
Recent Advances in Rapid Synthesis of Non-proteinogenic Amino Acids from Proteinogenic Amino Acids Derivatives via Direct Photo-Mediated C–H Functionalization



Abstract
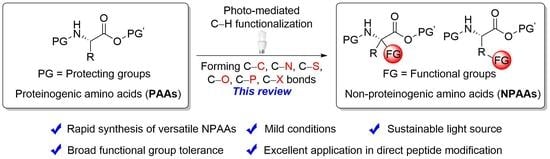
1. Introduction
2. C–C Formation
2.1. C–H Alkylation
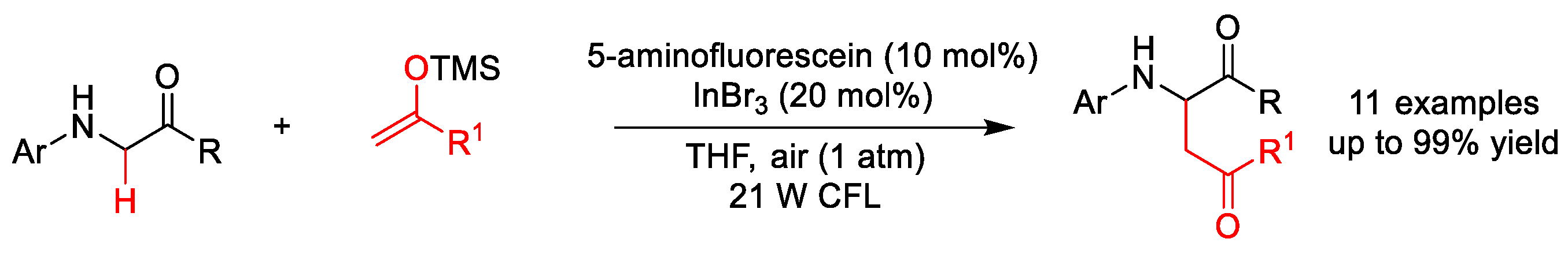
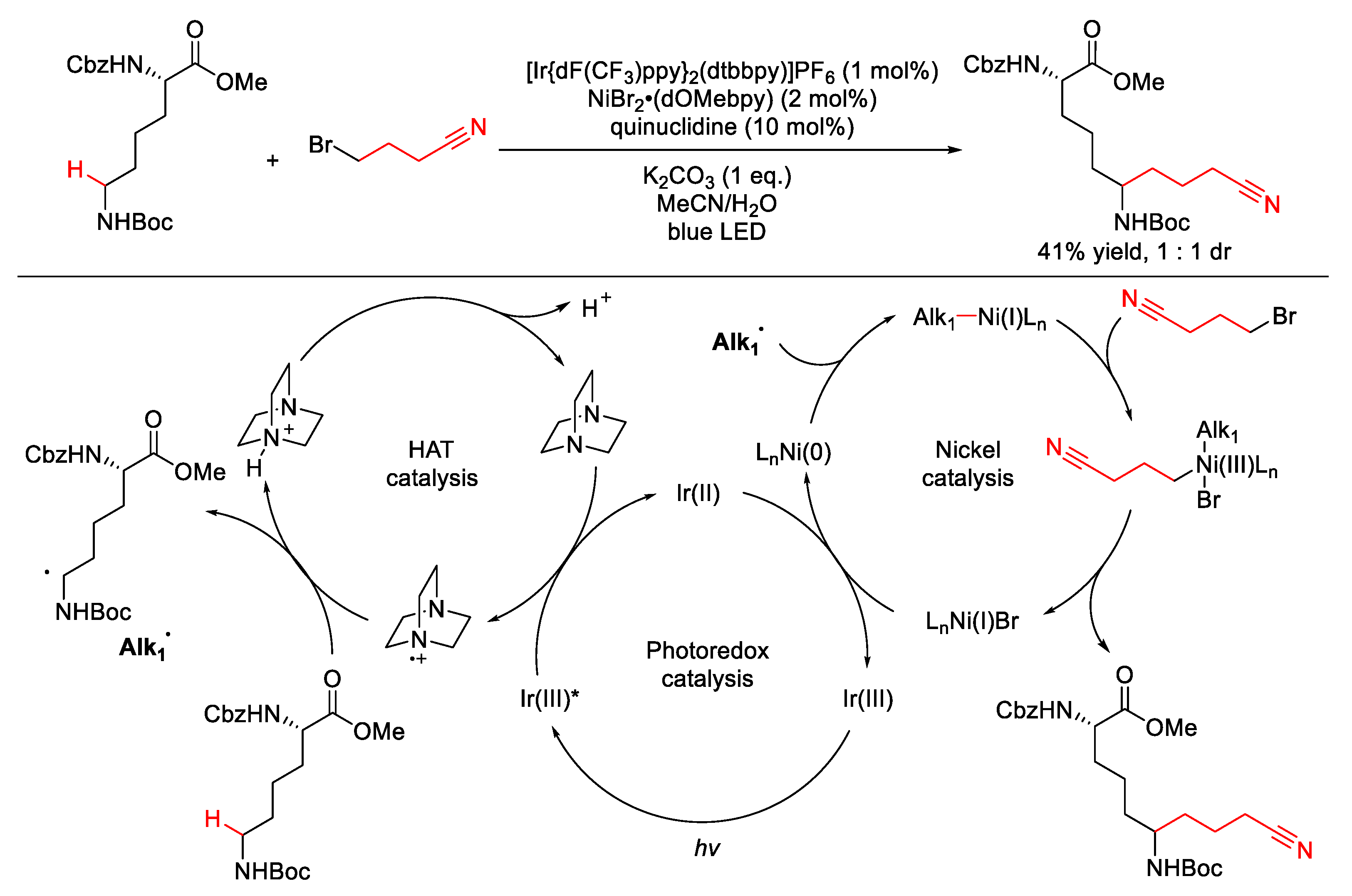
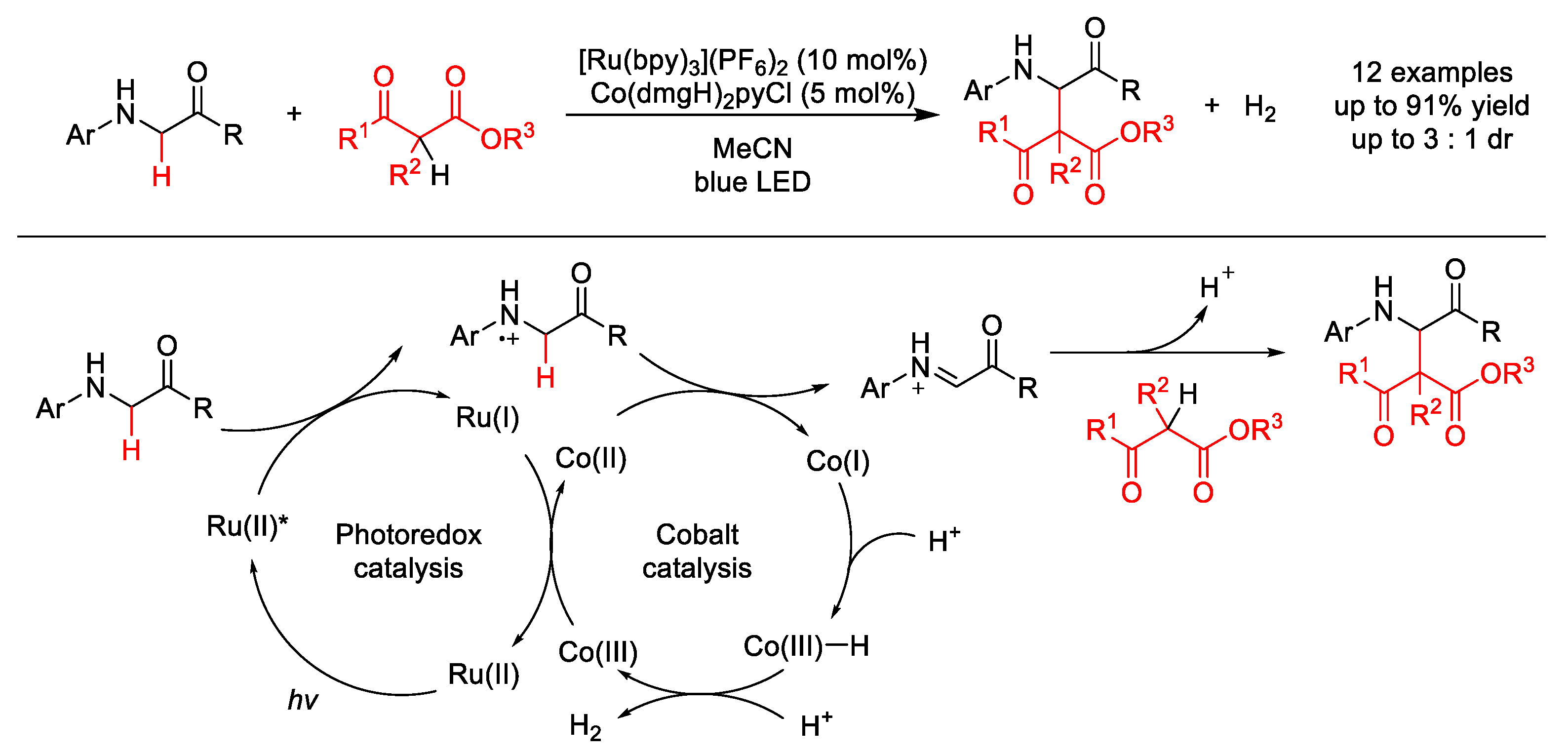
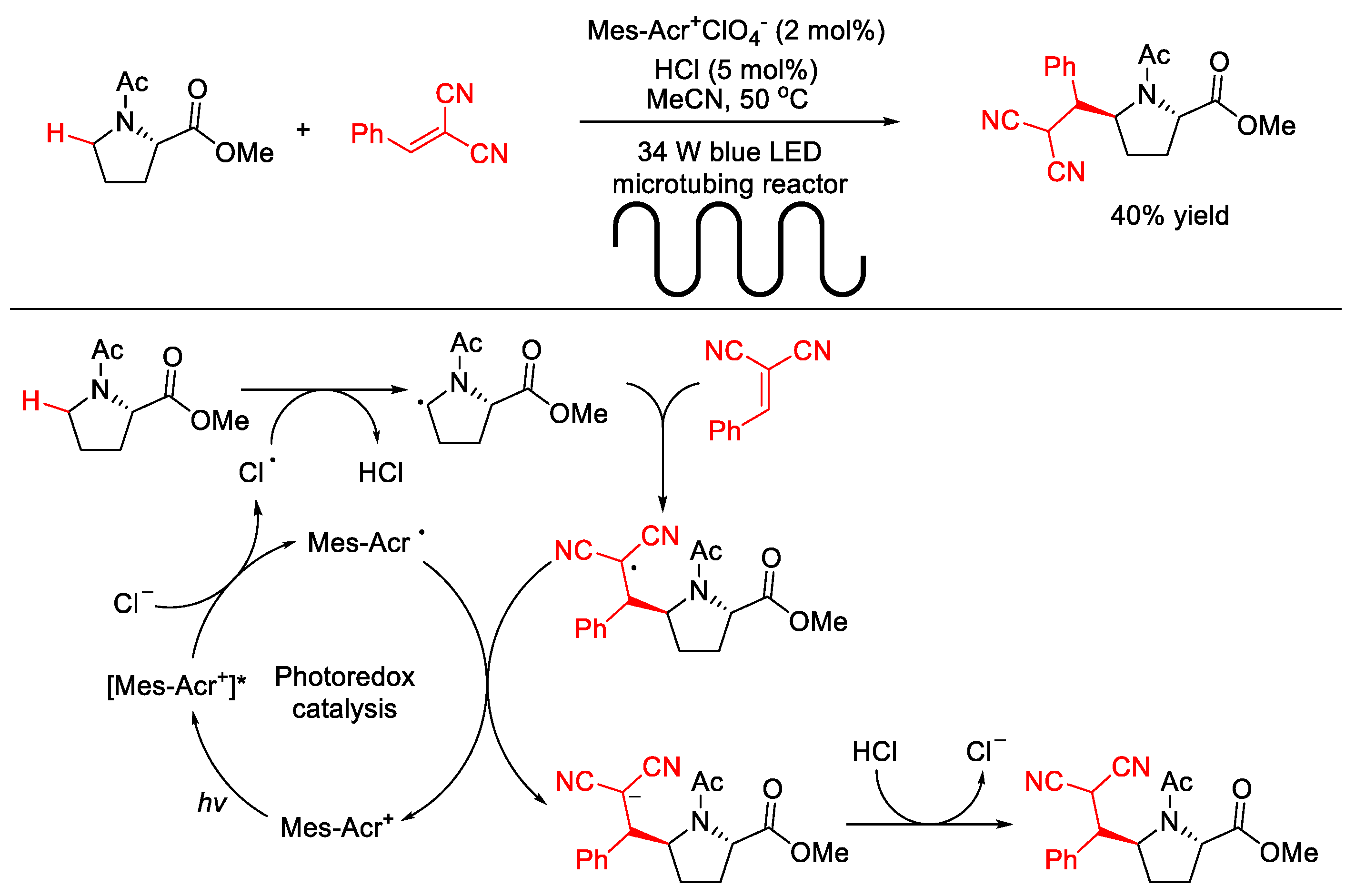
2.1.1. C(sp3)–H Alkylation
2.1.2. C(sp2)–H Alkylation
2.2. C–H Benzylation
2.2.1. C(sp3)–H Benzylation
2.2.2. C(sp2)–H Benzylation
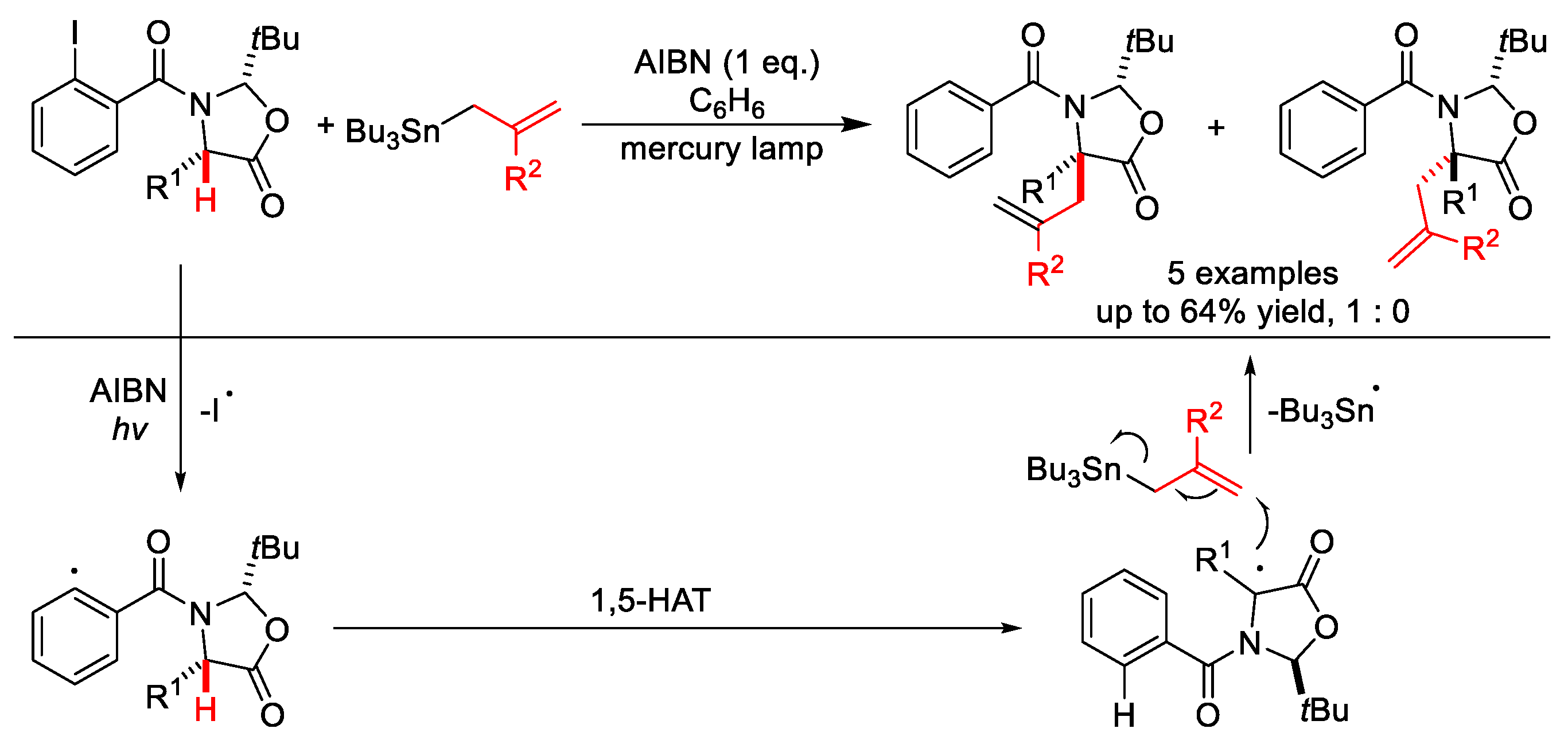
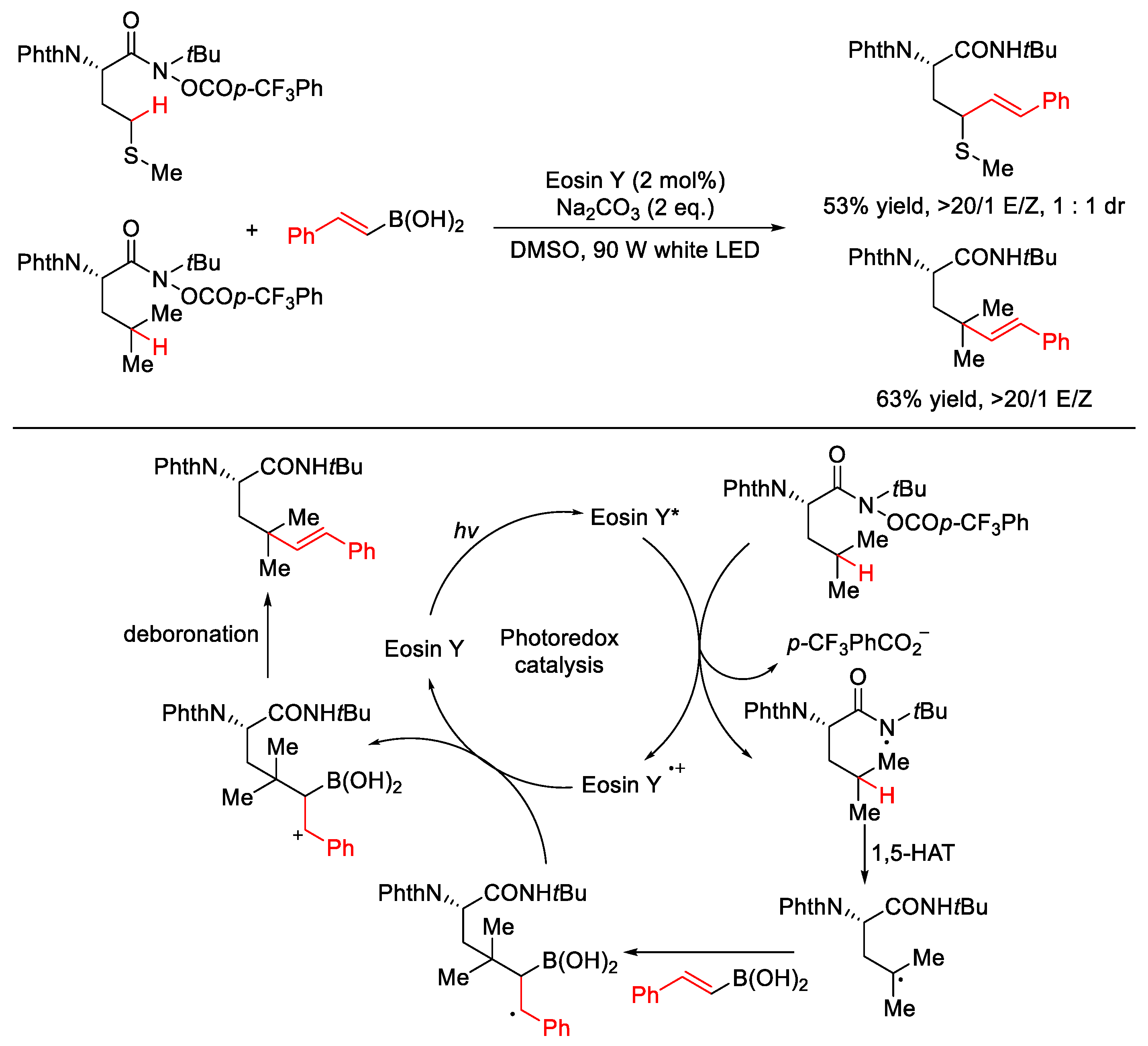
2.3. C–H Allylation
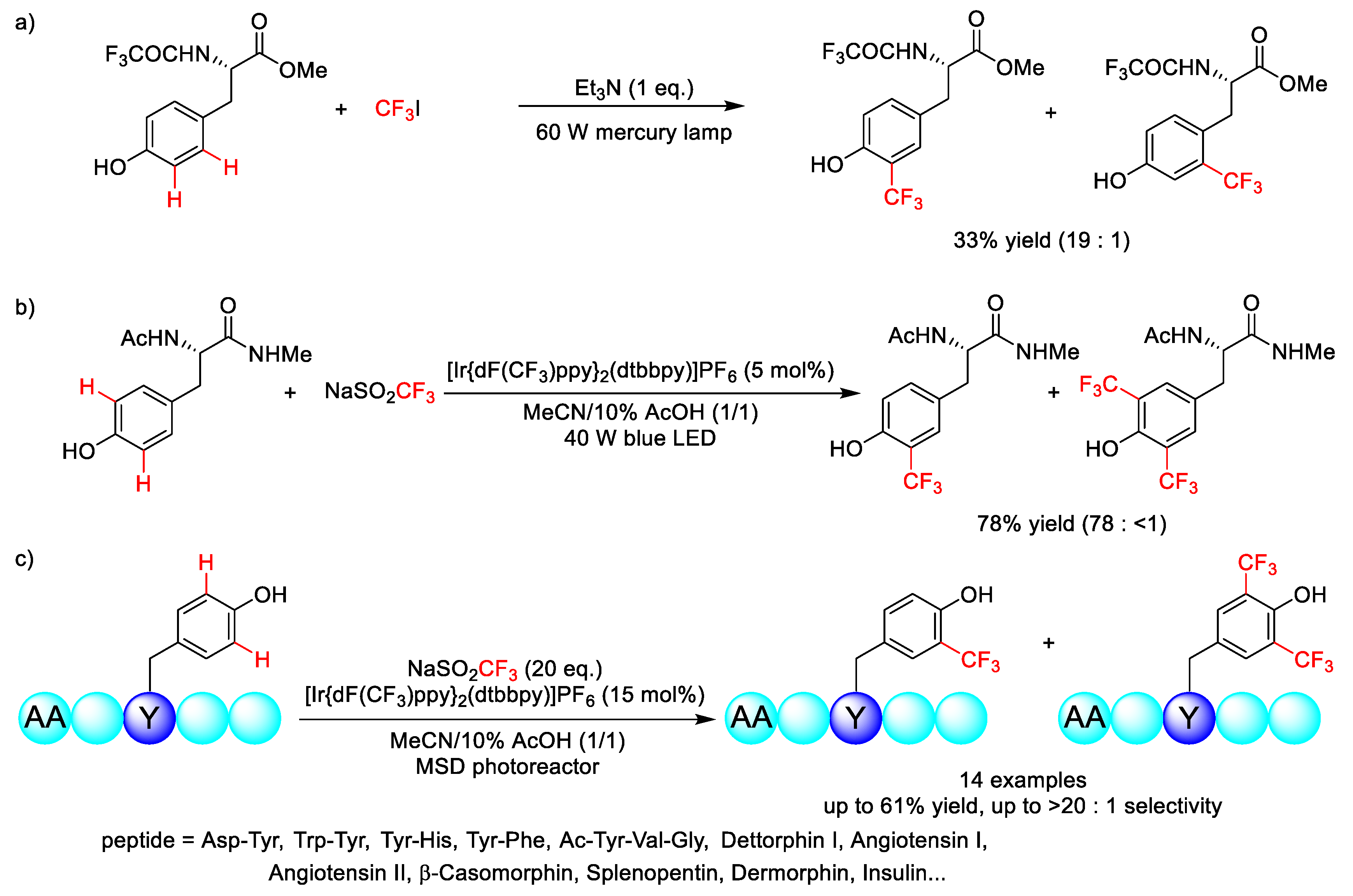
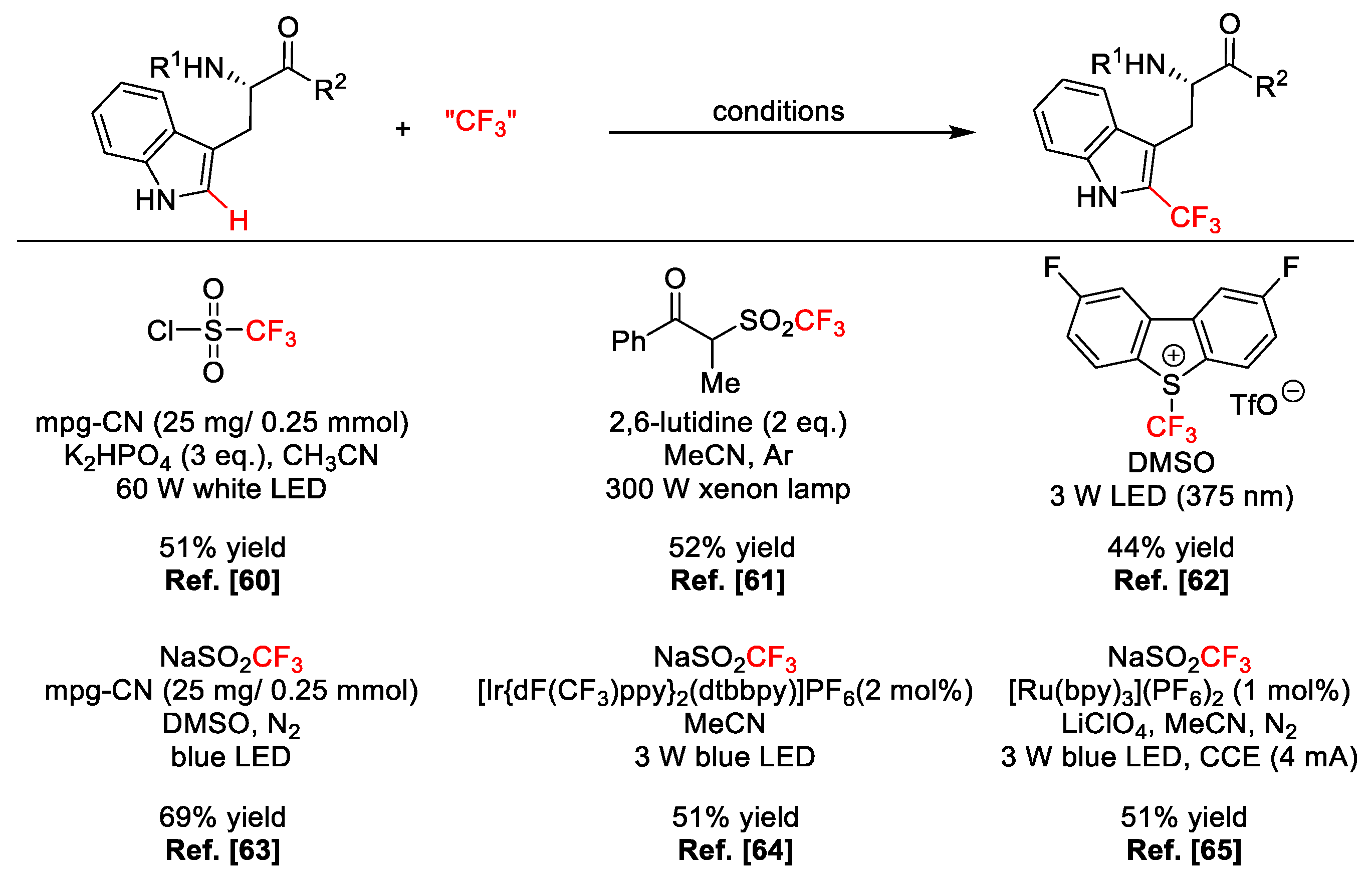
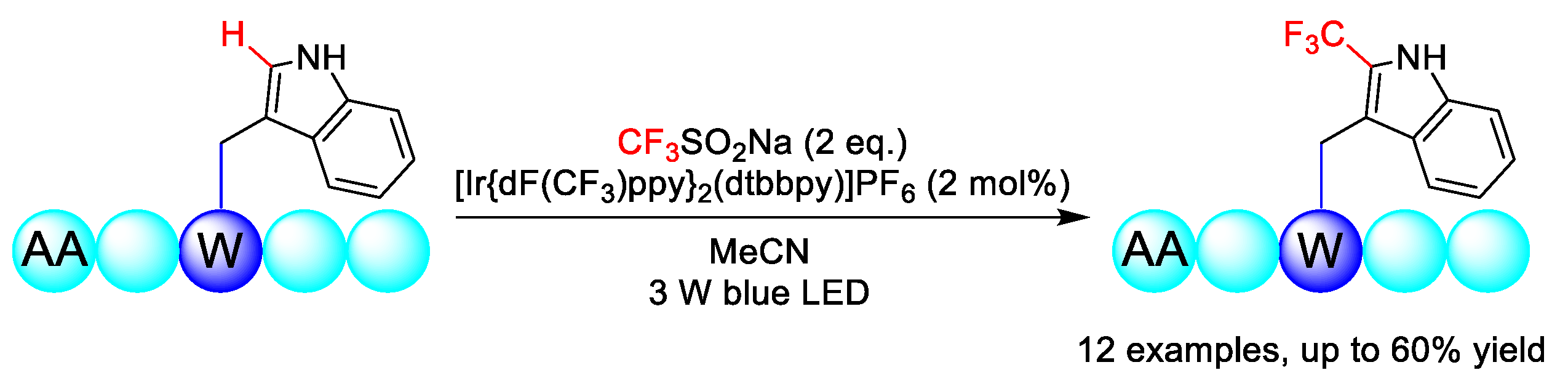
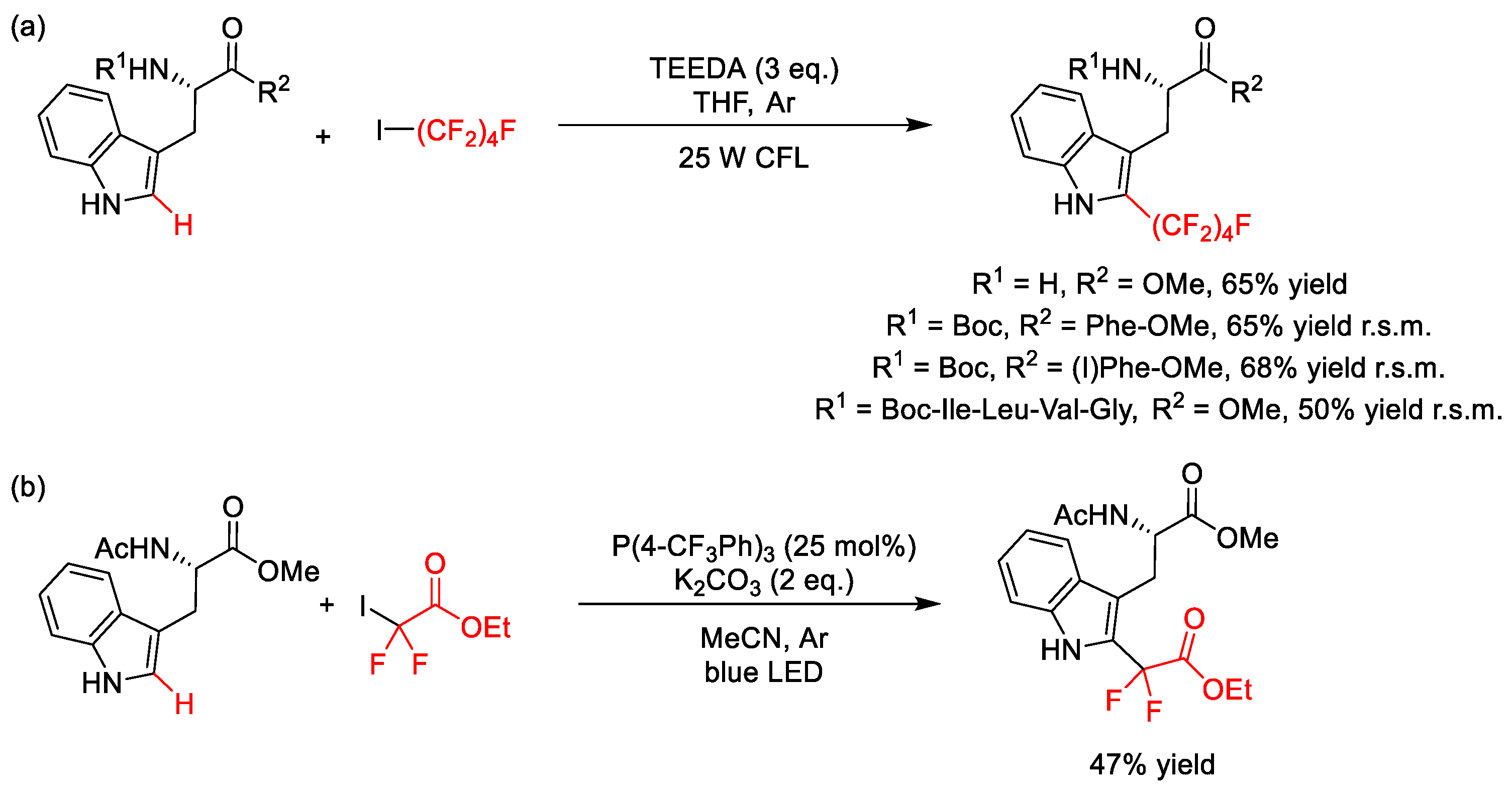
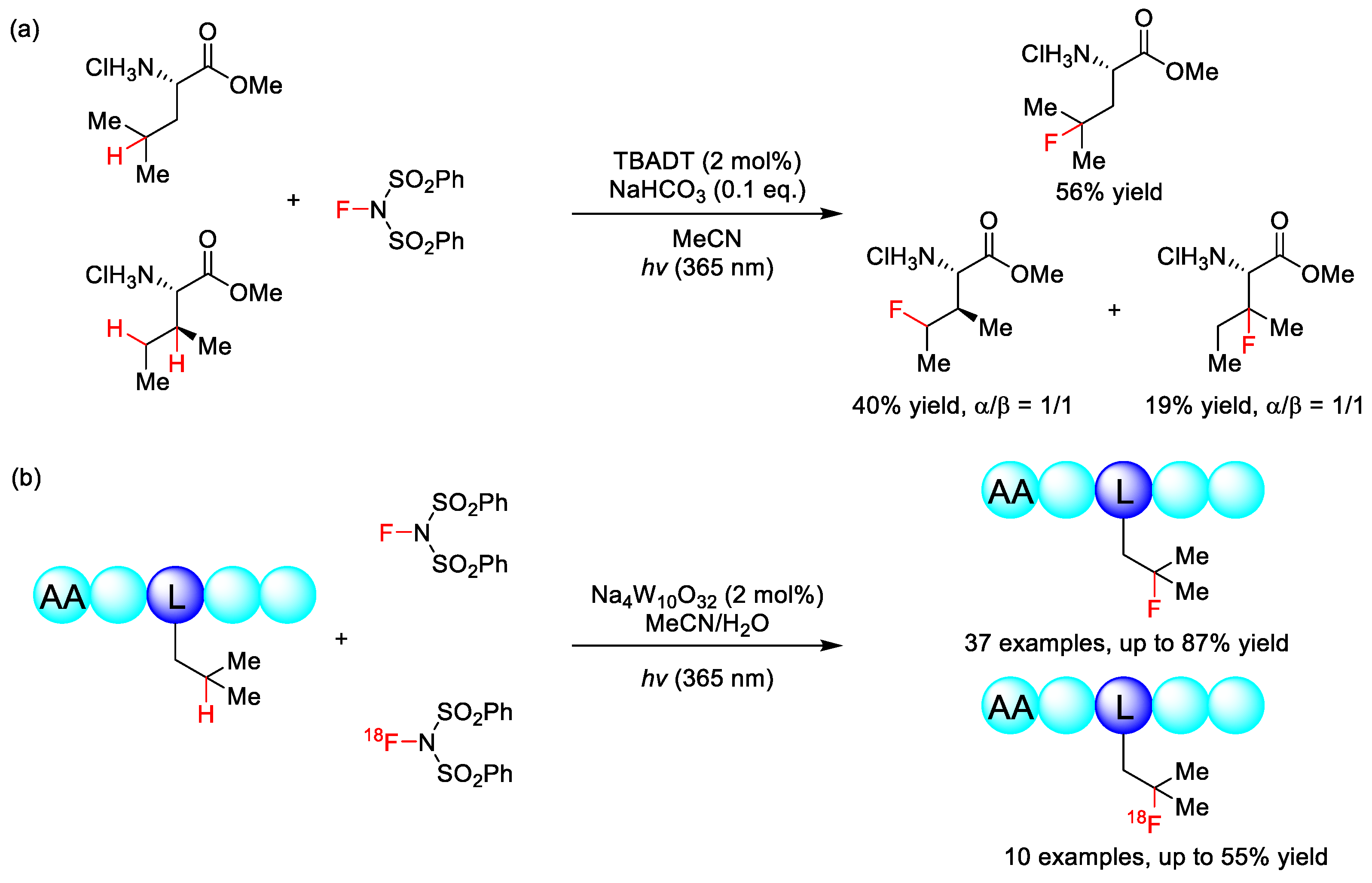
2.4. C–H Fluoroalkylation
2.5. Miscellaneous C–H Alkylation
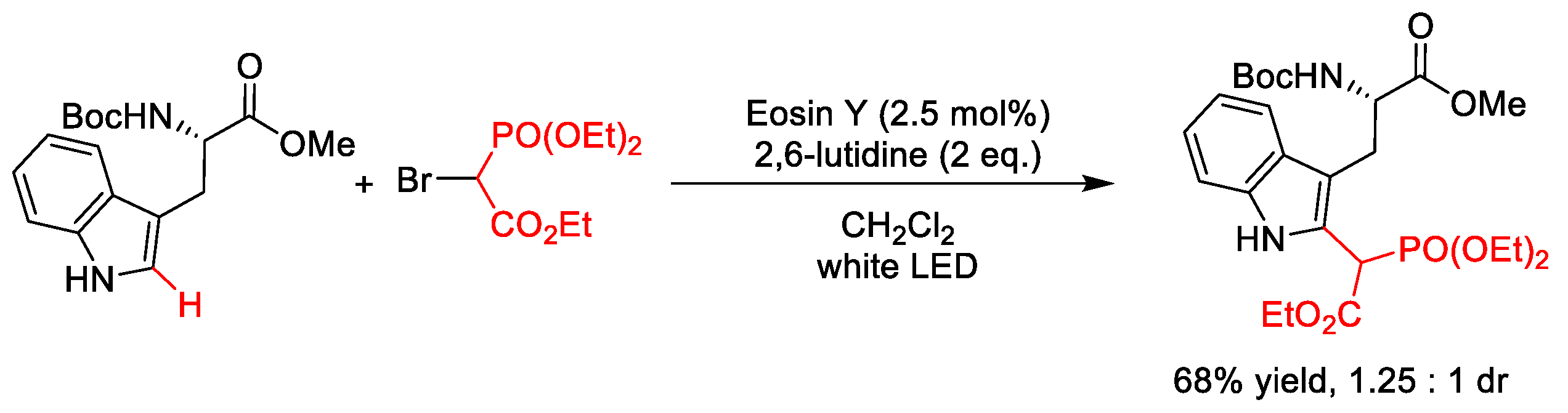
2.5.1. C(sp2)–Phosphonoacetylation
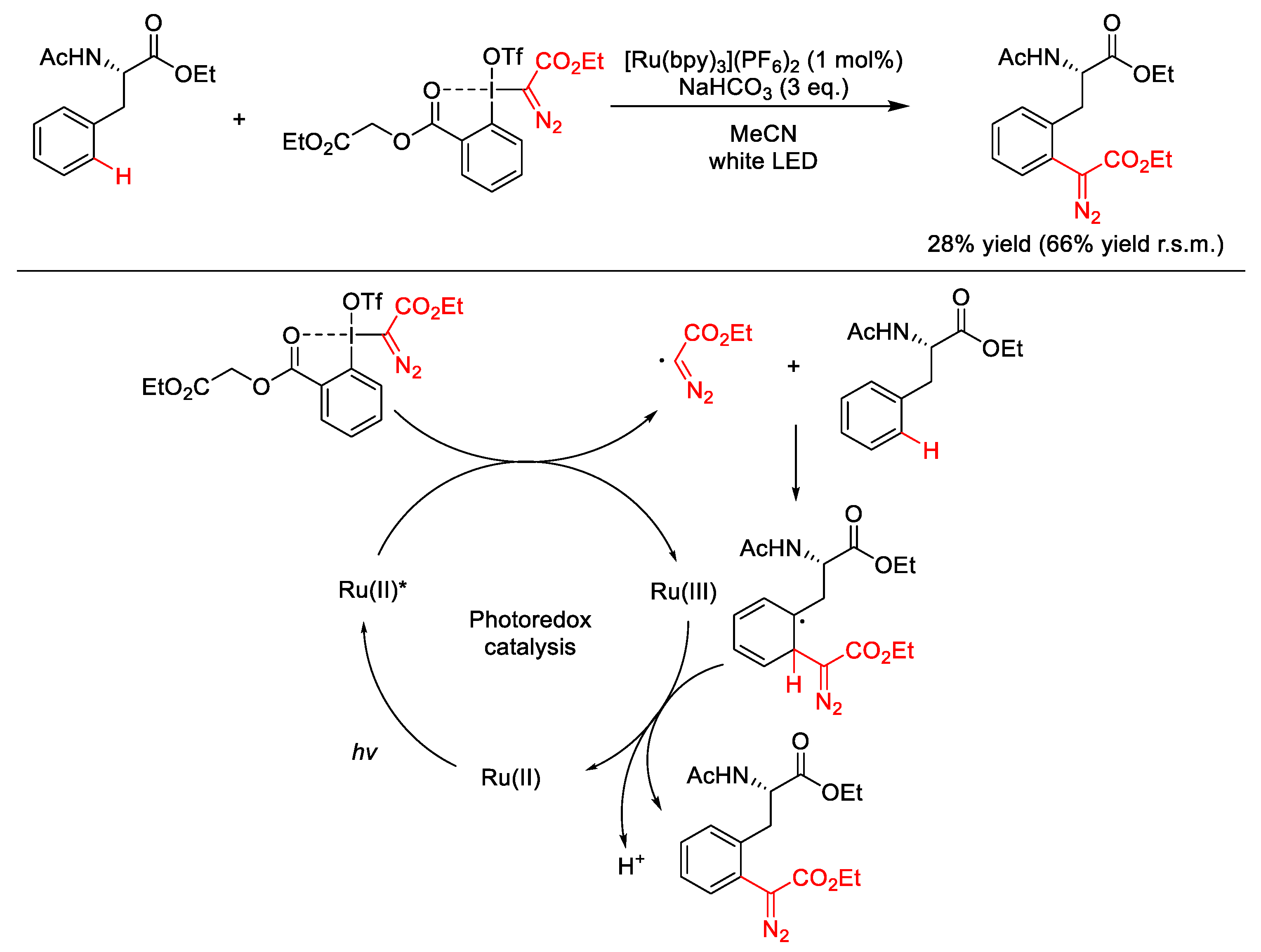
2.5.2. C(sp2)–Diazomethylation
2.6. C–H Alkenylation
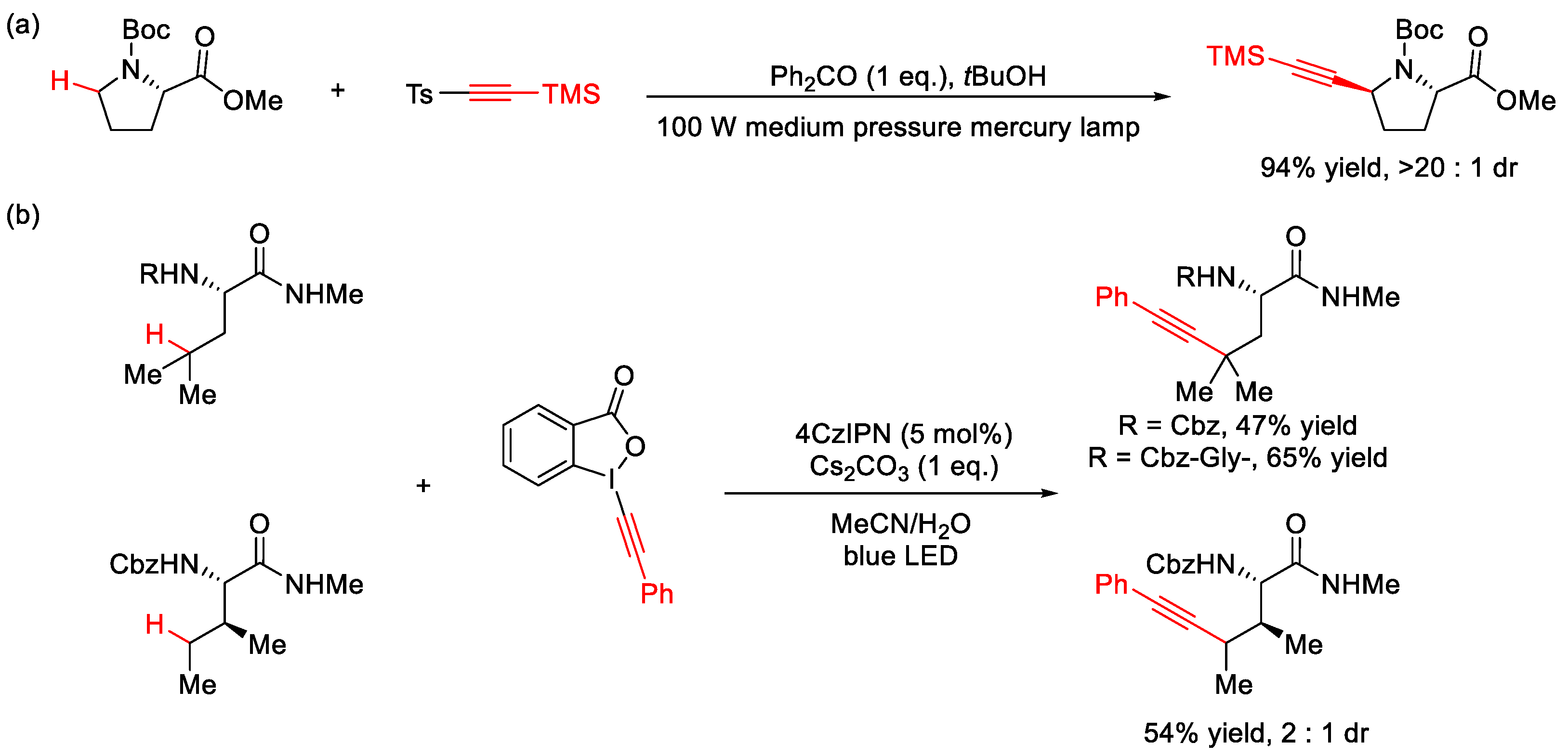
2.7. C–H Alkynylation
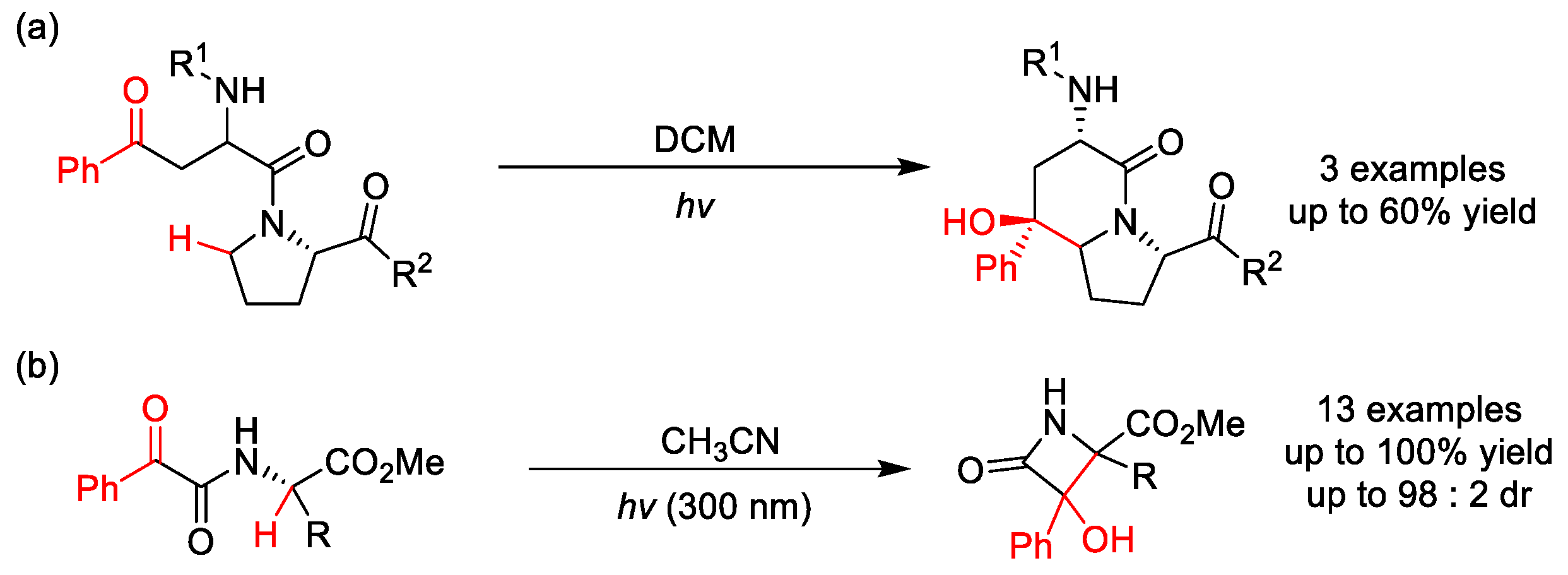
