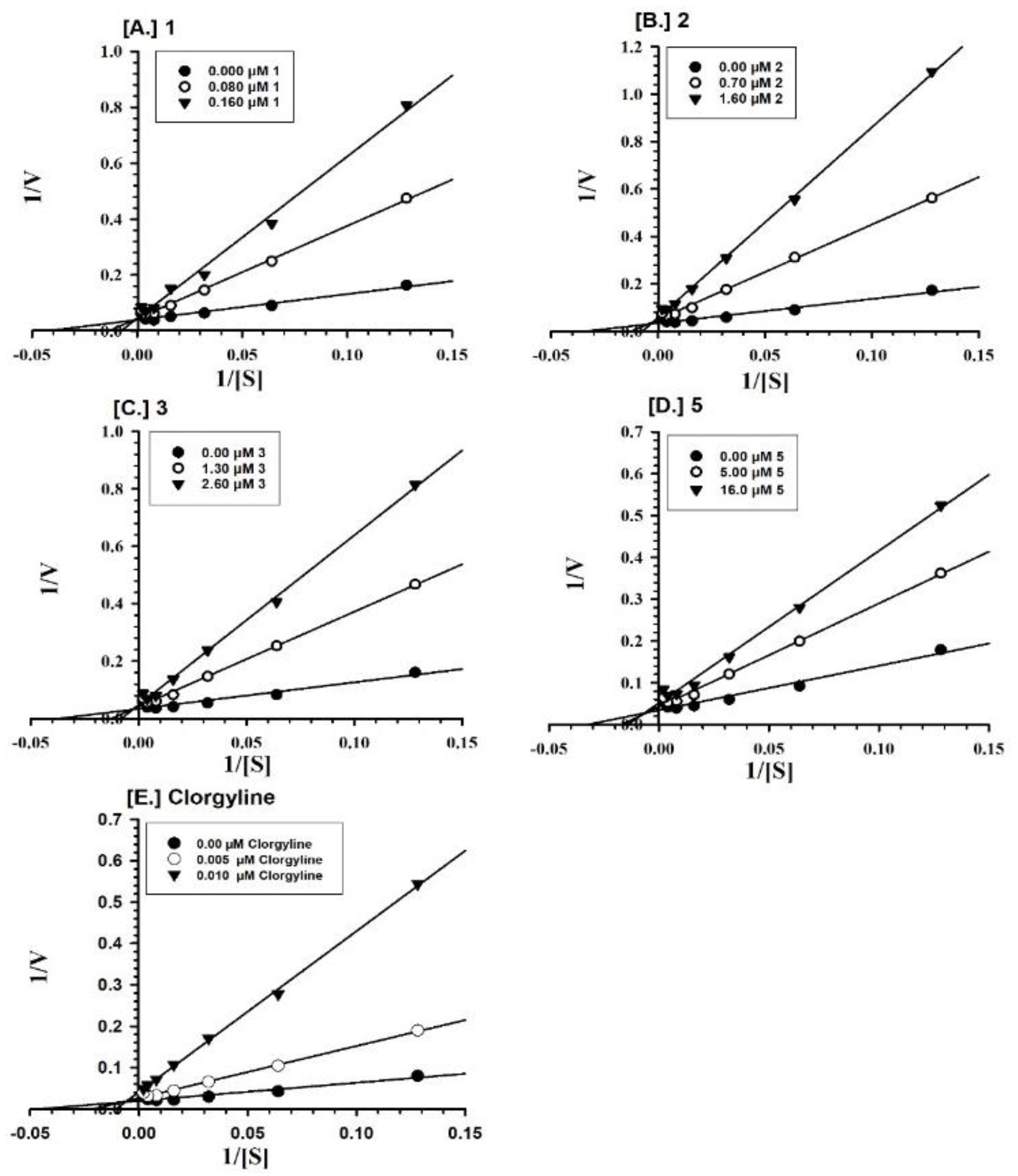
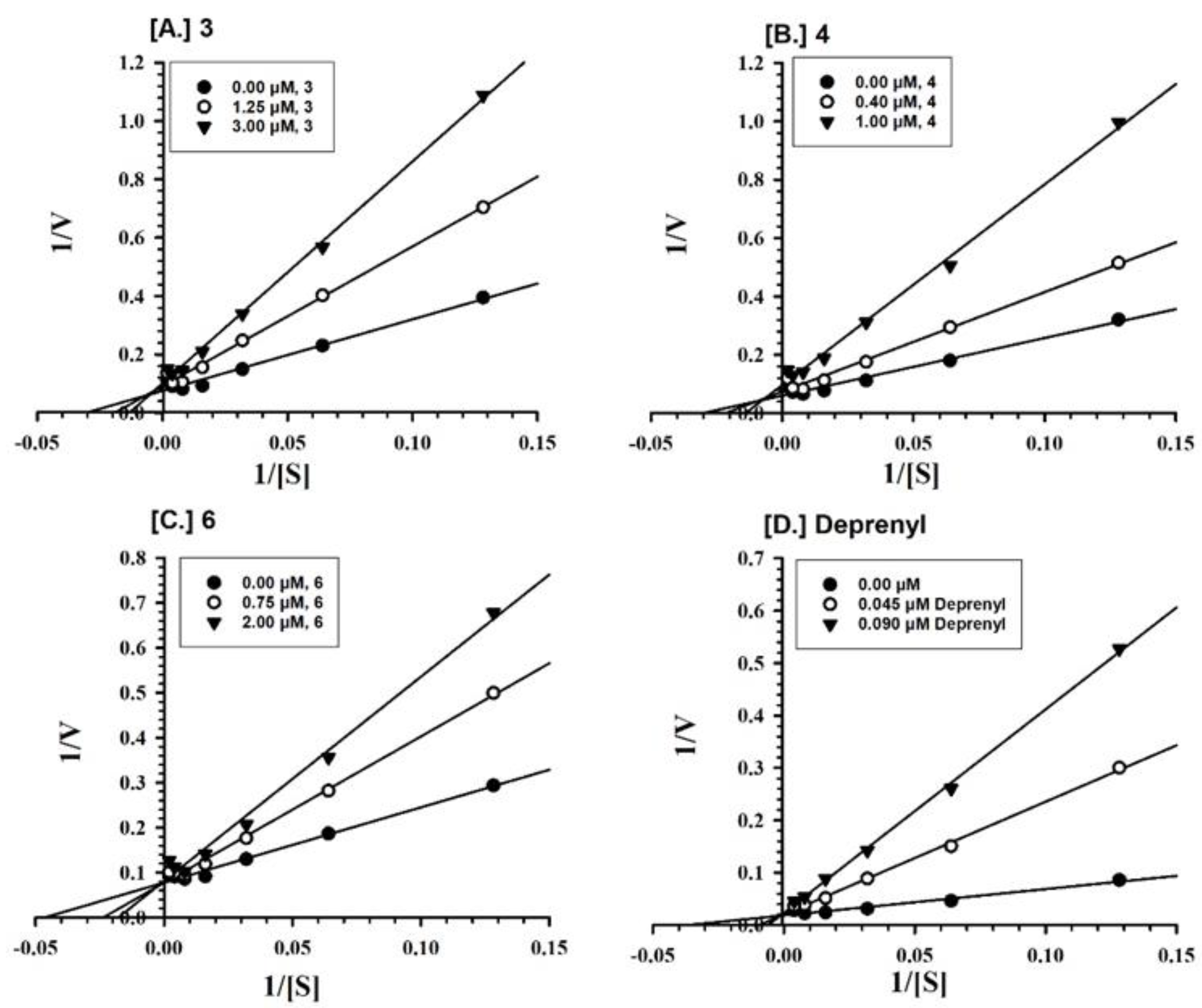
2.2. Enzyme Inhibition and Kinetics Mechanism of MAO-A and -B with Compounds 1–6
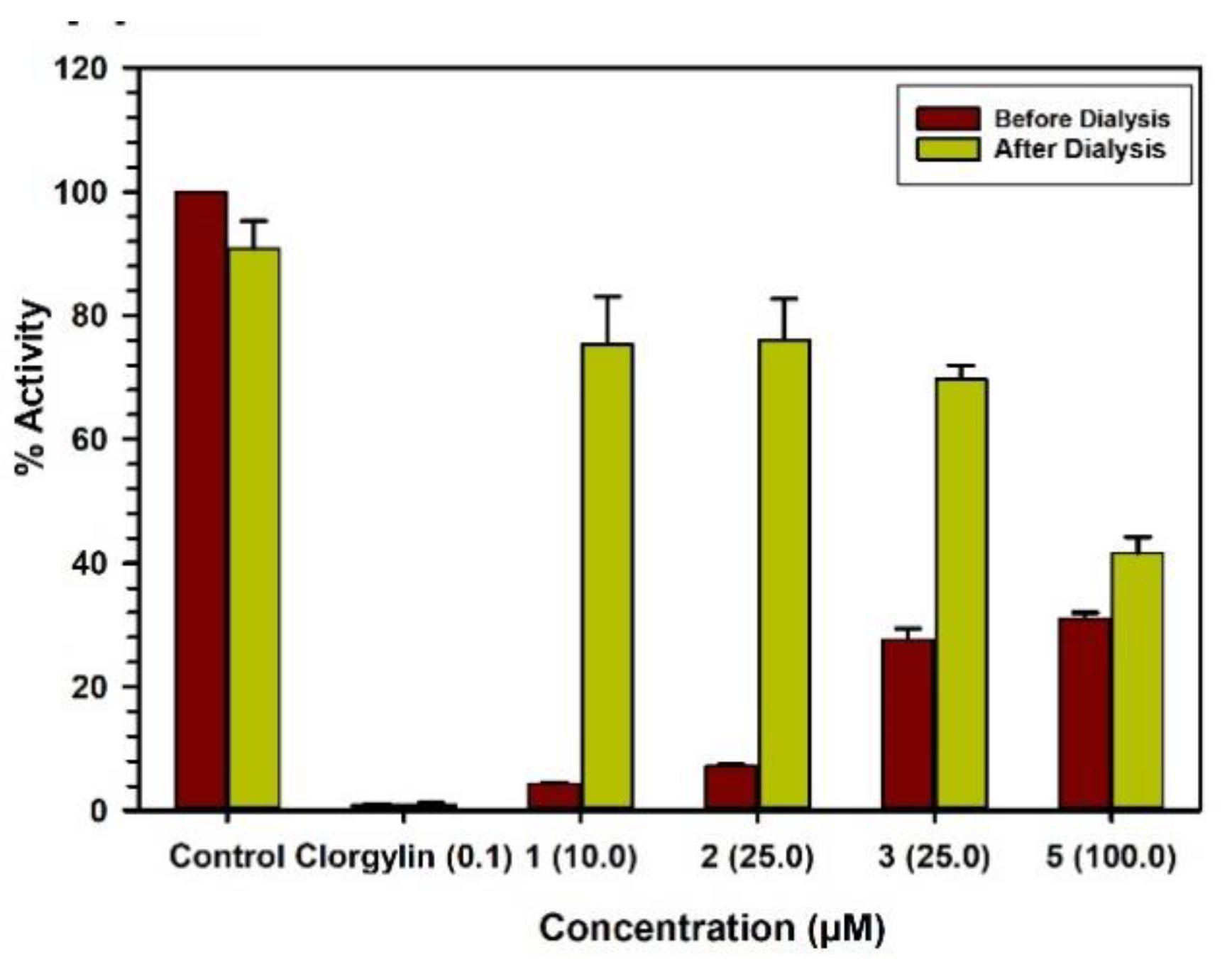
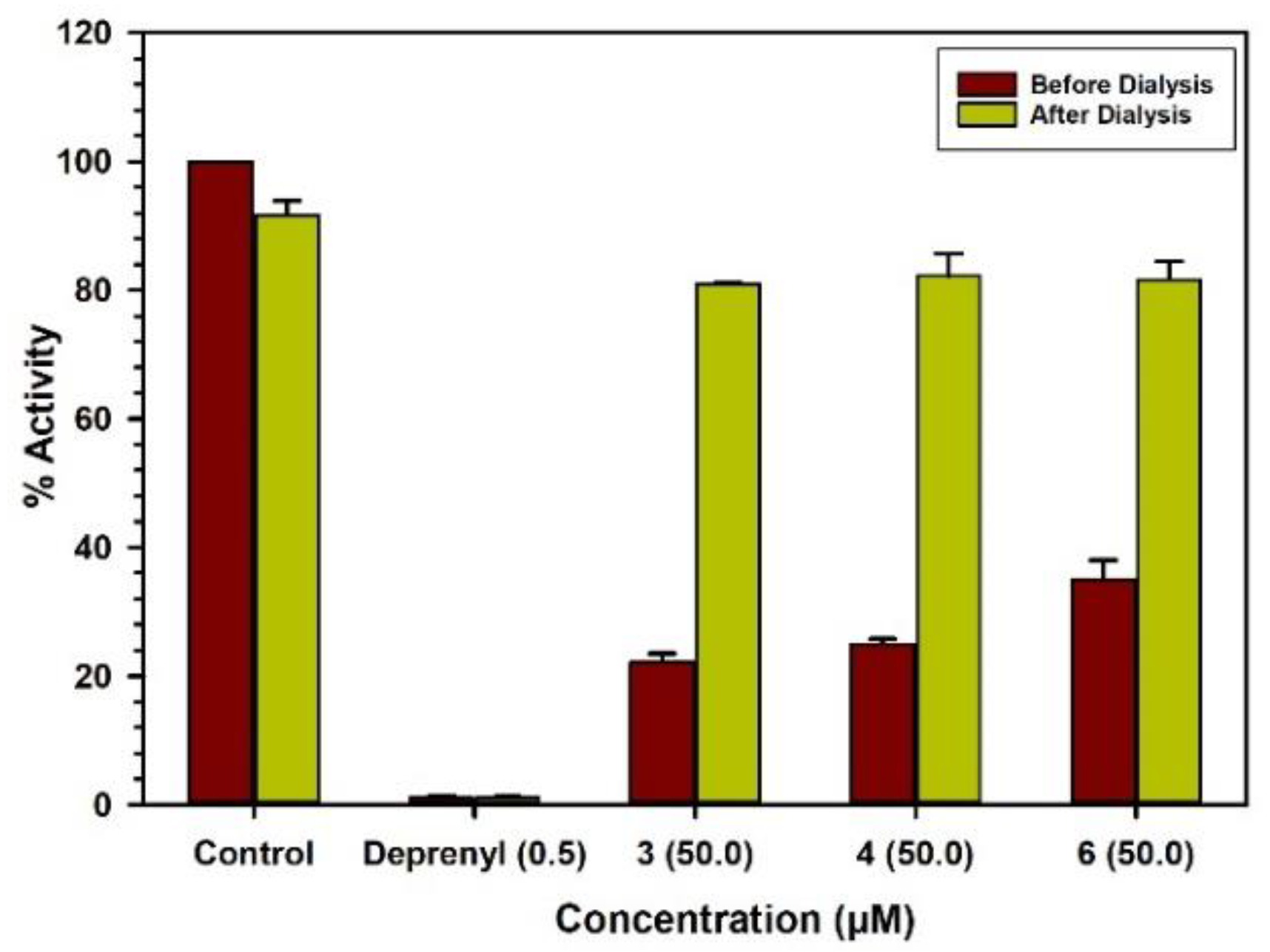
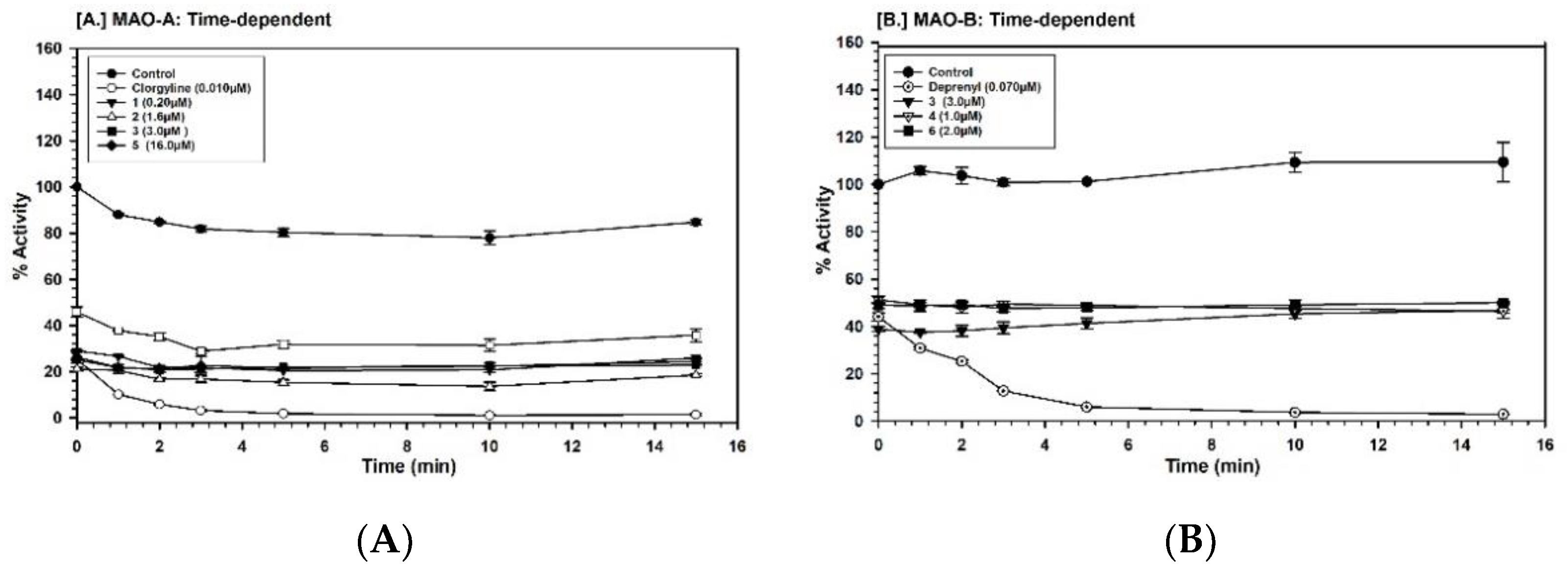
2.3. Binding and Time-Dependent Assays of MAO-A and -B with Compounds 1–6
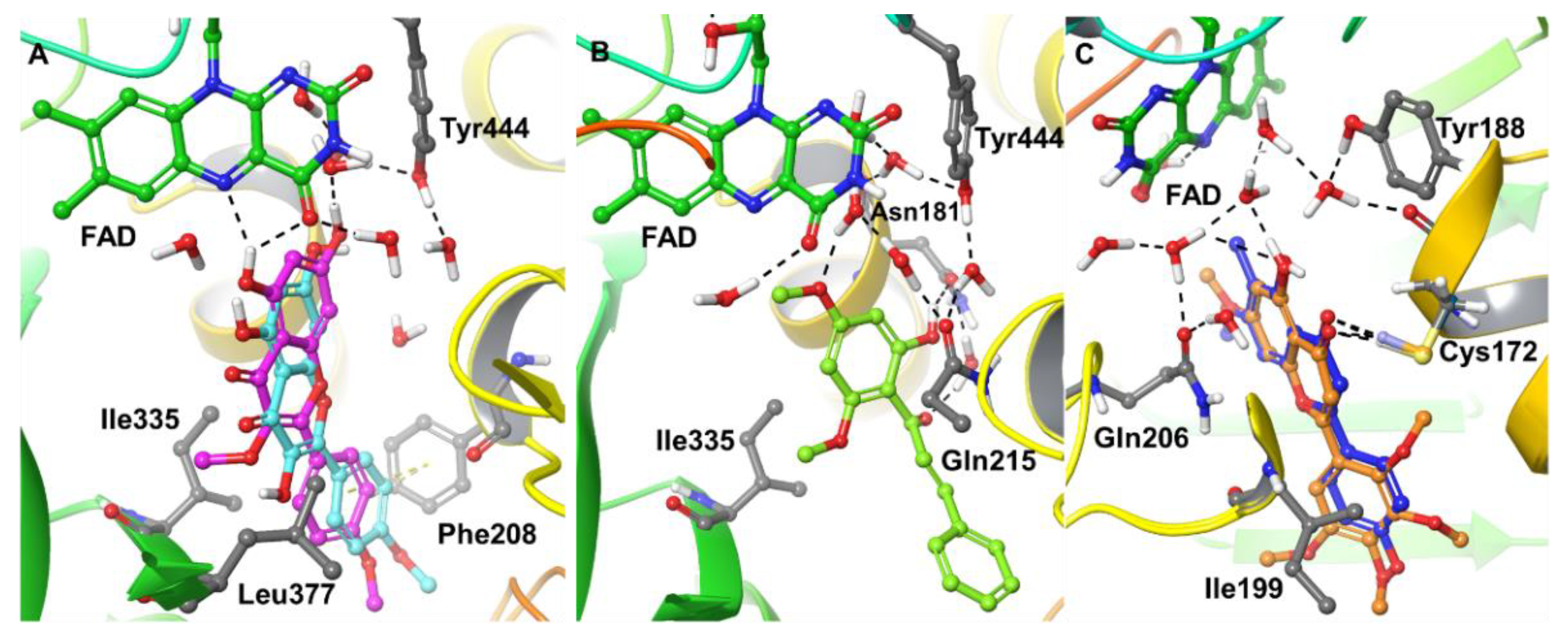
2.4. Computational Analysis of Enzyme–Inhibitor Interactions
3. Discussion
4. Materials and Methods
4.1. Enzymes
4.2. Isolation and Identification of Compounds 1–6
4.3. MAO Inhibition Assay
4.4. Determination of IC50 Values
4.5. Enzyme Kinetics and Mechanism Studies
4.6. Analysis of Binding of Inhibitor with The Enzymes
4.7. Time-Dependent Inhibition of the Enzyme
4.8. Computational Analysis of The Interaction of Test Compounds with MAO-A or -B
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Abell, C.W.; Kwan, S.W. Molecular characterization of monoamine oxidases A and B. Prog. Nucl. Acid Res. Mol. Biol. 2001, 65, 129–156. [Google Scholar]
- Carradori, S.; D’Ascenzio, M.; Chimenti, P.; Secci, D.; Bolasco, A. Selective MAO-B inhibitors: A lesson from natural products. Mol. Divers. 2014, 18, 219–243. [Google Scholar] [CrossRef]