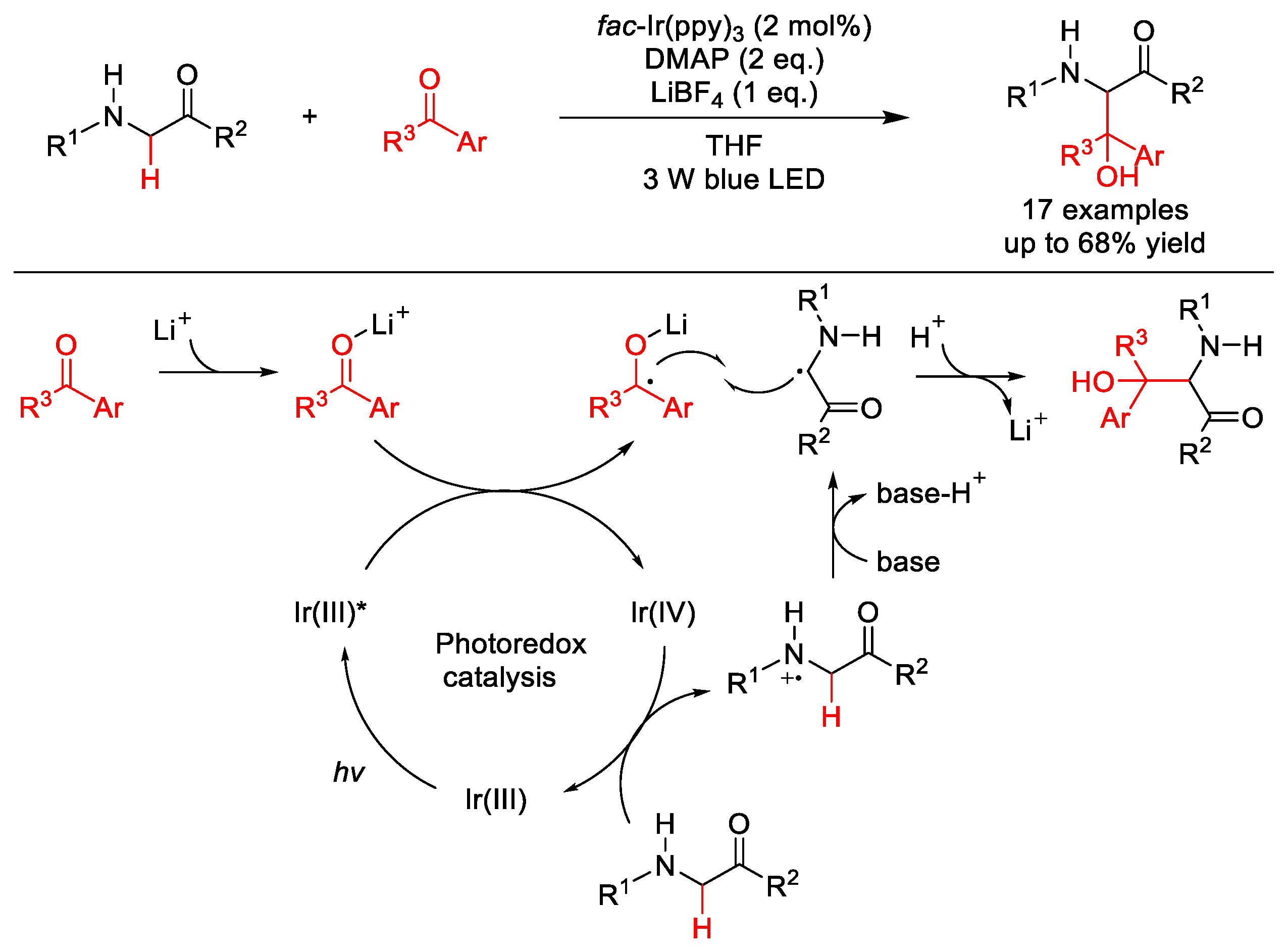
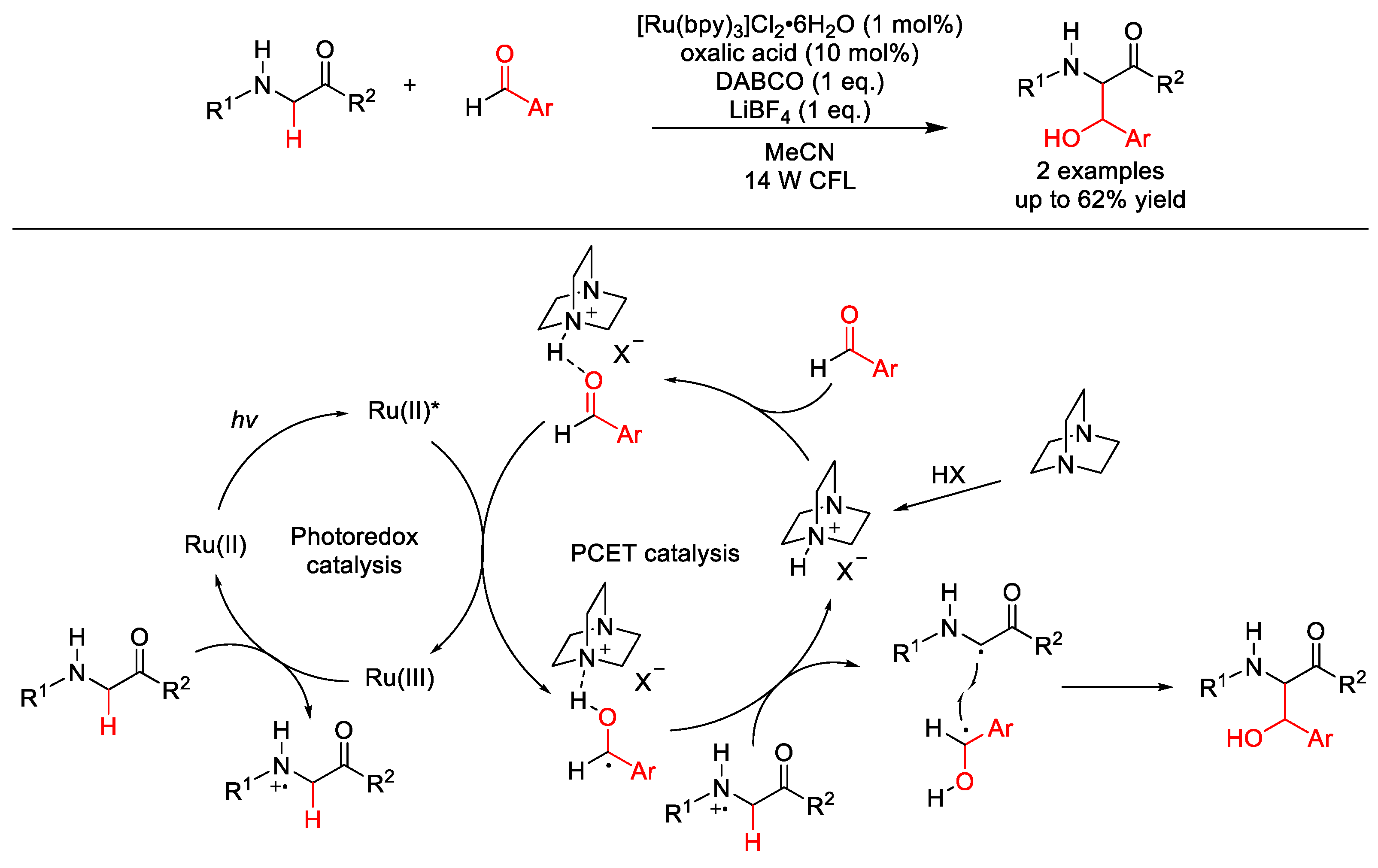
2.8. C–H Acylation
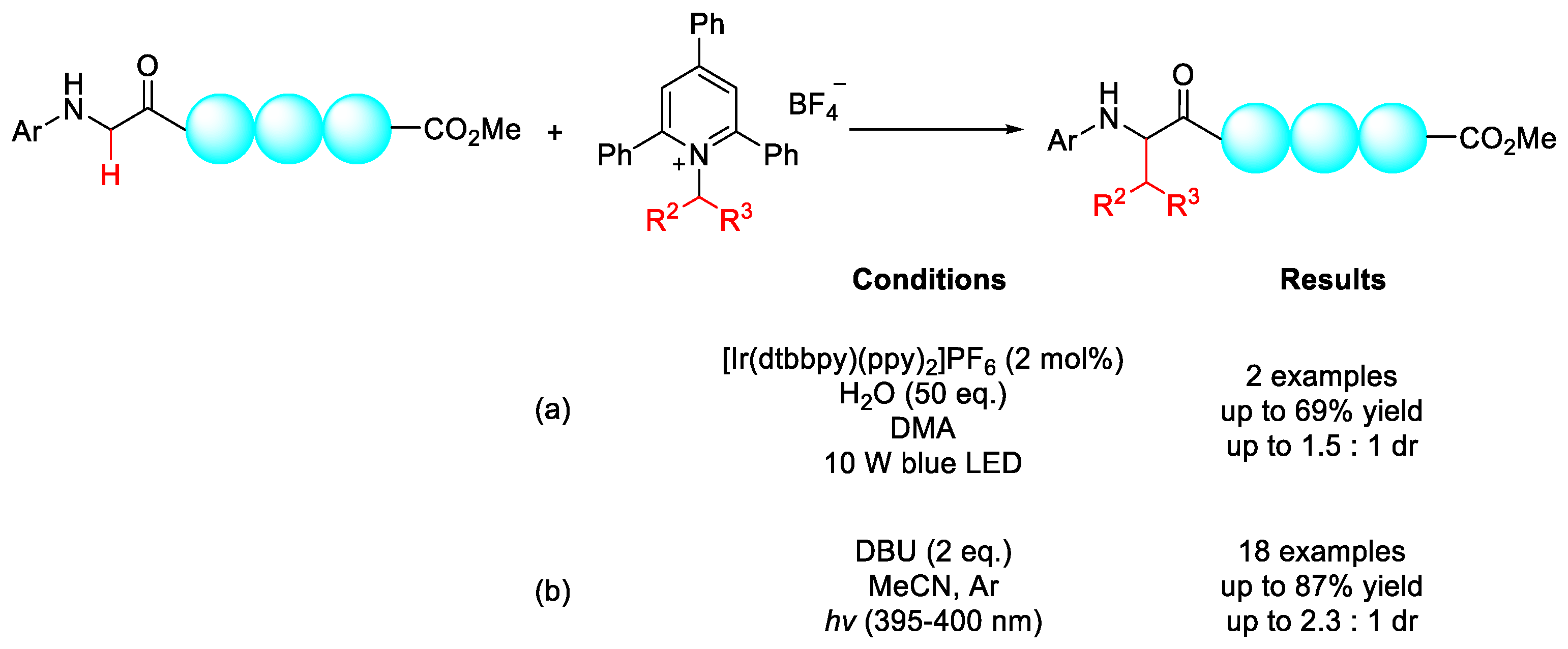
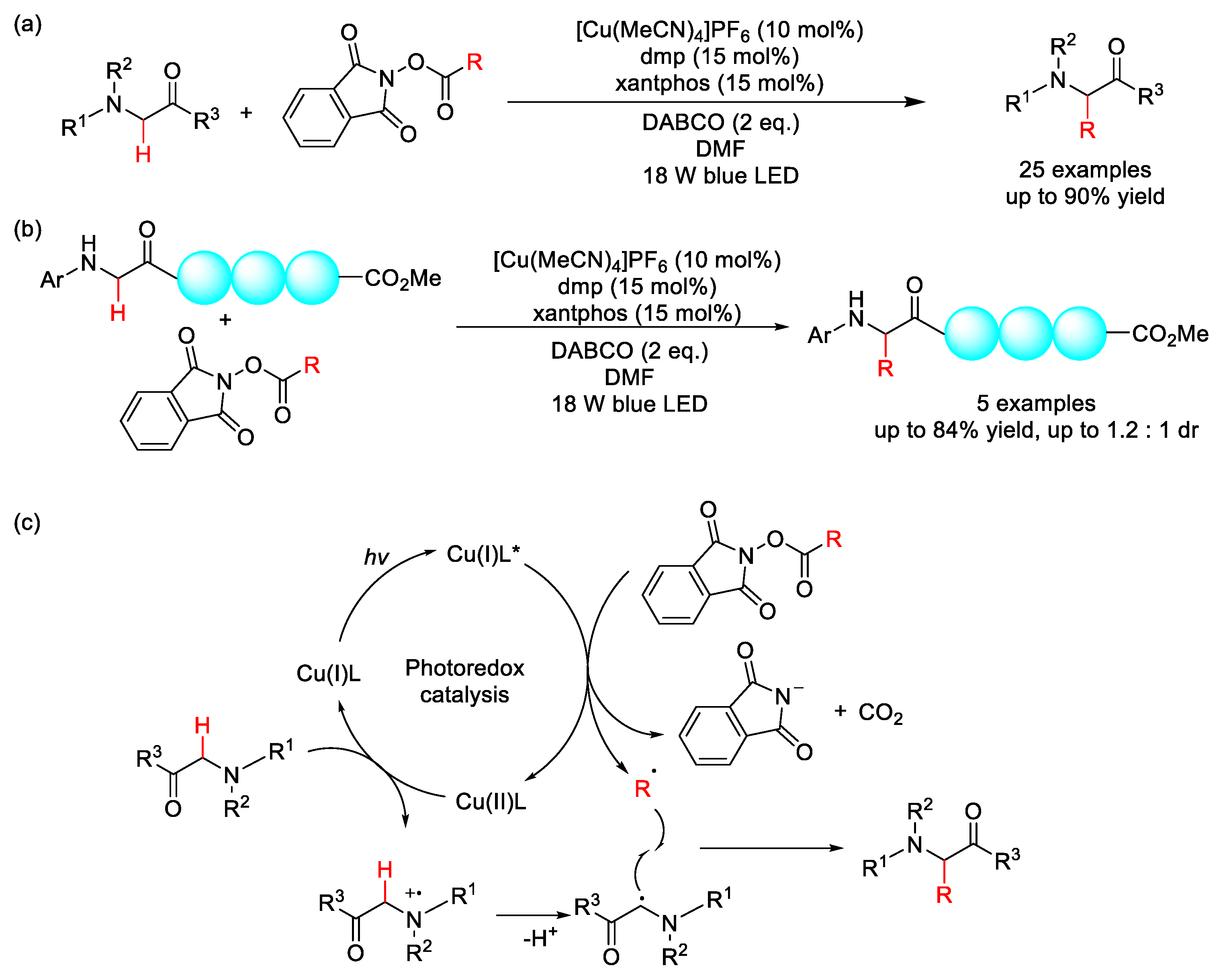
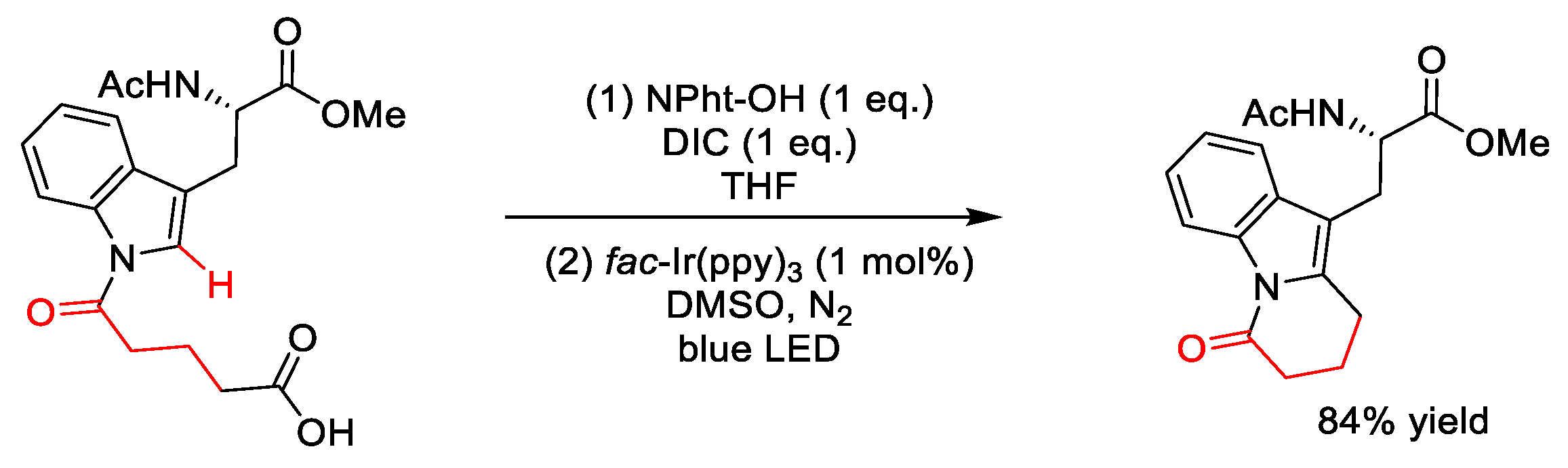
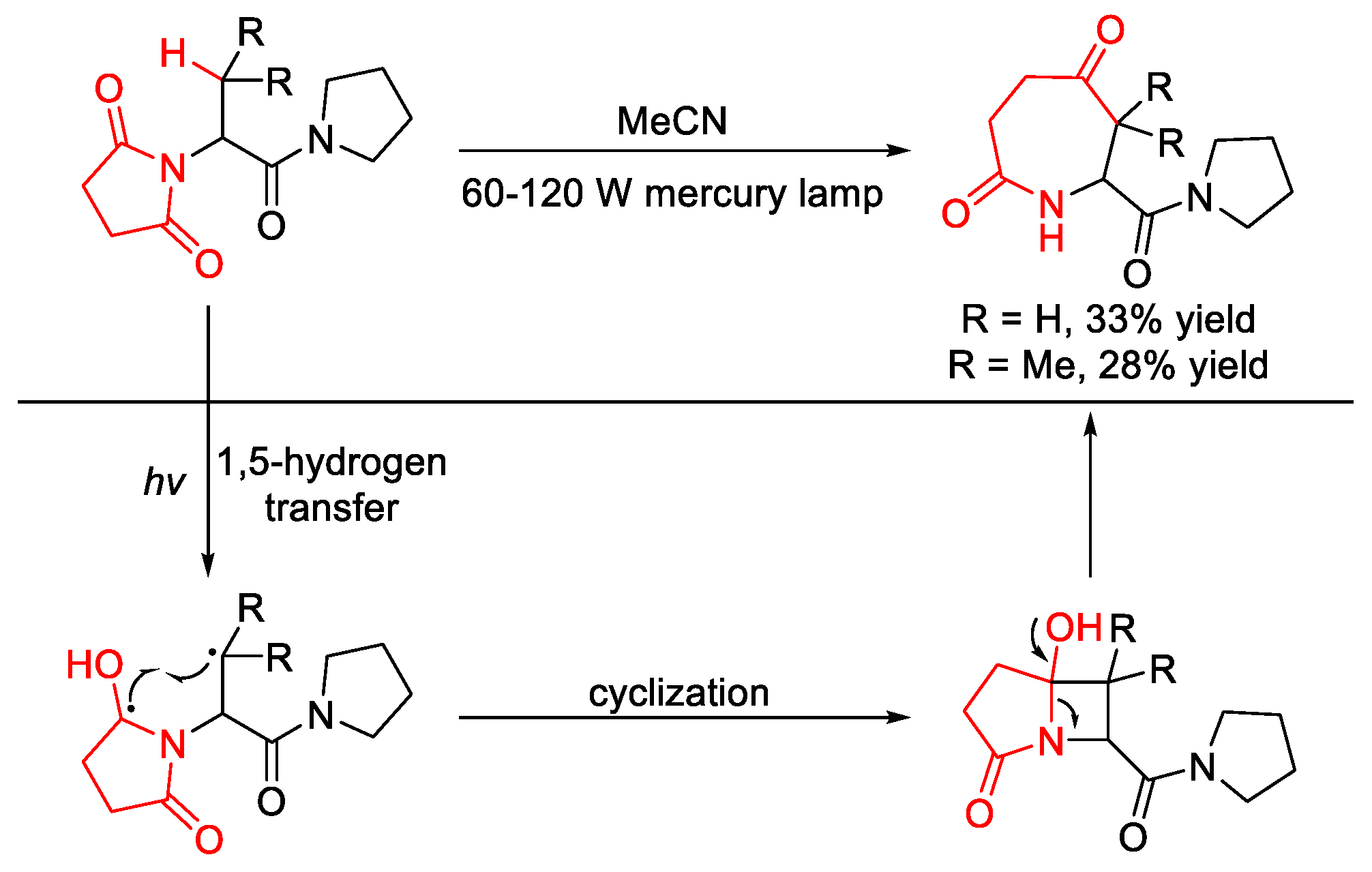
2.8.1. C(sp3)–H Acylation
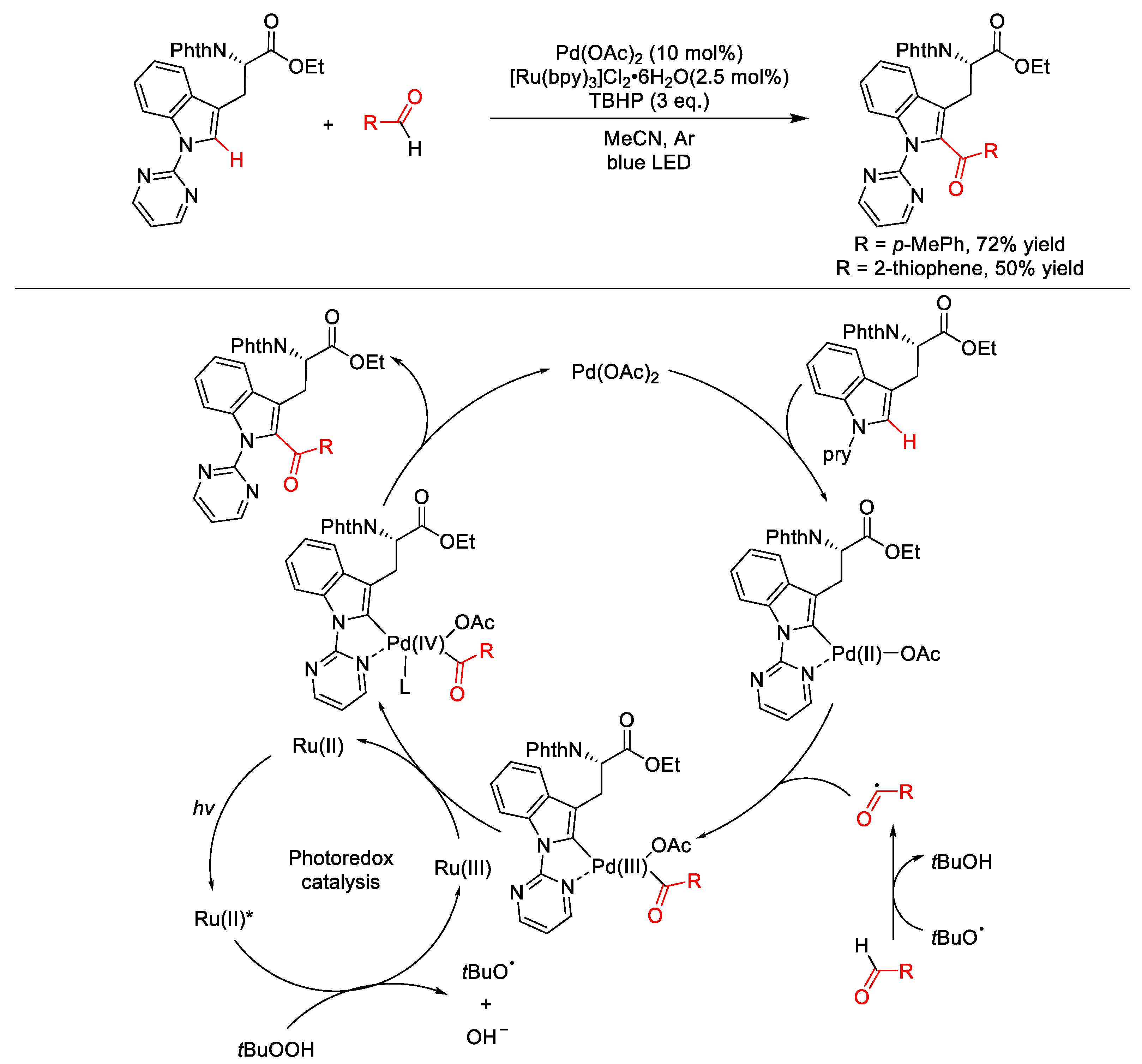
2.8.2. C(sp2)–H Acylation
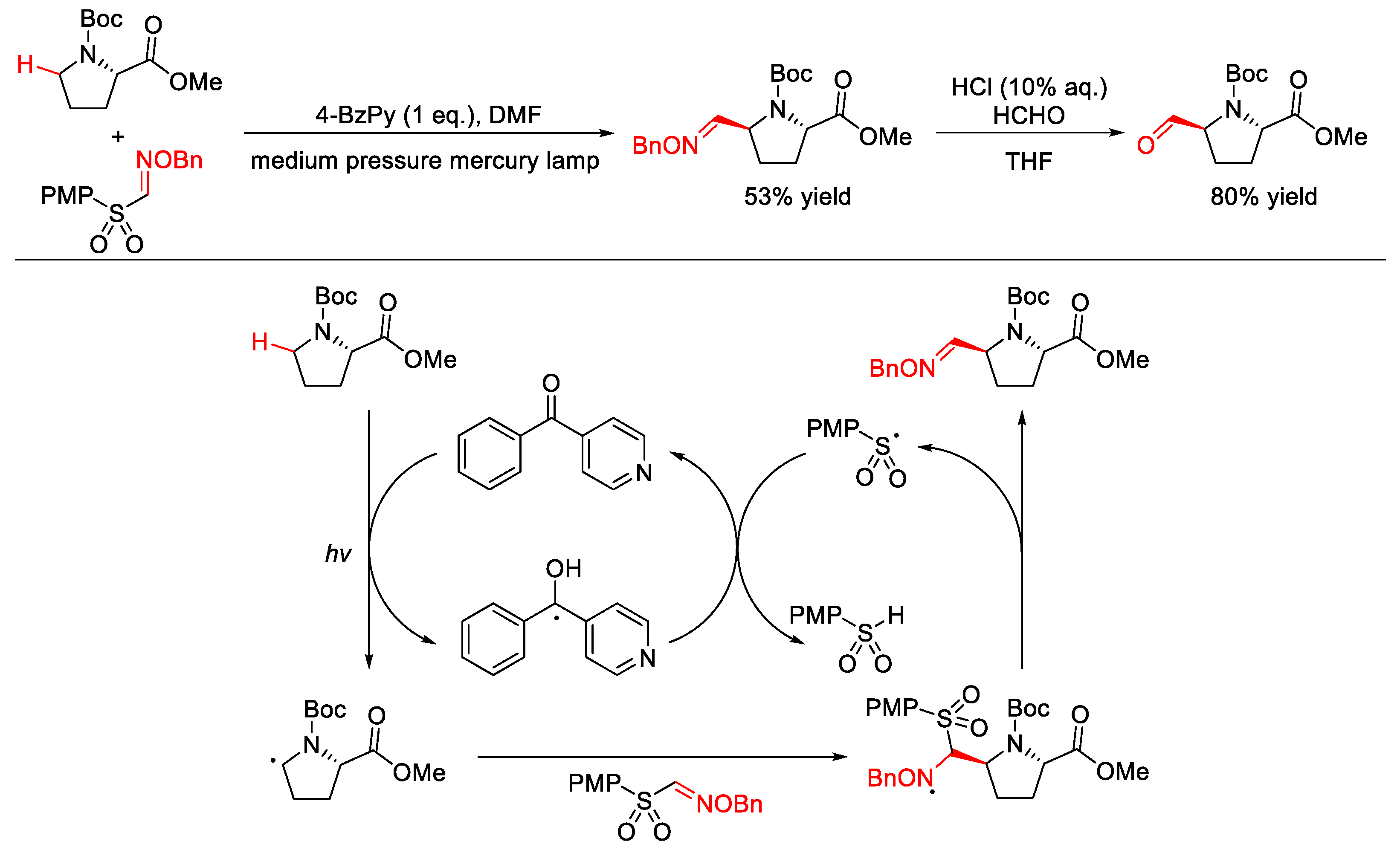
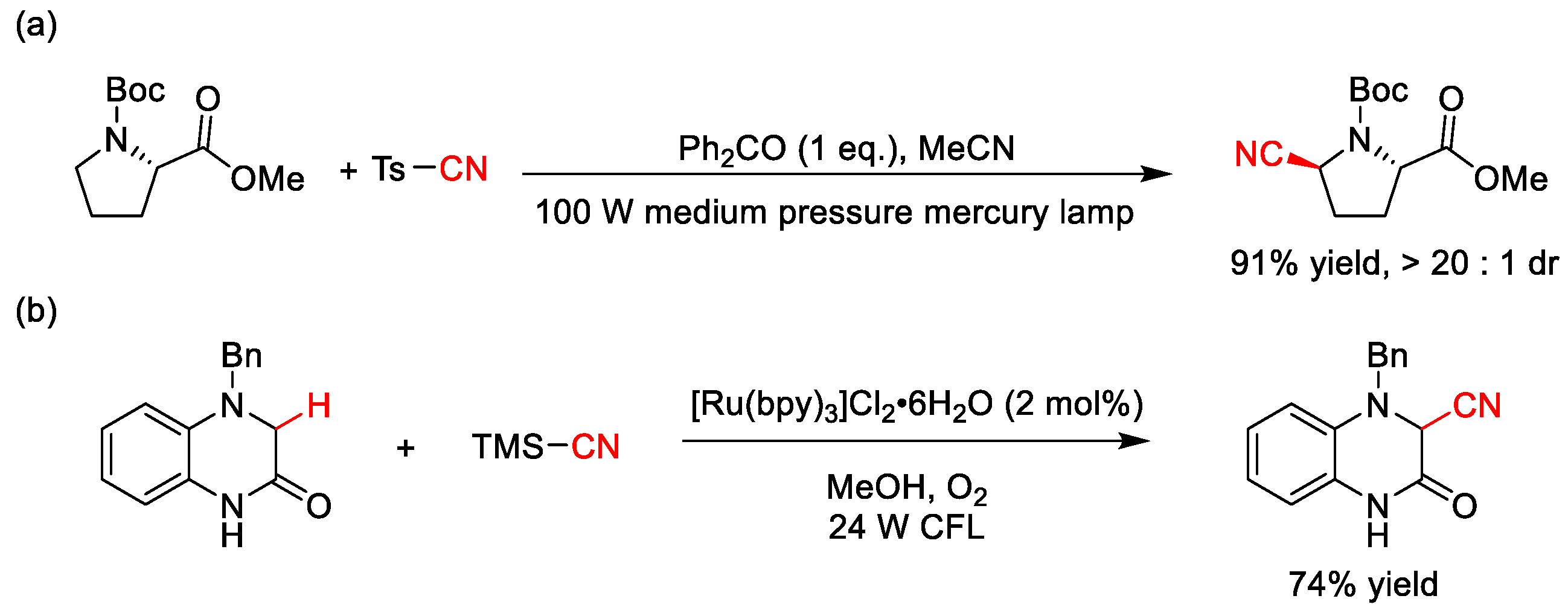
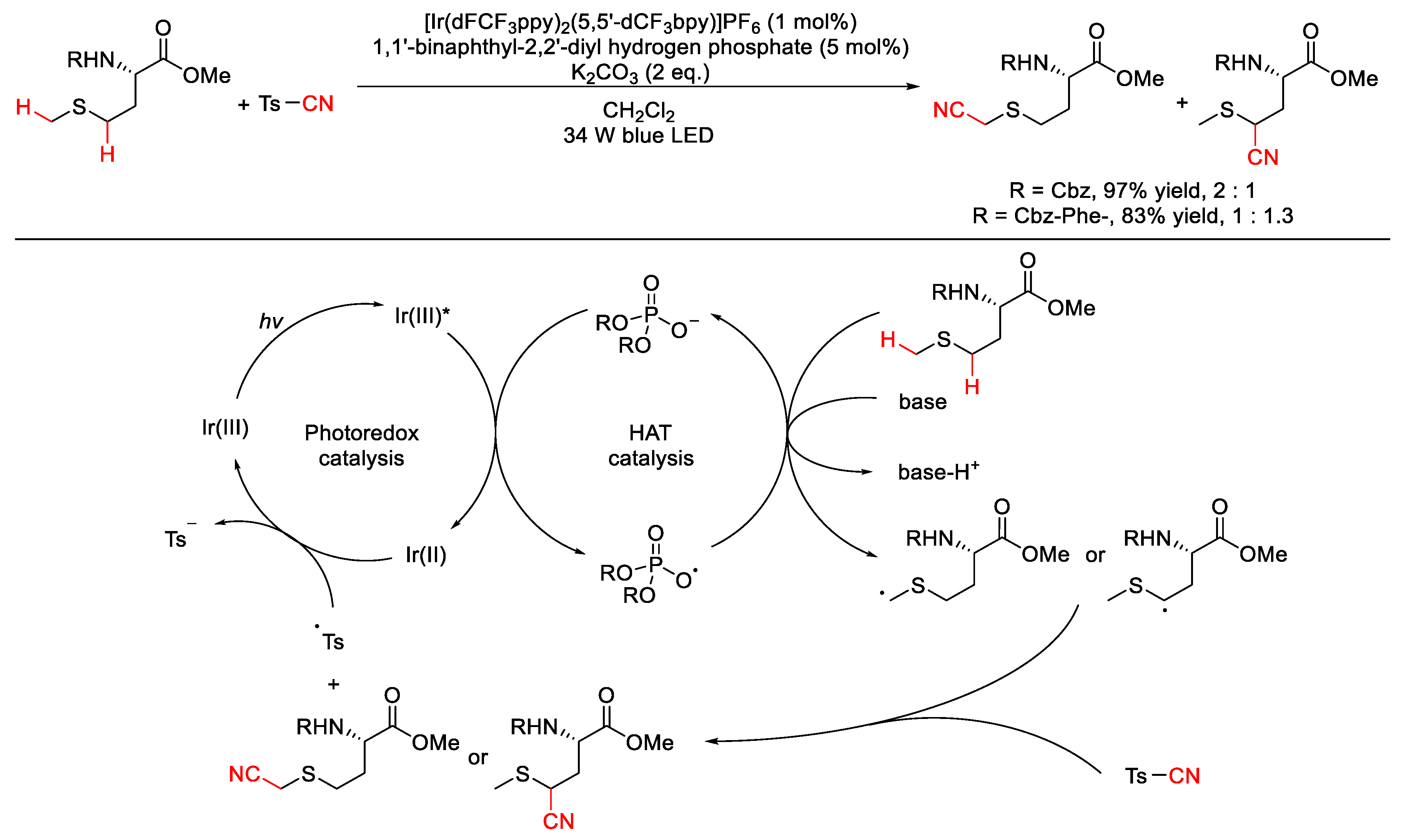
2.9. C–H Cyanation
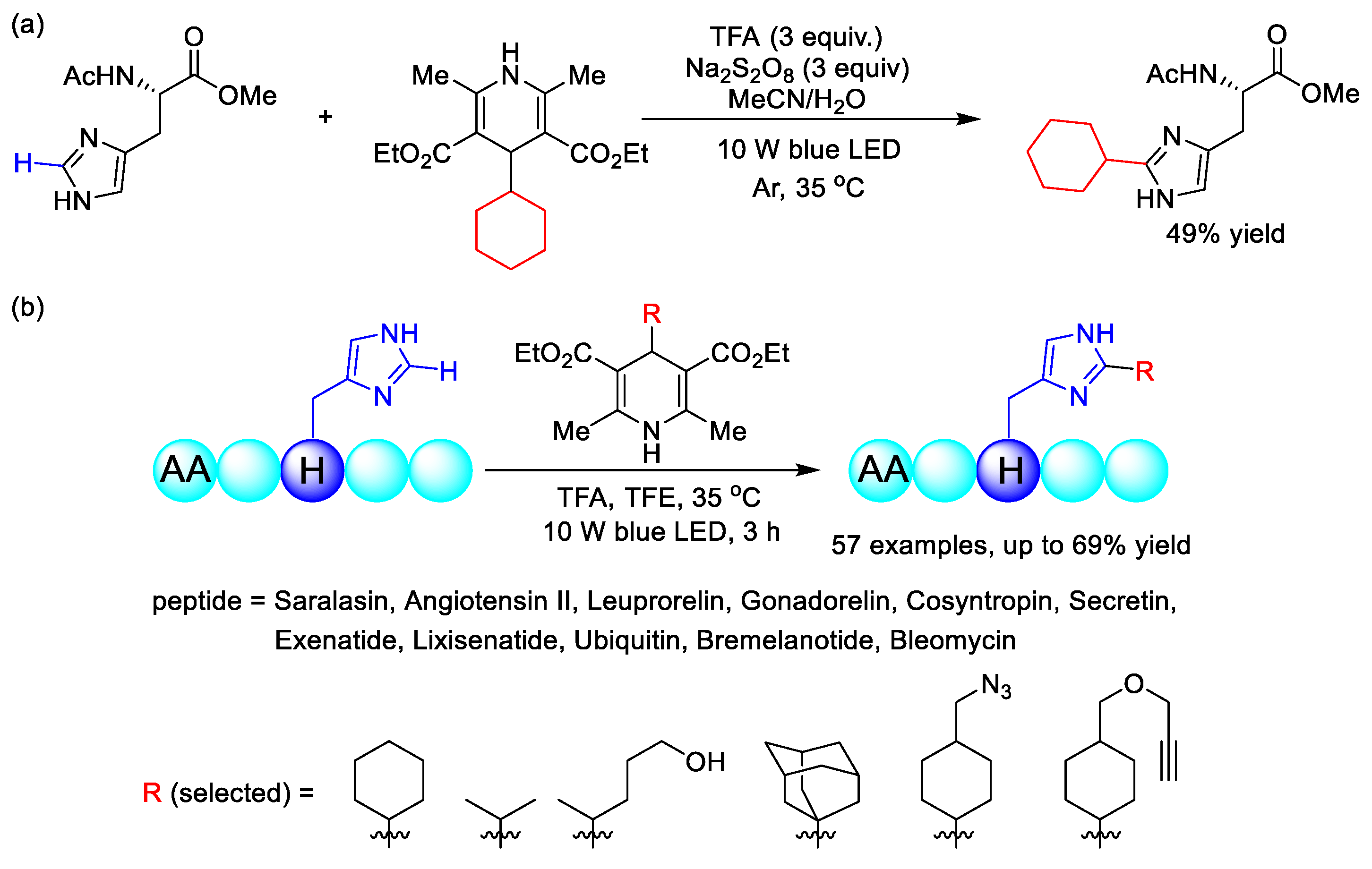
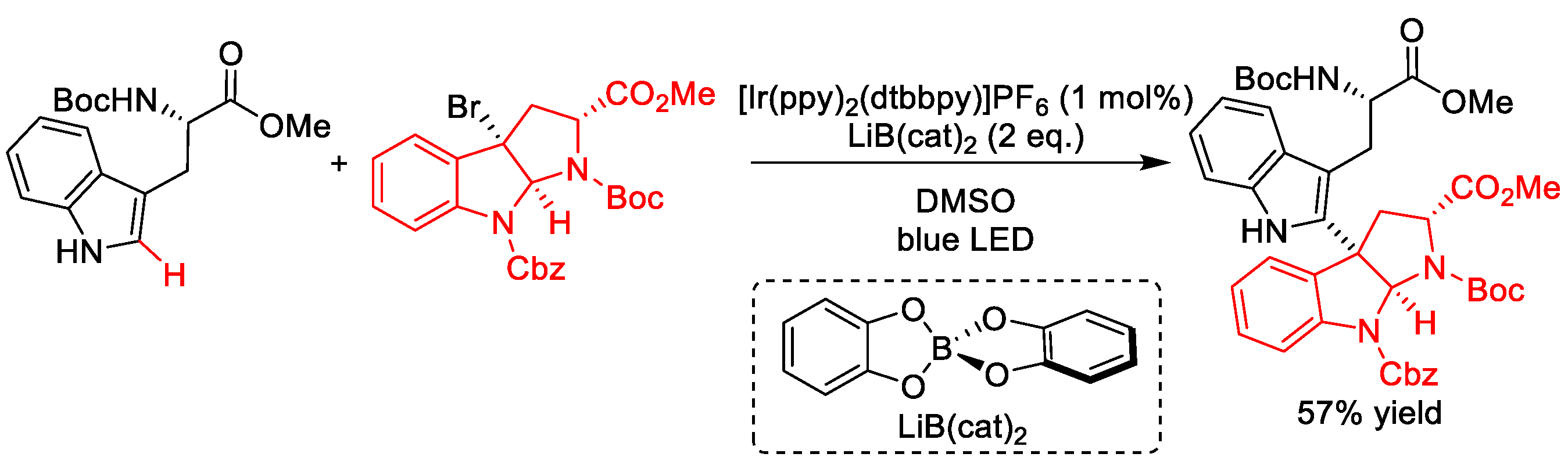
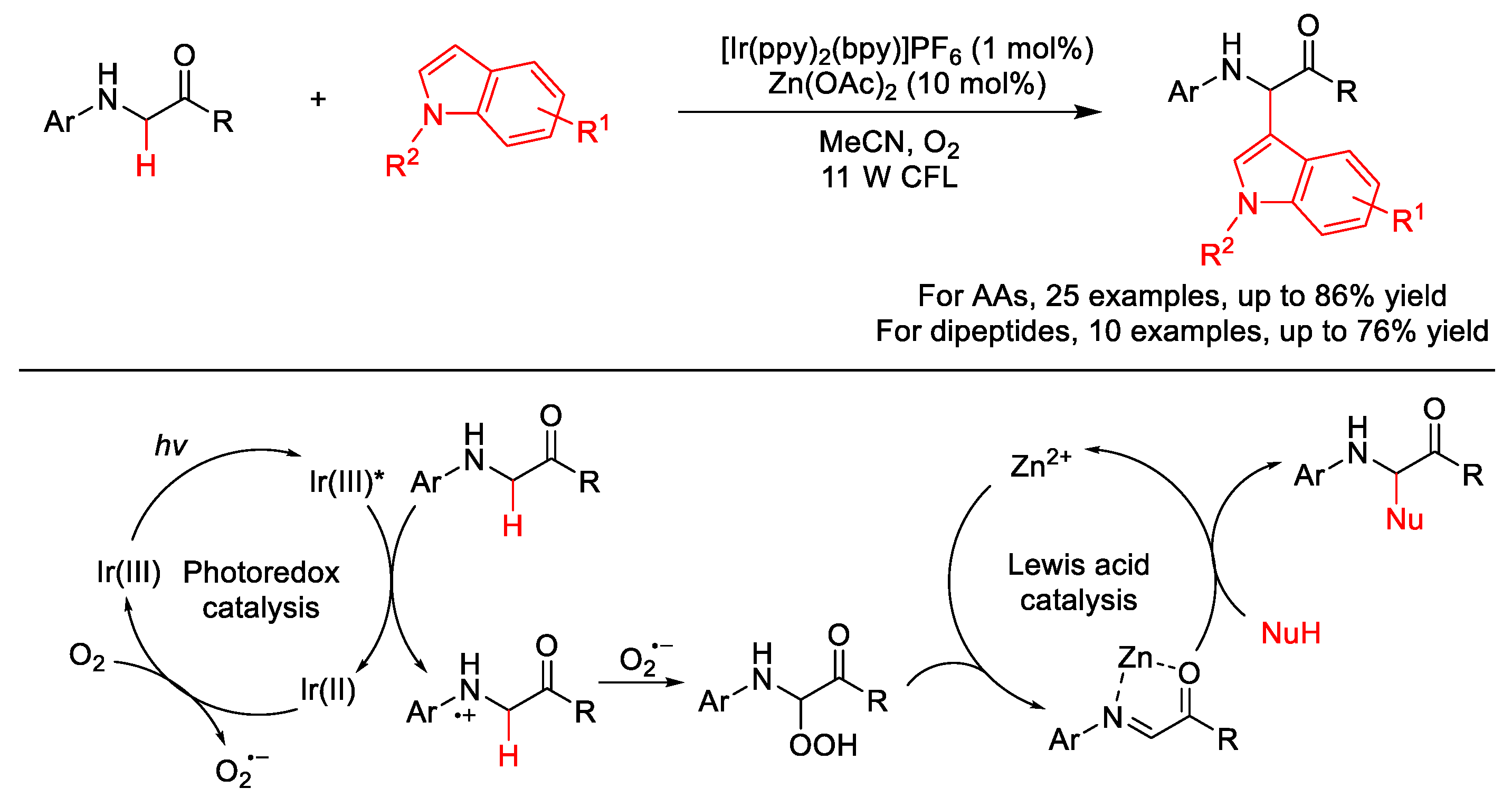
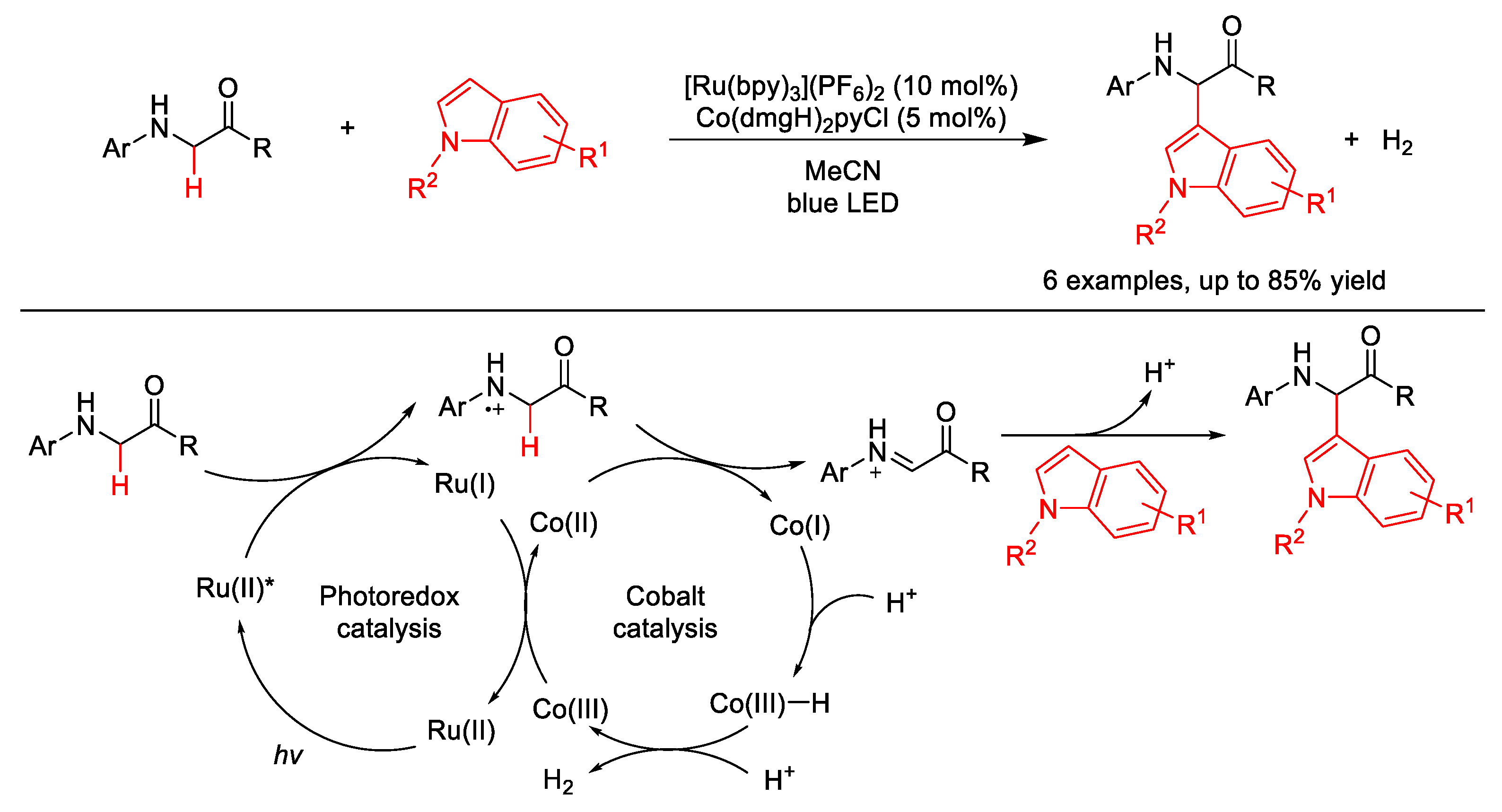
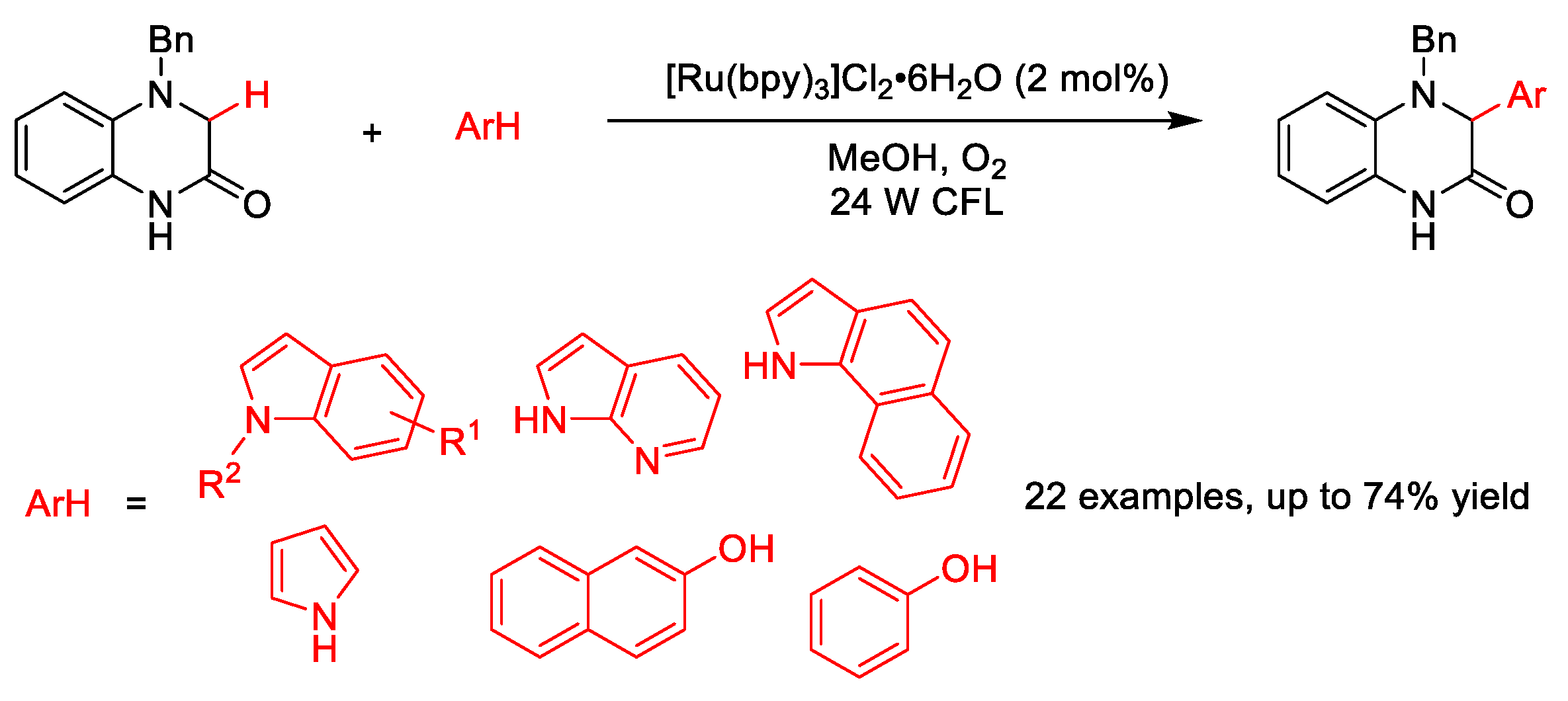
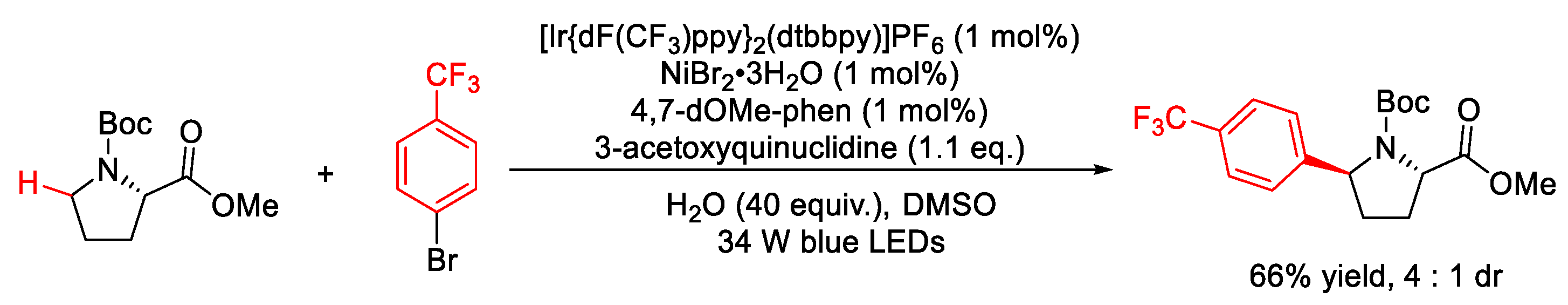
2.10. C–H (Hetero)arylation
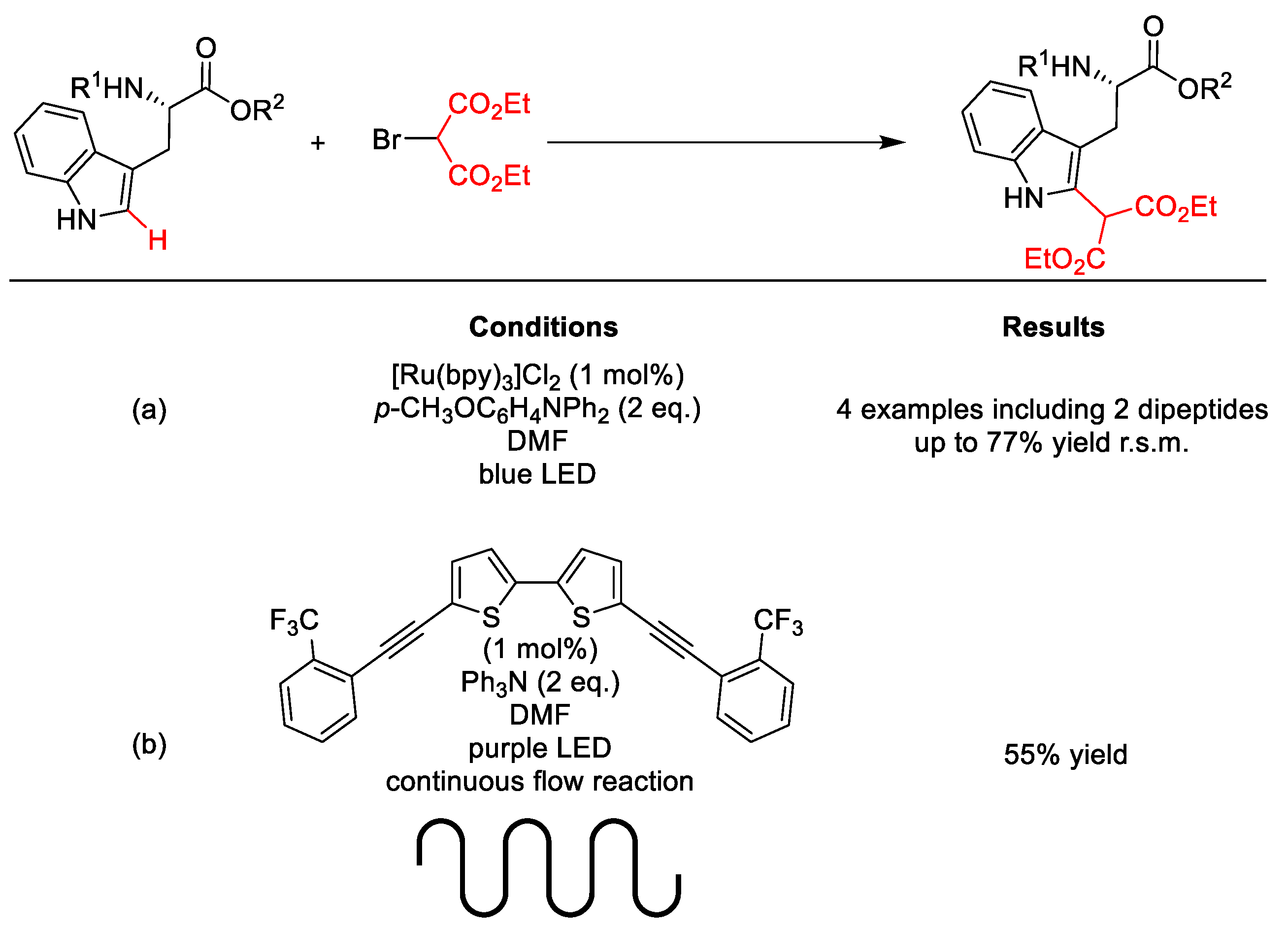
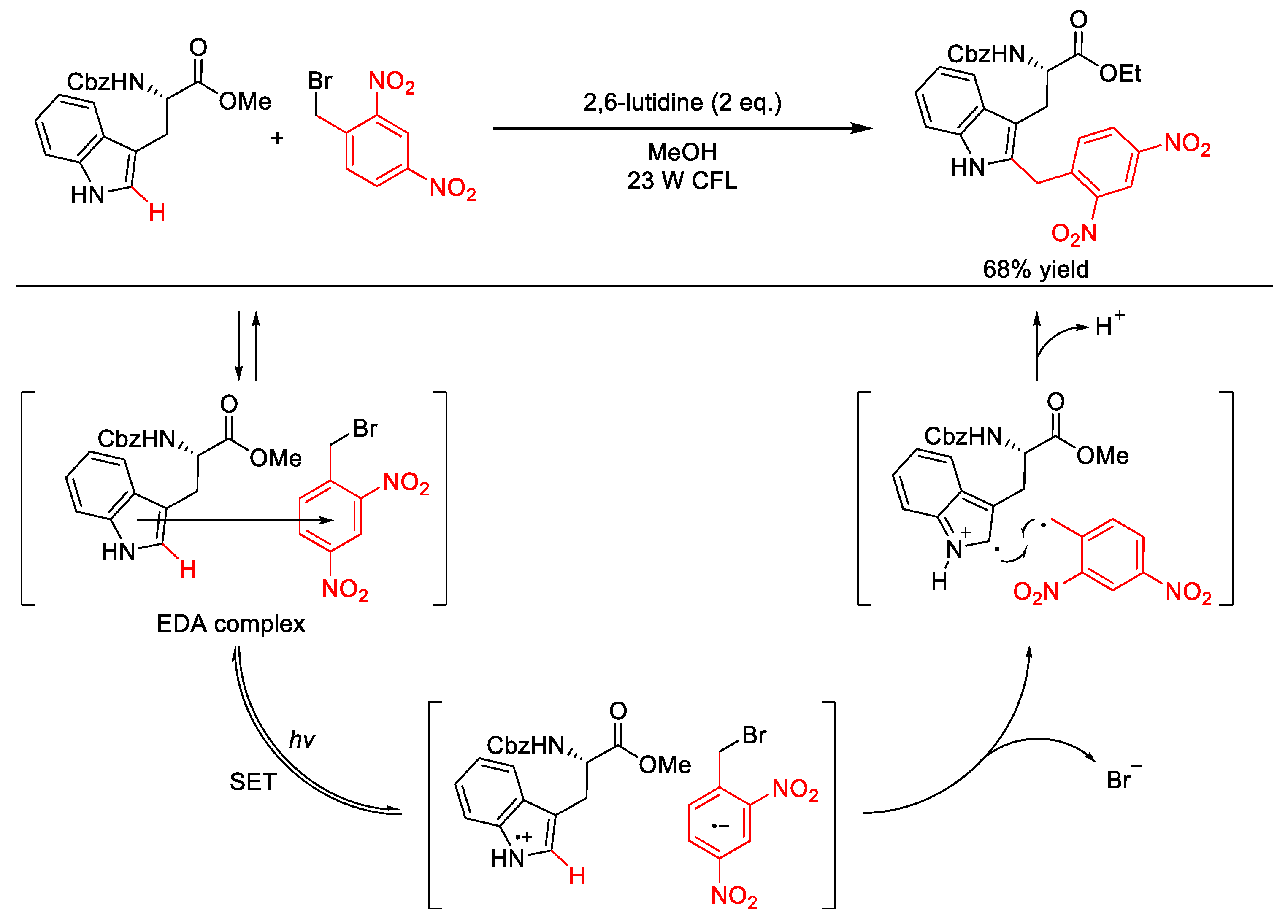
2.10.1. C(sp3)–H Arylation
2.10.2. C(sp2)–H Heteroarylation
3. C–N Formation
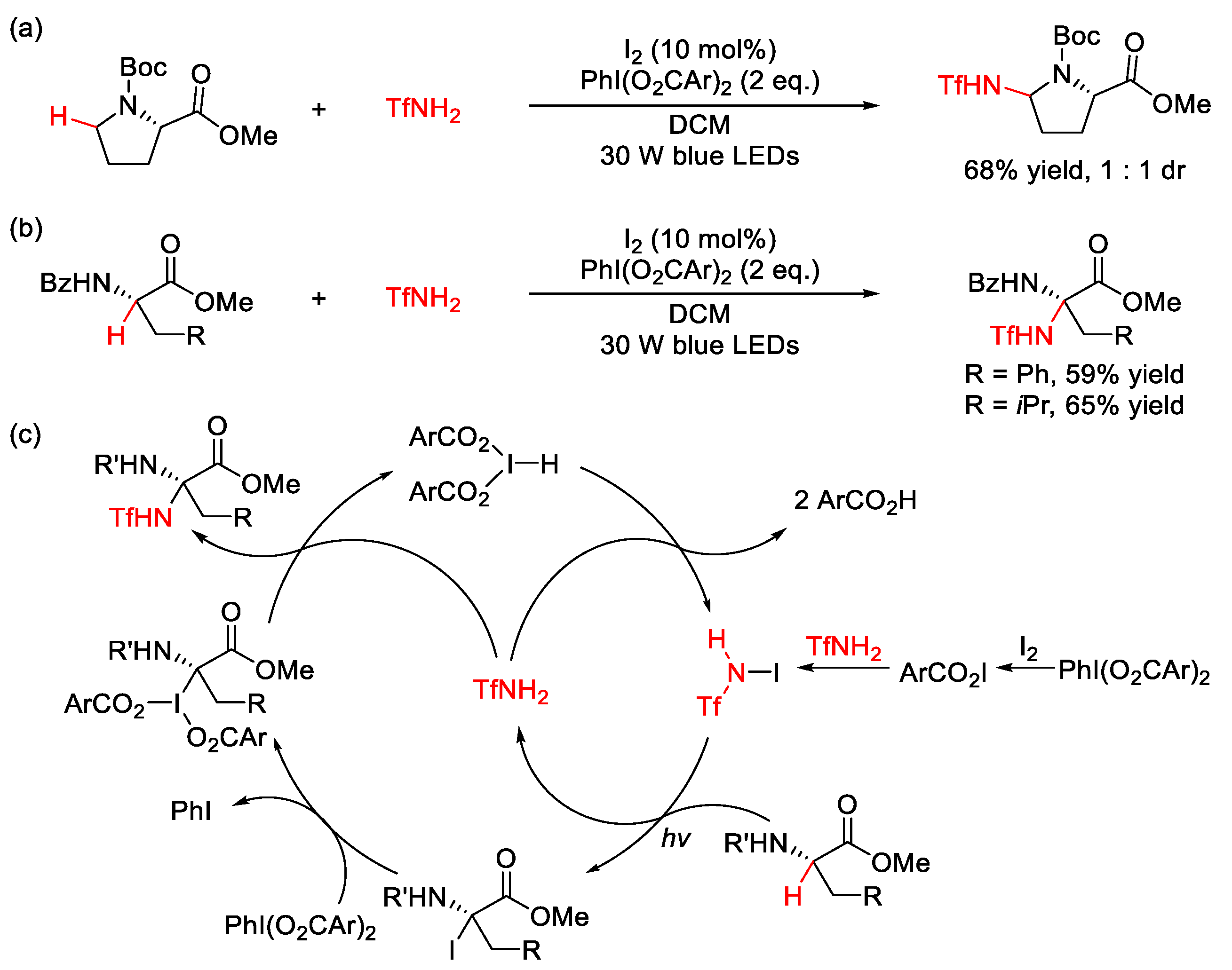
3.1. C(sp3)–N Formation
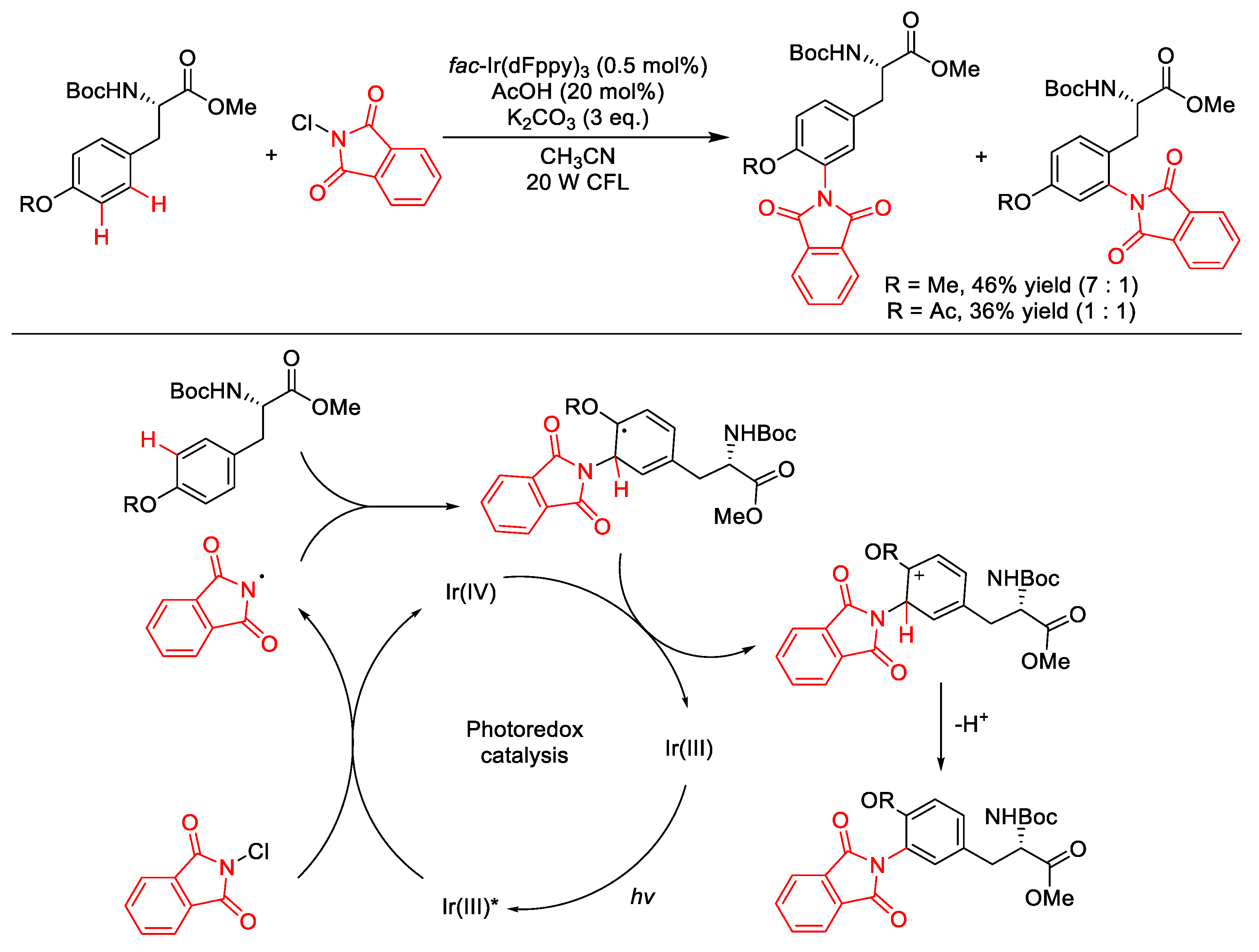
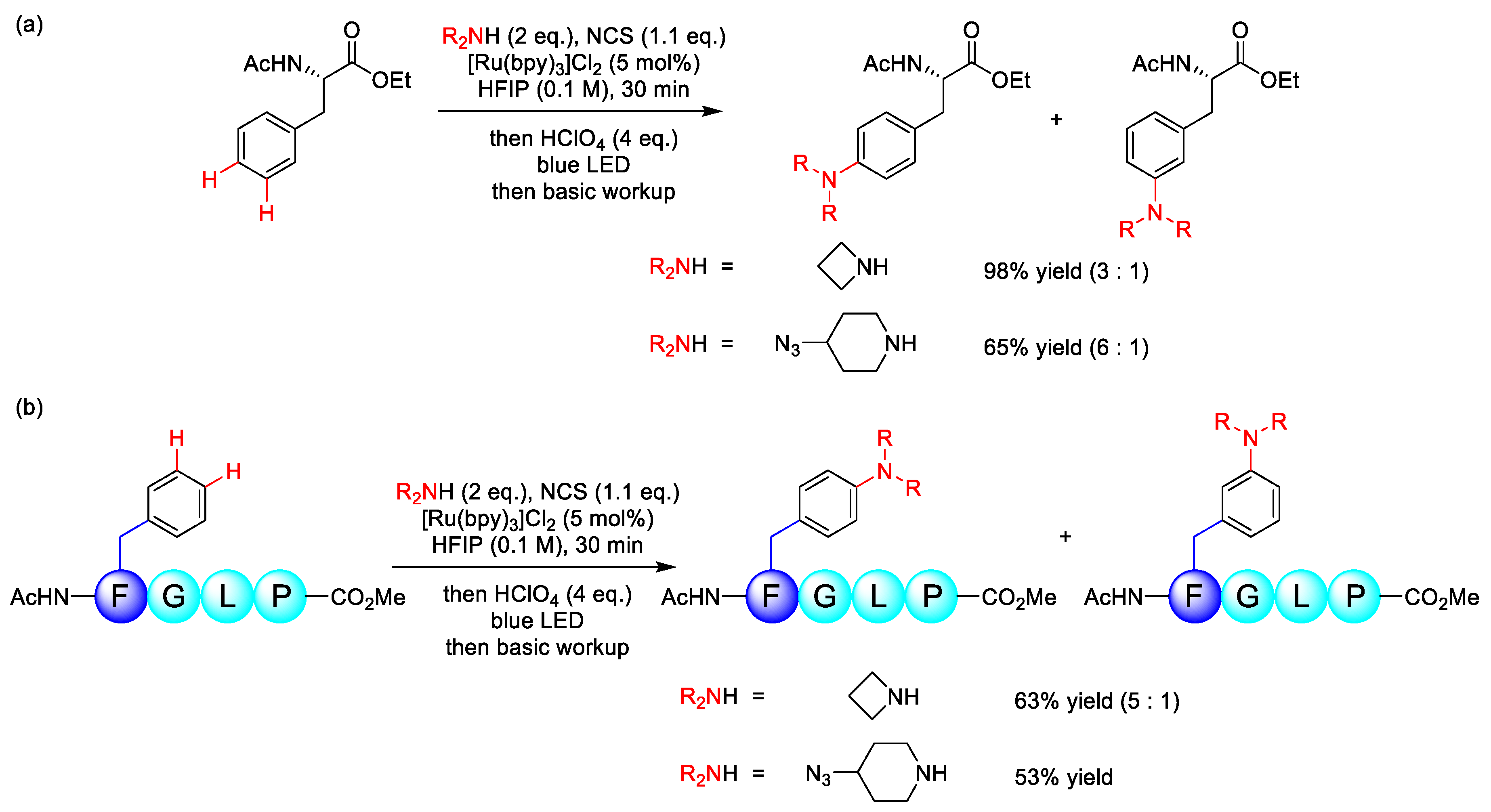
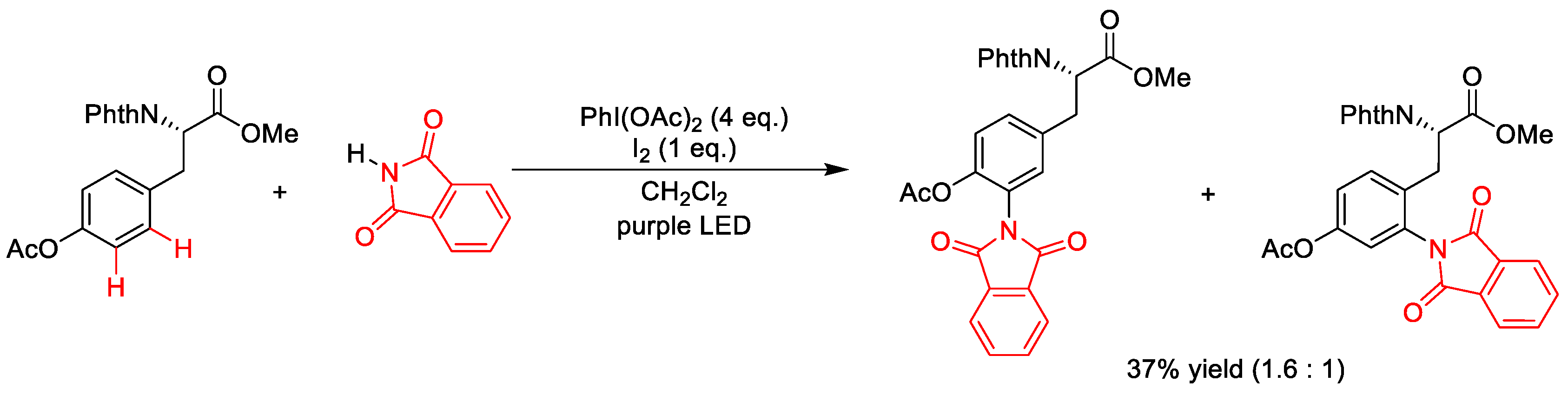
3.2. C(sp2)–N Formation
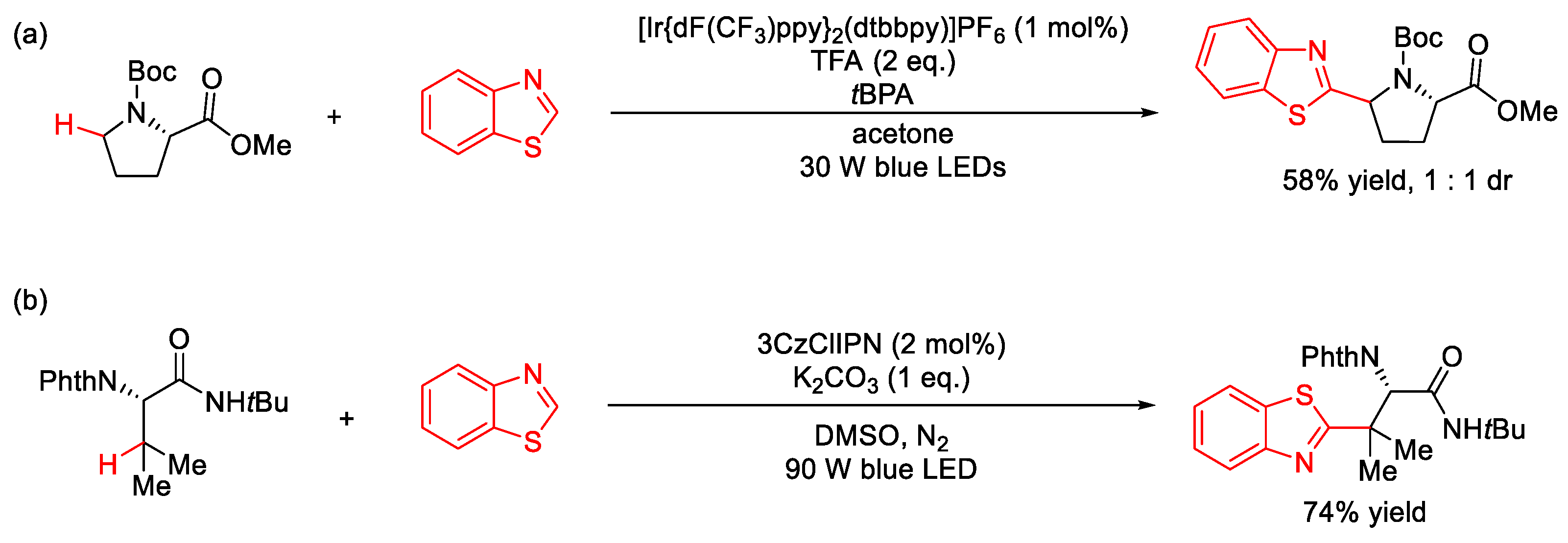