- Cesura, A.M.; Pletscher, A. The new generation of monoamine oxidase inhibitors. Prog. Drug Res. 1992, 38, 171–297. [Google Scholar] [PubMed]
- Yamada, M.; Yasuhara, H. Clinical pharmacology of MAO inhibitors: Safety and future. Neurotoxicology 2004, 25, 215–221. [Google Scholar] [CrossRef]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef]
- Youdim, M.B.; Fridkin, M.; Zheng, H. Novel bifunctional drugs targeting monoamine oxidase inhibition and iron chelation as an approach to neuroprotection in Parkinson’s disease and other neurodegenerative diseases. J. Neural Transm. 2004, 111, 1455–1471. [Google Scholar] [CrossRef]
- Youdim, M.B.; Bhakle, Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br. J. Pharmacol. Chemother. 2006, 147, S287–S296. [Google Scholar] [CrossRef]
- Finberg, J.P.M.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology. Front. Pharmacol. 2016, 7, 340–355. [Google Scholar] [CrossRef]
- Riederer, P.; Laux, G. MAO-inhibitors in Parkinson’s Disease. Exp. Neurobiol. 2011, 20, 1–17. [Google Scholar] [CrossRef]
- Cai, Z. Monoamine oxidase inhibitors: Promising therapeutic agents for Alzheimer’s disease. Mol. Med. Rep. 2014, 9, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Muller, T. Monoamine oxidase-B inhibitors in the treatment of Parkinson’s disease: Clinical-pharmacological aspects. J. Neural Transm. 