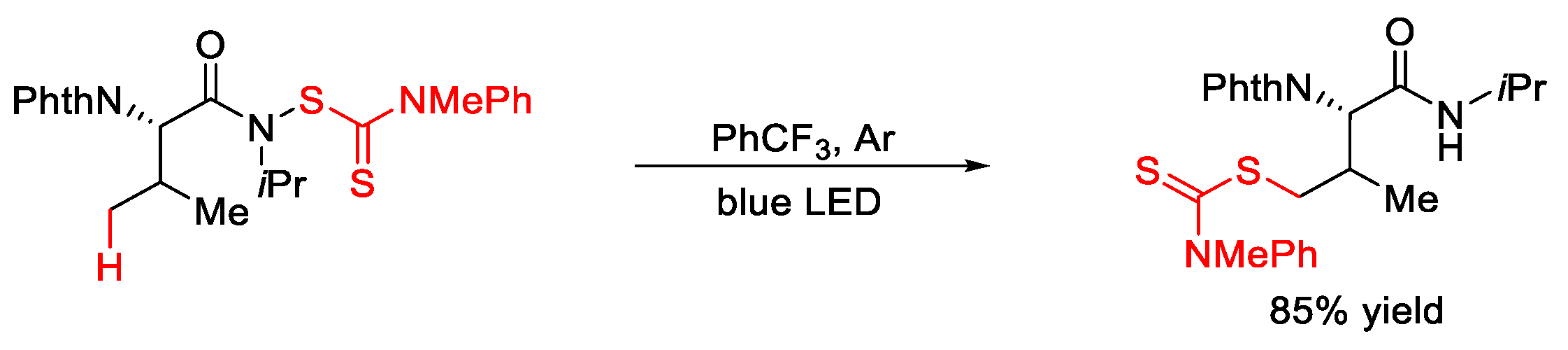
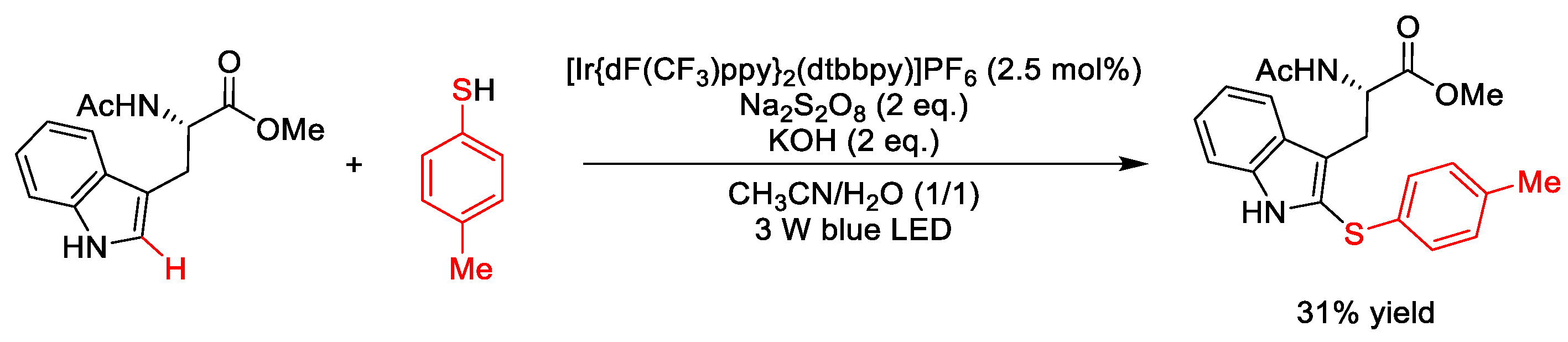
4. C–S Formation
4.1. C(sp3)–S Formation
4.2. C(sp2)–S Formation
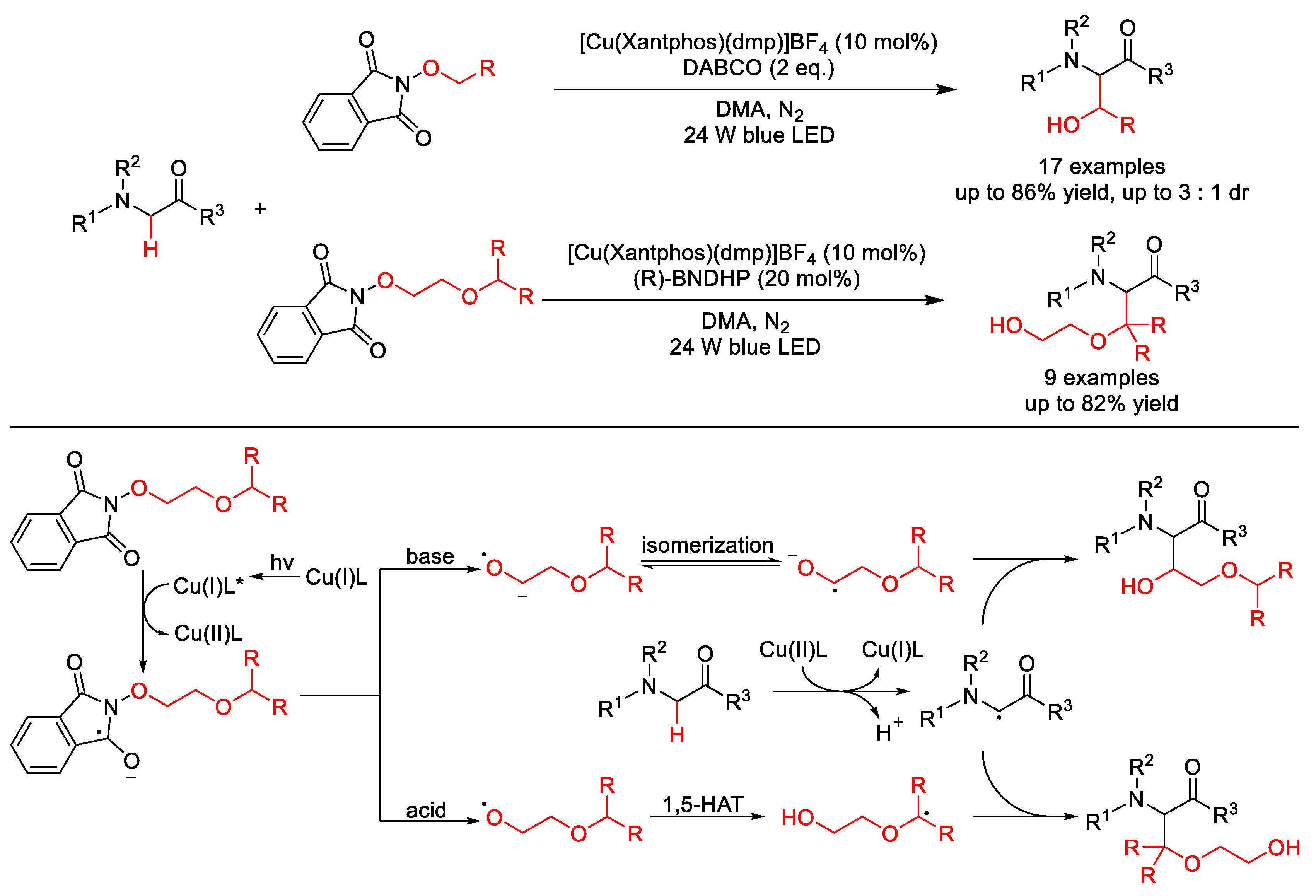
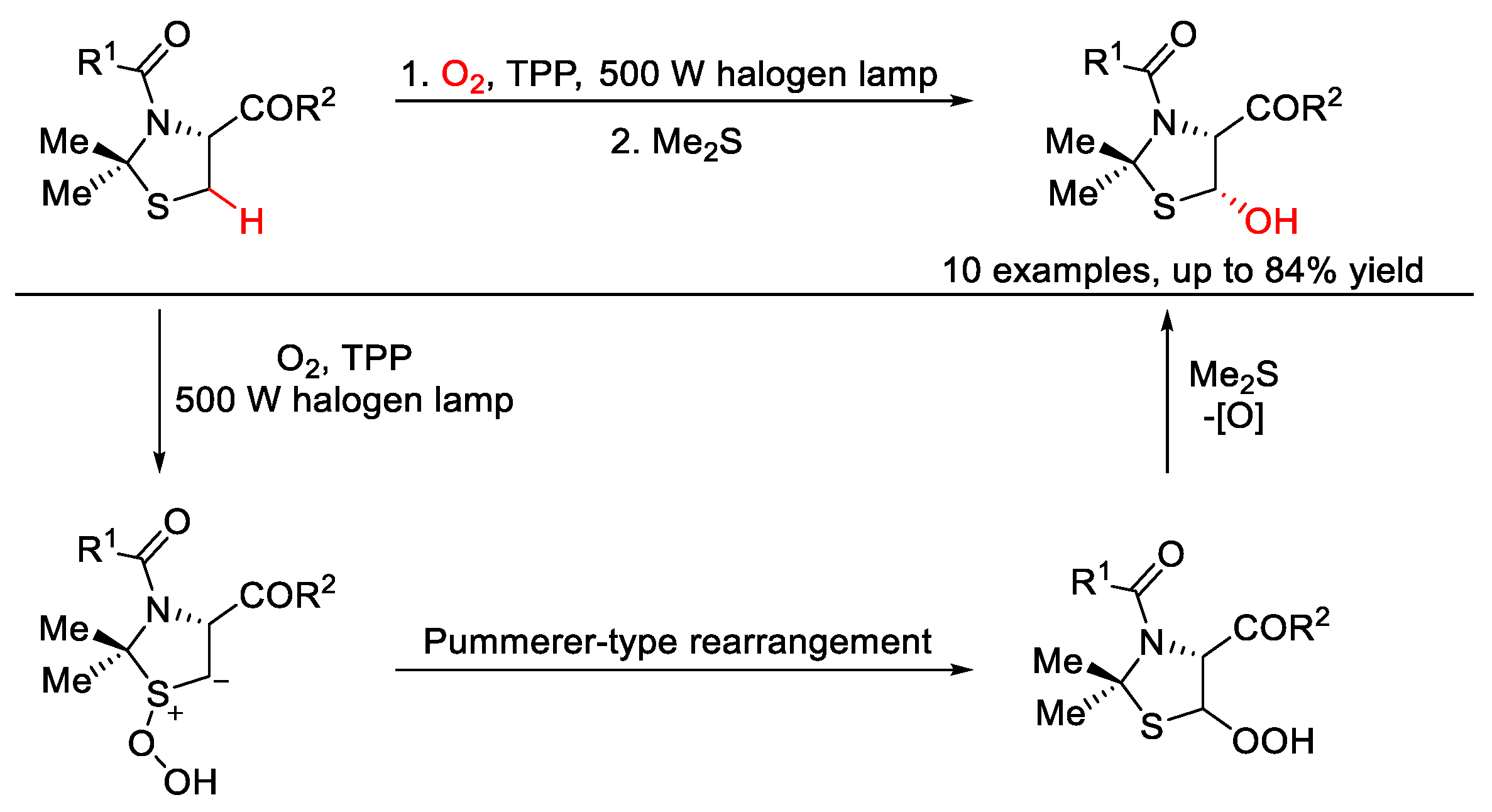
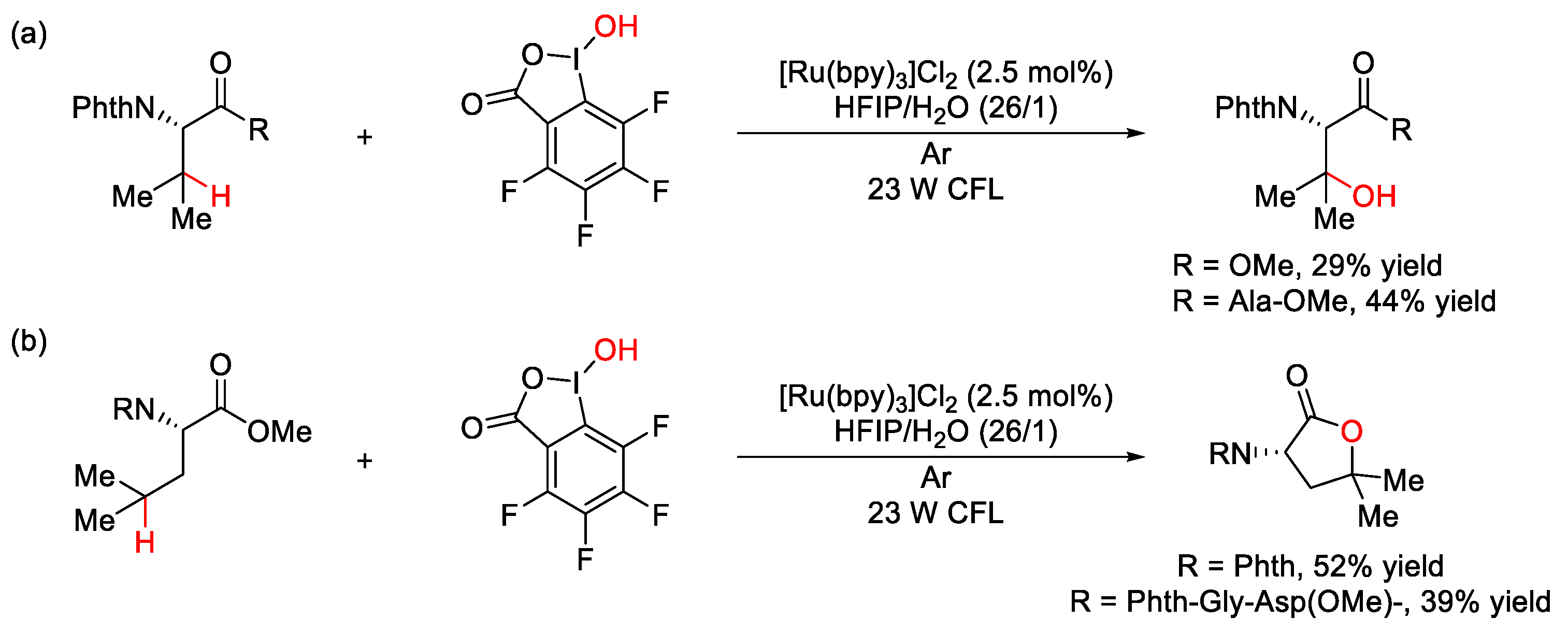
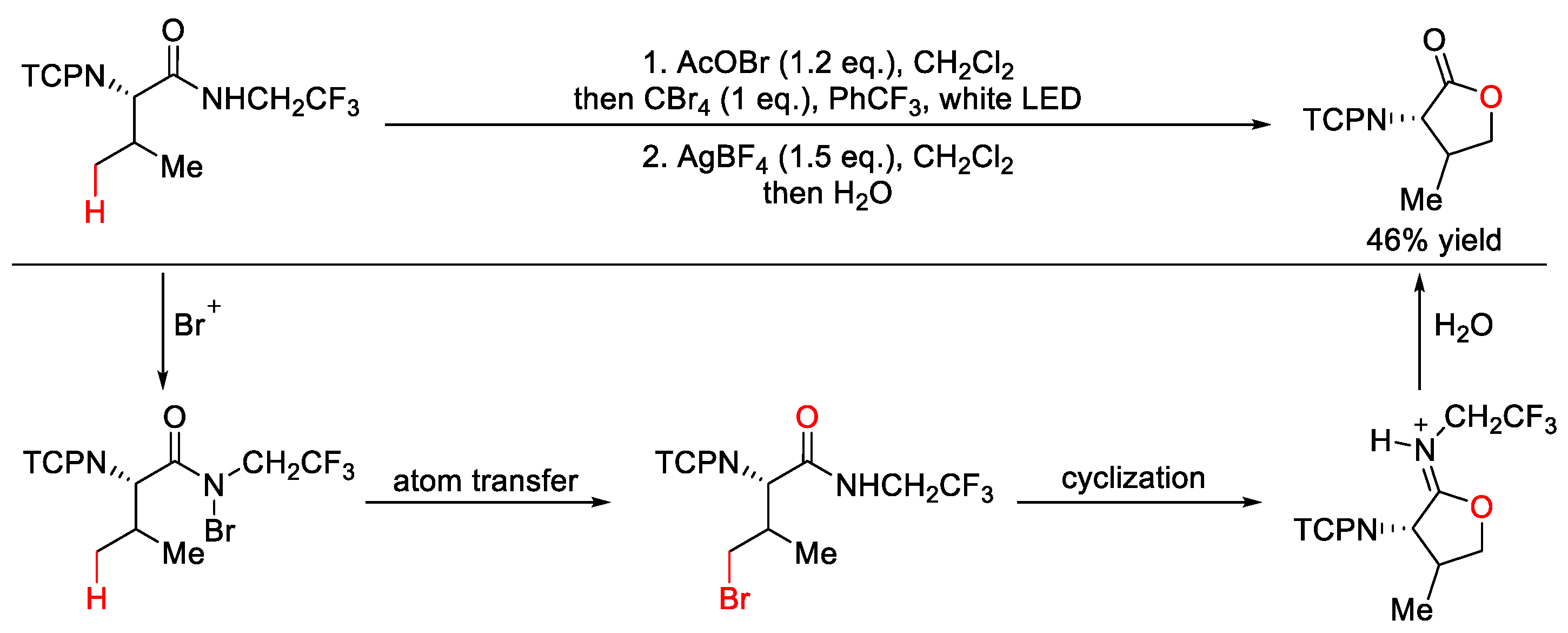
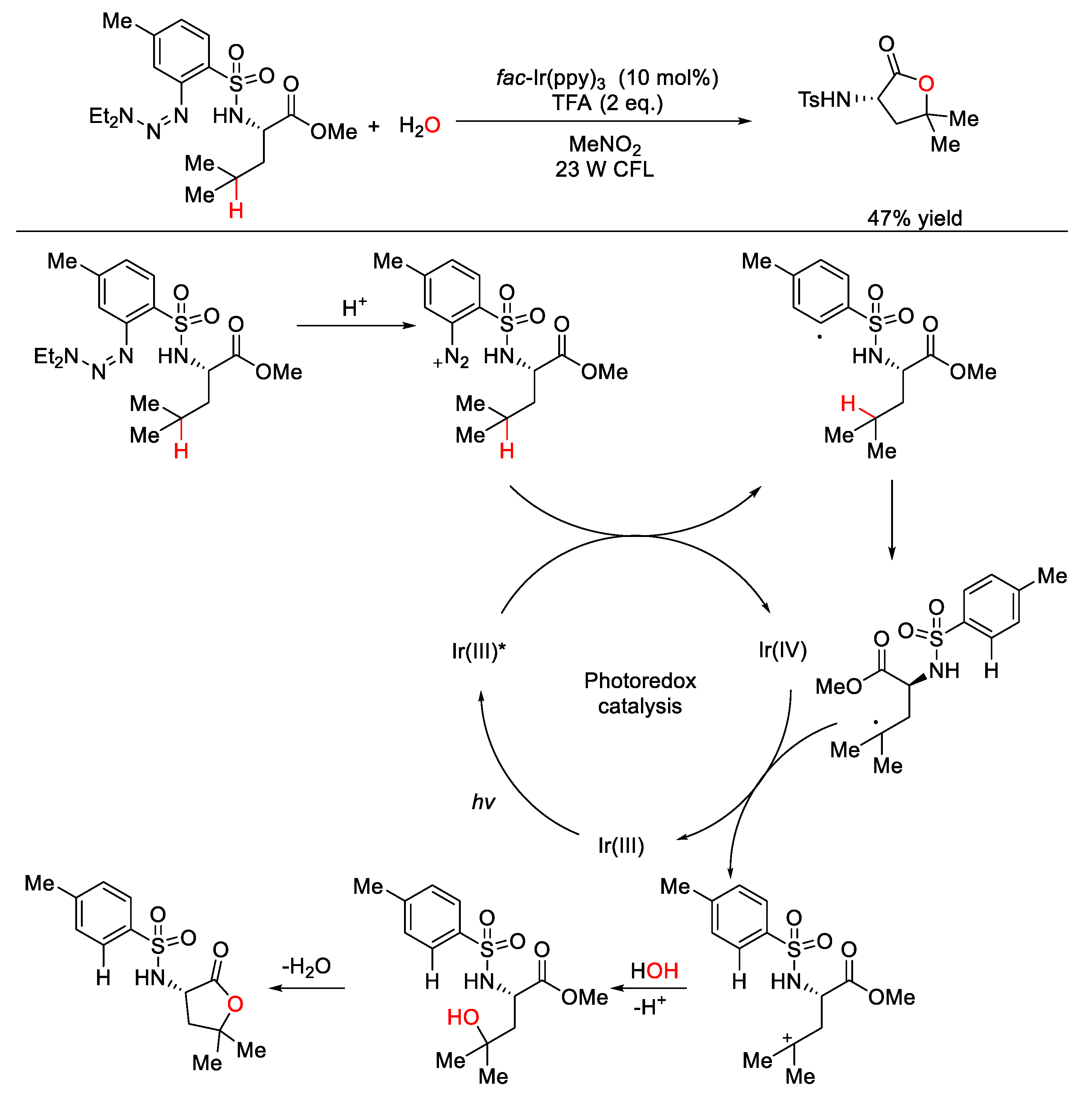
5. C–O Formation
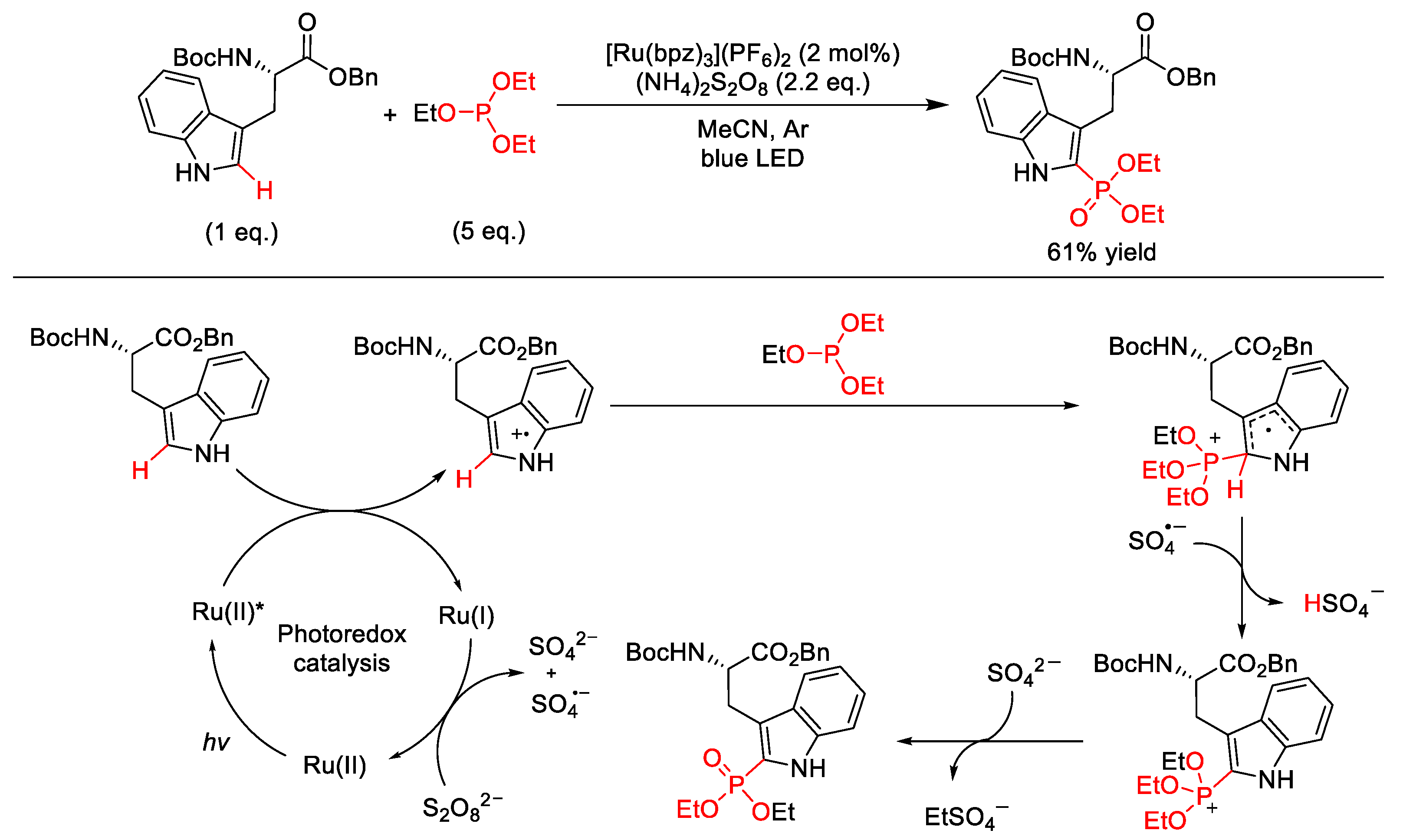
6. C–P Formation
7. C–X Formation
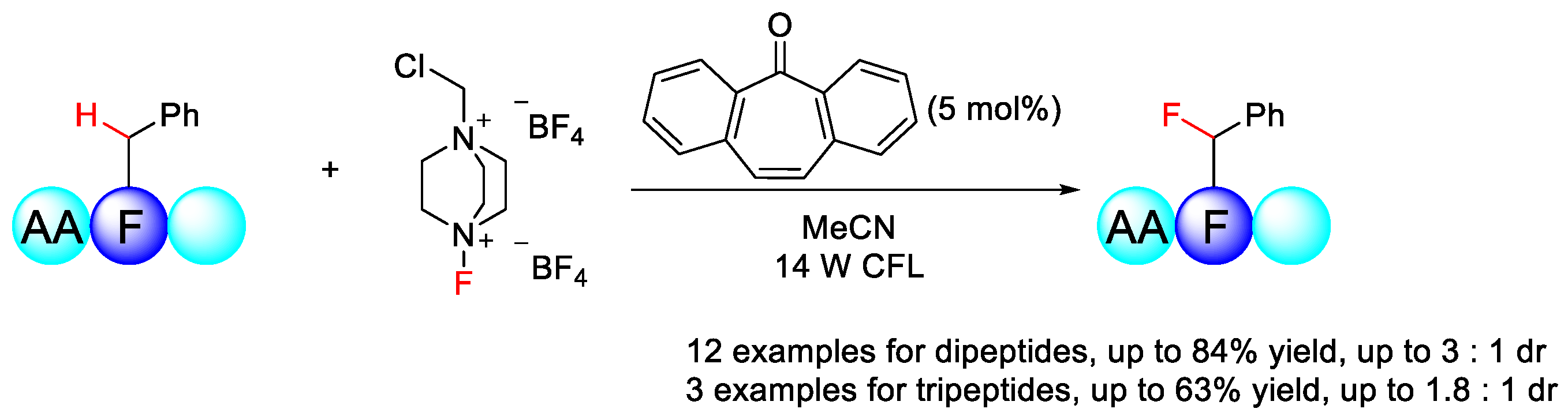
7.1. C–H Fluorination
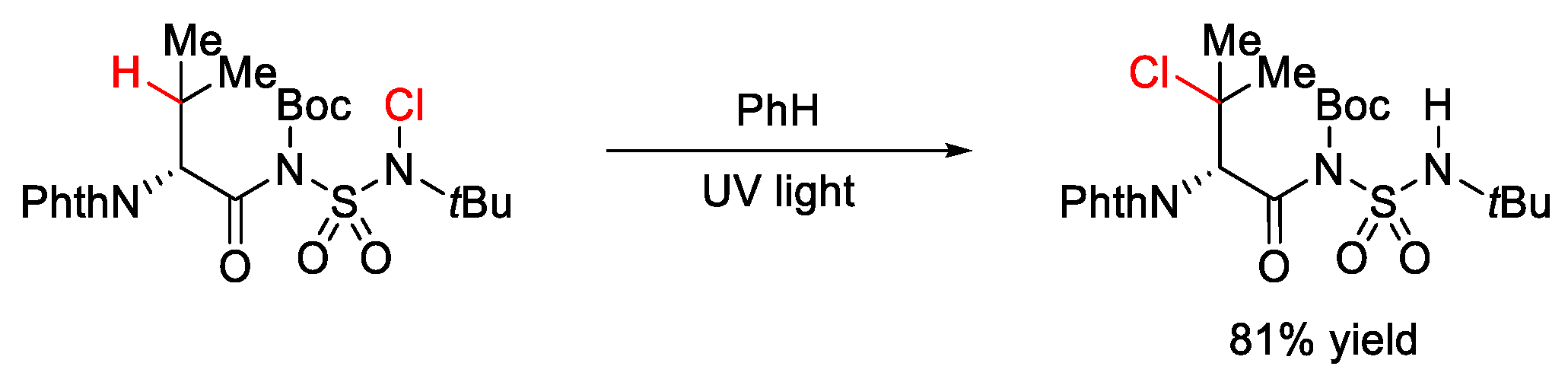
7.2. C–H Chlorination and Bromination
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sadovnikova, M.S.; Belikov, V.M. Industrial Applications of Amino-acids. Russ. Chem. Rev. 1978, 47, 199–212. [Google Scholar] [CrossRef]
- Albericio, F.; Kruger, H.G. Therapeutic peptides. Future Med. Chem. 2012, 4, 1527–1531. [Google Scholar] [CrossRef]
- Fujino, T.; Murakami, H. In Vitro Selection Combined with Ribosomal Translation Containing Non-proteinogenic Amino Acids. Chem. Rec. 2016, 16, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Nödling, A.R.; Spear, L.A.; Williams, T.L.; Luk, L.Y.P.; Tsai, Y.-H. Using genetically incorporated unnatural amino acids to control protein functions in mammalian cells. Essays Biochem. 2019, 63, 237–266. [Google Scholar] [CrossRef] [PubMed]
- Narancic, T.; Almahboub, S.A.; O’Connor, K.E. Unnatural amino acids: Production and biotechnological potential. World J. Microbiol. Biotechnol. 2019, 35, 67. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Guo, J. Expanding the chemistry of fluorescent protein biosensors through genetic incorporation of unnatural amino acids. Mol. Biosyst. 2013, 9, 2961–2970. [Google Scholar] [CrossRef]
- Agirre, M.; Arrieta, A.; Arrastia, I.; Cossío, F.P. Organocatalysts Derived from Unnatural α-Amino Acids: Scope and Applications. Chem. Asian J. 2019, 14, 44–66. [Google Scholar] [CrossRef]
- Lu, X.; Xiao, B.; Shang, R.; Liu, L. Synthesis of unnatural amino acids through palladium-catalyzed C(sp3)–H functionalization. Chin. Chem. Lett. 2016, 27, 305–311. [Google Scholar] [CrossRef]
- Ohfune, Y.; Shinada, T. Enantio- and diastereoselective construction of α,α-disubstituted α-amino acids for the synthesis of biologically active compounds. Eur. J. Org. Chem. 2005, 2005, 5127–5143. [Google Scholar] [CrossRef]
- Luo, Y.-C.; Zhang, H.-H.; Wang, Y.; Xu, P.-F. Synthesis of α-Amino Acids Based on Chiral Tricycloiminolactone Derived from Natural (+)-Camphor. Acc. Chem. Res. 2010, 43, 1317–1330. [Google Scholar] [CrossRef]
- Wei, L.; Xiao, L.; Hu, Y.; Wang, Z.; Tao, H.; Wang, C. Recent Advances in Metallated Azomethine Ylides for the Synthesis of Chiral Unnatural α-Amino Acids. Chin. J. Org. Chem. 2019, 39, 2119–2130. [Google Scholar] [CrossRef]
- Cowart, M.; Kowaluk, E.A.; Daanen, J.F.; Kohlhaas, K.L.; Alexander, K.M.; Wagenaar, F.L.; Kerwin, J.F. Nitroaromatic amino acids as inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 1998, 41, 2636–2642. [Google Scholar] [CrossRef]
- Fan, M.; Zhou, W.; Jiang, Y.; Ma, D. CuI/Oxalamide Catalyzed Couplings of (Hetero)aryl Chlorides and Phenols for Diaryl Ether Formation. Angew. Chem. Int. Ed. 2016, 55, 6211–6215. [Google Scholar] [CrossRef] [PubMed]
- Bottecchia, C.; Rubens, M.; Gunnoo, S.B.; Hessel, V.; Madder, A.; Noël, T. Visible-Light-Mediated Selective Arylation of Cysteine in Batch and Flow. Angew. Chem. Int. Ed. 2017, 56, 12702–12707. [Google Scholar] [CrossRef] [PubMed]
- Chinn, A.J.; Kim, B.; Kwon, Y.; Miller, S.J. Enantioselective Intermolecular C–O Bond Formation in the Desymmetrization of Diarylmethines Employing a Guanidinylated Peptide-Based Catalyst. J. Am. Chem. Soc. 2017, 139, 18107–18114. [Google Scholar] [CrossRef] [PubMed]