2018, 125, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, P.; Carradorri, S.; Ammazzalorso, A.; Secci, D. Novel approaches to the discovery of selective human monoamine oxidase-B inhibitors: Is there room for improvement. Expert Opin. Drug Discov. 2019, 14, 995–1035. [Google Scholar] [CrossRef] [PubMed]
- Orhan, I.E. Potential of Natural Products of Herbal Origin as Monoamine Oxidase Inhibitors. Curr. Pharm. Des. 2016, 22, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Suresh, J.; Mathew, G.E.; Parasuraman, R.; Abdulla, N. Plant secondary metabolites- potent inhibitors of monoamine oxidase isoforms. Cent. Nerv. Syst. Agents. Med. Chem. 2014, 14, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, M.; Perumal, E. Potential plant-derived catecholaminergic activity enhancers for neuropharmacological approaches: A review. Phytomedicine 2019, 55, 148–164. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, M.; Lankatillake, C.; Dias, D.A.; Docea, A.O.; Mahomoodally, M.F.; Lobine, D.; Chazot, P.L.; Kurt, B.; Tumer, T.B.; Moreira, A.C.; et al. Impact of Natural Compounds on Neurodegenerative Disorders: From Preclinical to Pharmacotherapeutics. J. Clin. Med. 2020, 9, 1061. [Google Scholar] [CrossRef]