- Vara, B.A.; Li, X.; Berritt, S.; Walters, C.R.; Petersson, E.J.; Molander, G.A. Scalable thioarylation of unprotected peptides and biomolecules under Ni/photoredox catalysis. Chem. Sci. 2018, 9, 336–344. [Google Scholar] [CrossRef]
- Noisier, A.F.M.; Brimble, M.A. C–H functionalization in the synthesis of amino acids and peptides. Chem. Rev. 2014, 114, 8775–8806. [Google Scholar] [CrossRef]
- Mondal, S.; Chowdhury, S. Recent Advances on Amino Acid Modifications via C–H Functionalization and Decarboxylative Functionalization Strategies. Adv. Synth. Catal. 2018, 360, 1884–1912. [Google Scholar] [CrossRef]
- Pereira, C.F.; Simoes, M.M.; Tome, J.P.; Almeida Paz, F.A. Porphyrin-Based Metal-Organic Frameworks as Heterogeneous Catalysts in Oxidation Reactions. Molecules 2016, 21, 1348. [Google Scholar] [CrossRef]
- Bayer, P.; Pérez-Ruiz, R.; Von Wangelin, A.J. Stereoselective Photooxidations by the Schenck Ene Reaction. Chemphotochem 2018, 2, 559–570. [Google Scholar] [CrossRef]