- Chaurasiya, N.D.; Ibrahim, M.A.; Muhammad, I.; Walker, L.A.; Tekwani, B.L. Monoamine oxidase inhibitory constituents of propolis: Kinetics and mechanism of inhibition of recombinant human MAO-A and MAO-B. Molecules 2014, 19, 18936–18952. [Google Scholar] [CrossRef]
- Chaurasiya, N.D.; Zhao, J.; Pandey, P.; Doerksen, R.J.; Muhammad, I.; Tekwani, B.L. Selective Inhibition of Human Monoamine Oxidase B by Acacetin 7-Methyl Ether Isolated from Turnera diffusa (Damiana). Molecules 2019, 24, 810. [Google Scholar] [CrossRef]
- Chaurasiya, N.D.; Gogineni, V.; Elokely, K.M.; León, J.F.; Núñez, M.J.; Klein, M.L.; Walker, L.A.; Cutler, S.J.; Tekwani, B.L. Isolation of Acacetin from Calea urticifolia with Inhibitory Properties against Human Monoamine Oxidase-A and -B. J. Nat. Prod. 2016, 79, 2538–2544. [Google Scholar] [CrossRef]
- Jalili-Baleh, L.; Babaei, E.; Abdpour, S.; Nasir Abbas Bukhari, S.; Foroumadi, A.; Ramazani, A.; Sharifzadeh, M.; Abdollahi, M.; Khoobi, M. A review on flavonoid-based scaffolds as multi-target-directed ligands (MTDLs) for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 152, 570–589. [Google Scholar] [CrossRef] [PubMed]
- Larit, F.; Elokely, K.M.; Chaurasiya, N.D.; Benyahia, S.; Nael, M.A.; León, F.; Abu-Darwish, M.S.; Efferth, T.; Wang, Y.H.; Belouahem-Abed, D.; et al. Inhibition of human monoamine oxidase A and B by flavonoids isolated from two Algerian medicinal plants. Phytomedicine 2018, 40, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Chimenti, P.; Secci, D.; Rossi, F.; Yáñez, M.; Orallo, F.; Ortuso, F.; Alcaro, S. Chalcones: A valid scaffold for monoamine oxidases inhibitors. J. Med. Chem. 2009, 52, 2818–2824. [Google Scholar] [CrossRef] [PubMed]
- Kerubo, L.; Midiwo, J.O.; Derese, S.; Langat, M.K.; Akala, H.M.; Waters, N.C.; Peter, M.; Heydenreich, M. Antiplasmodial activity of compounds from surface exudates of Senecio roseiflorus. Nat. Prod. Commun. 2013, 8, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Midiwo, J.O.; Matasi, J.J.; Wanjau, O.M.; Mwangi, R.W.; Waterman, P.G.; Wollenweber, E. Anti-feedant effects of surface accumulated flavonoids of Polygonum senegalense. Bull. Chem. Soc. Ethiop. 1990, 4, 123. [Google Scholar]
- Cardona, M.; Seoan, E. Flavonoid and xanthonolignoids of Hypericum ericoides. Phytochemistry 1982, 21, 2759–2760. [Google Scholar] [CrossRef]
- Awas, L.K.; Omosa, J.O.; Midiwo, A.; Ndakala, J. Anti-oxidant Activities of flavonoid aglycones from Kenyan Gardenia ternifolia Schum and Thonn. J. Pharm. Biol. Sci. 2016, 11, 136–141. [Google Scholar]
- Bernard, F.; Yenesev, A.J.; Midiwo, J.O.; Waterman, P.G. Flavones and phenypropanoids in exudate of Psiadia punctulata. Phytochemistry 2001, 57, 571–574. [Google Scholar]