- Zhang, H.; Lei, A. Visible-Light-Induced C–H Functionalization and C–C/C–X Bond-Forming Oxidative Cross-Coupling Reactions. Asian J. Org. Chem. 2018, 7, 1164–1177. [Google Scholar] [CrossRef]
- Li, Z.; Jin, J.; Huang, S. Recent Advances in Transition Metal-Catalyzed Cross-Coupling Reactions Directly Promoted by Visible Light. Chin. J. Org. Chem. 2020, 40, 563–574. [Google Scholar] [CrossRef]
- Uygur, M.; Mancheño, O.G. Visible light-mediated organophotocatalyzed C–H bond functionalization reactions. Org. Biomol. Chem. 2019, 17, 5475–5489. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Q.; Shatskiy, A.; Matsuura, B.S.; Kärkäs, M.D. Recent Advances in Photoredox Catalysis Enabled Functionalization of α-Amino Acids and Peptides: Concepts, Strategies and Mechanisms. Synthesis 2019, 51, 2759–2791. [Google Scholar] [CrossRef]
- Dong, Z.; Ren, Z.; Thompson, S.J.; Xu, Y.; Dong, G. Transition-Metal-Catalyzed C–H Alkylation Using Alkenes. Chem. Rev. 2017, 117, 9333–9403. [Google Scholar] [CrossRef]
- Wegmann, M.; Henkel, M.; Bach, T. C–H alkylation reactions of indoles mediated by Pd(II) and norbornene: Applications and recent developments. Org. Biomol. Chem. 2018, 16, 5376–5385. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, Z.-J.; Liu, Z.-P.; Lou, H. Visible-light-mediated de-aminative alkylation of N-arylamines with alkyl Katritzky salts. Org. Chem. Front. 2019, 6, 3902–3905. [Google Scholar] [CrossRef]