- Tripathi, R.K.P.; Ayyannam, S.R. Monoamine oxidase-B inhibitors as potential neurotherapeutic agents: An overview and update. Med. Res. Rev. 2019, 39, 1603–1706. [Google Scholar] [CrossRef]
- Carradori, S.; Secci, D.; Petzer, J.P. MAO inhibitors and their wider applications: A patent review. Expert. Opin. Ther. Pat. 2018, 28, 211–226. [Google Scholar] [CrossRef]
- Lee, H.W.; Ryu, H.W.; Baek, S.C.; Kang, M.G.; Park, O.D.; Han, H.Y.; An, J.H.; Oh, S.R.; Kim, H. Potent inhibitions of monoamine oxidase A and B by acacetin and its 7-O-(6-O-malonylglucoside) derivative from Agastache rugosa. Int. Biol. Macromol. 2017, 104, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Koirala, N.; Thuan, N.H.; Ghimire, G.P.; Van Thang, D.; Sohng, J.K. Methylation of flavonoids: Chemical structures, bioactivities, progress and perspectives for biotechnological production. Enzym. Microb. Technol. 2016, 86, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Schroder, G.; Wehinger, E.; Lukacin, R.; Wellmann, F.; Seefelder, W.; Schwab, W.; Schröder, J. Flavonoid methylation: A novel 4′-O-methyltransferase from Catharanthus roseus, and evidence that partially methylated flavanones are substrates of four different flavonoid dioxygenases. Phytochemistry 2004, 65, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouységu, L. Plant polyphenols: Chemical properties, biological activities, and synthesis. Angew. Chem. Int. Ed. Engl. 2011, 50, 586–621. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Alhusban, M.; Chaurasiya, N.D.; Tekwani, B.L.; Chittiboyina, A.G.; Khan, I.A. Isoform selectivity of harmine-conjugated 1,2,3-triazoles against human monoamine oxidase. Future Med. Chem. 2018, 10, 1435–1448. [Google Scholar] [CrossRef]
- Chaurasiya, N.D.; Leon, F.; Ding, Y.; Gomez-Betancur, I.; Benjumea, D.; Walker, L.A.; Cutler, S.J.; Tekwani, B.L. Interactions of Desmethoxyyangonin, a Secondary Metabolite from Renealmia alpinia, with Human Monoamine Oxidase-A and Oxidase-B. Evid. Complem. Altern. Med. eCAM 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Albreht, A. Kinetics, mechanism, and inhibition of monoamine oxidase. J. Neural Transm. 2018, 125, 1659–1683. [Google Scholar] [CrossRef]
- Binda, C.; Aldeco, M.; Geldenhuys, W.J.; Tortorici, M.; Mattevi, A.; Edmondson, D.E. Molecular insights into human monoamine oxidase B inhibition by the glitazone antidiabetes drugs. ACS Med. Chem. Lett. 2011, 3, 39–42. [Google Scholar] [CrossRef]
- Maestro, Version 10.6; Schrödinger; LLC: New York, NY, USA, 2016.
- Schrödinger, Version 2016-2; Schrödinger; LLC: New York, NY, USA, 2016.
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 1281–1296. [Google Scholar] [CrossRef]
- LigPrep, Version 3.8; Schrödinger; LLC: New York, NY, USA, 2016.
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors after the execution of Intellectual Property and Material Transfer Agreements. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | MAO-A IC50 (µM) a | MAO-B IC50 (µM) a | SI b | SI c |
|---|---|---|---|---|
| 1 | 0.033 ± 0.042 | 9.667 ± 2.309 | 0.0034 | 292.93 |
| 2 | 0.407 ± 0.075 | 5.933 ± 0.833 | 0.082 | 14.57 |
| 3 | 1.167 ± 0.513 | 2.700 ± 0.794 | 0.432 | 2.31 |
| 4 | 5.167 ± 1.106 | 0.800 ± 0.180 | 6.451 | 0.154 |
| 5 | 1.350 ± 0.198 | >100 | - | >74.07 |
| 6 | 87.501 ± 3.536 | 0.875 ± 0.035 | 100.0 | 0.009 |
| Clorgyline b | 0.0065 ± 0.001 | - | - | - |
| Deprenyl c | - | 0.043 ± 0.011 | - | - |
| Compounds | Monoamine Oxidase-A | Monoamine Oxidase-B | ||
|---|---|---|---|---|
| Ki (μM) a | Type of Inhibition | Ki (μM) a | Type of Inhibition | |
| 1 | 0.0379 ± 0.0008 | Competitive/Reversible | - | - |
| 2 | 0.339 ± 0.219 | Competitive/Reversible | - | - |
| 3 | 0.633 ± 0.107 | Competitive/Reversible | 1.242 ± 0.600 | Competitive/Reversible |
| 4 | - | - | 0.809 ± 0.093 | Mixed/Reversible |
| 5 | 3.531 ± 0.265 | Mixed/Partially Reversible | - | - |
| 6 | - | - | 0.874 ± 0.069 | Competitive/Reversible |
| Clorgyline b | 0.0018 ± 0.0003 | Mixed/Irreversible | - | - |
| Deprenyl b | - | - | 0.0101 ± 0.0034 | Mixed/Irreversible |
| Compounds | Experimental IC50 (µM) a | GlideScore (kcal/mol) | Binding Free-Energy (kcal/mol) | |||
|---|---|---|---|---|---|---|
| MAO-A | MAO-B | MAO-A | MAO-B | MAO-A | MAO-B | |
| 1 | 0.033 ± 0.04 | 9.667 ± 2.309 | −11.667 | −10.028 b | −57.522 | −76.353 b |
| 2 | 0.407 ± 0.075 | 5.933 ± 0.833 | −10.686 | −9.999 | −47.724 | −28.119 |
| 3 | 1.167 ± 0.513 | 2.700 ± 0.794 | −9.951 | −10.579 | −37.683 | −51.309 |
| 4 | 5.167 ± 1.106 | 0.800 ± 0.180 | −9.664 | −10.225 | −35.043 | −53.574 |
| 5 | 1.350 ± 0.198 | >100 | −10.567 | ND c | −47.035 | ND c |
| 6 | 87.501 ± 3.536 | 0.875 ± 0.035 | −7.239 | −11.191 | −34.651 | −68.053 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaurasiya, N.D.; Midiwo, J.; Pandey, P.; Bwire, R.N.; Doerksen, R.J.; Muhammad, I.; Tekwani, B.L. Selective Interactions of O-Methylated Flavonoid Natural Products with Human Monoamine Oxidase-A and -B. Molecules 2020, 25, 5358. https://doi.org/10.3390/molecules25225358
Chaurasiya ND, Midiwo J, Pandey P, Bwire RN, Doerksen RJ, Muhammad I, Tekwani BL. Selective Interactions of O-Methylated Flavonoid Natural Products with Human Monoamine Oxidase-A and -B. Molecules. 2020; 25(22):5358. https://doi.org/10.3390/molecules25225358
Chicago/Turabian StyleChaurasiya, Narayan D., Jacob Midiwo, Pankaj Pandey, Regina N. Bwire, Robert J. Doerksen, Ilias Muhammad, and Babu L. Tekwani. 2020. "Selective Interactions of O-Methylated Flavonoid Natural Products with Human Monoamine Oxidase-A and -B" Molecules 25, no. 22: 5358. https://doi.org/10.3390/molecules25225358
APA StyleChaurasiya, N. D., Midiwo, J., Pandey, P., Bwire, R. N., Doerksen, R. J., Muhammad, I., & Tekwani, B. L. (2020). Selective Interactions of O-Methylated Flavonoid Natural Products with Human Monoamine Oxidase-A and -B. Molecules, 25(22), 5358. https://doi.org/10.3390/molecules25225358