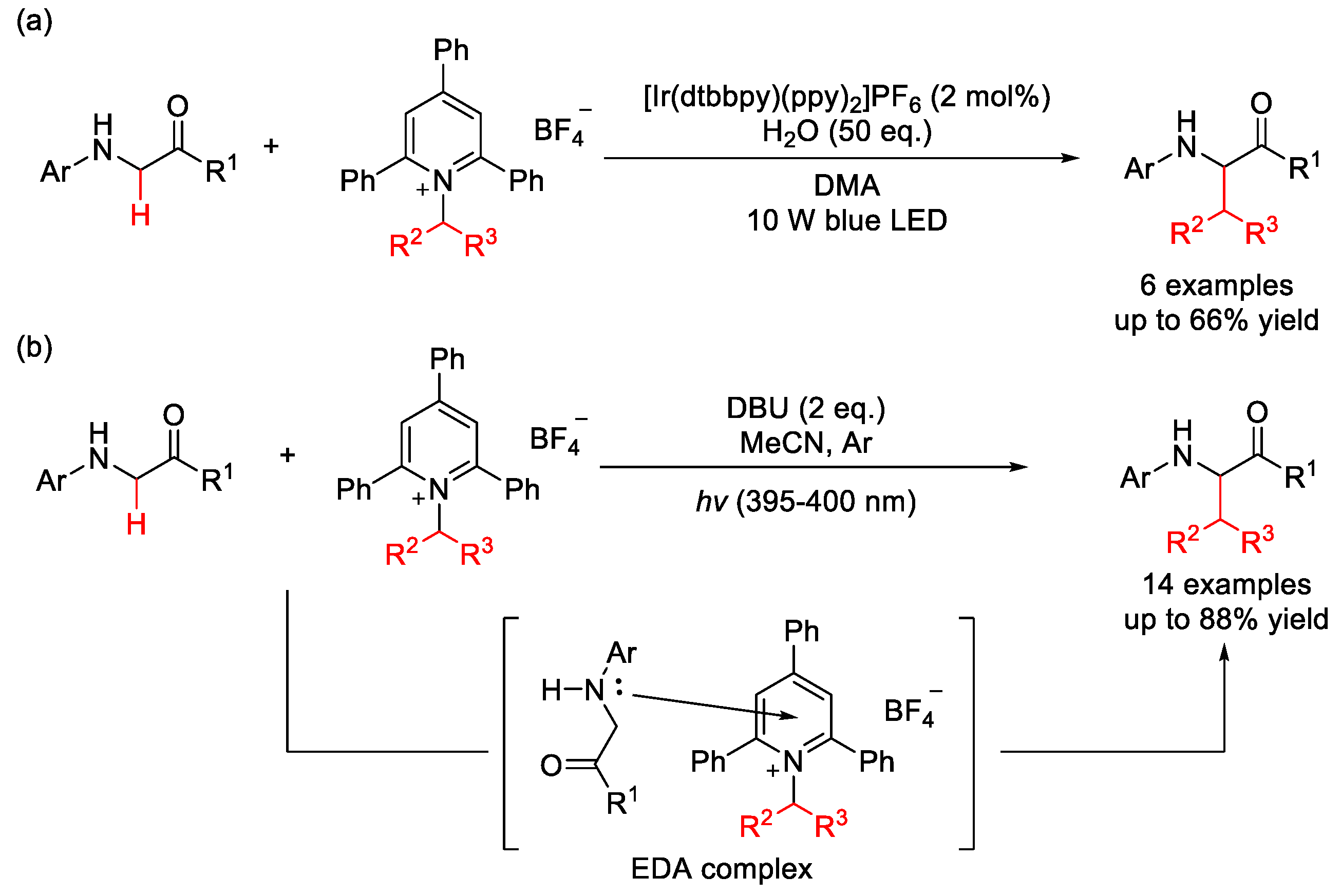
- Wang, C.; Qi, R.; Xue, H.; Shen, Y.; Chang, M.; Chen, Y.; Wang, R.; Xu, Z. Visible-Light-Promoted C(sp3)–H Alkylation by Intermolecular Charge Transfer: Preparation of Unnatural α-Amino Acids and Late-Stage Modification of Peptides. Angew. Chem. Int. Ed. 2020, 59, 7461–7466. [Google Scholar] [CrossRef]
- Wang, C.; Guo, M.; Qi, R.; Shang, Q.; Liu, Q.; Wang, S.; Zhao, L.; Wang, R.; Xu, Z. Visible-Light-Driven, Copper-Catalyzed Decarboxylative C(sp3)–H Alkylation of Glycine and Peptides. Angew. Chem. Int. Ed. 2018, 57, 15841–15846. [Google Scholar] [CrossRef]
- Wang, C.; Yu, Y.; Liu, W.-L.; Duan, W.-L. Site-Tunable Csp3–H Bonds Functionalization by Visible-Light-Induced Radical Translocation of N-Alkoxyphthalimides. Org. Lett. 2019, 21, 9147–9152. [Google Scholar] [CrossRef]
- Okamura, I.; Park, S.; Han, J.H.; Notsu, S.; Sugiyama, H. A Combination of Visible-light Photoredox and Metal Catalysis for the Mannich-type Reaction of N-Aryl Glycine Esters. Chem. Lett. 2017, 46, 1597–1600. [Google Scholar] [CrossRef]
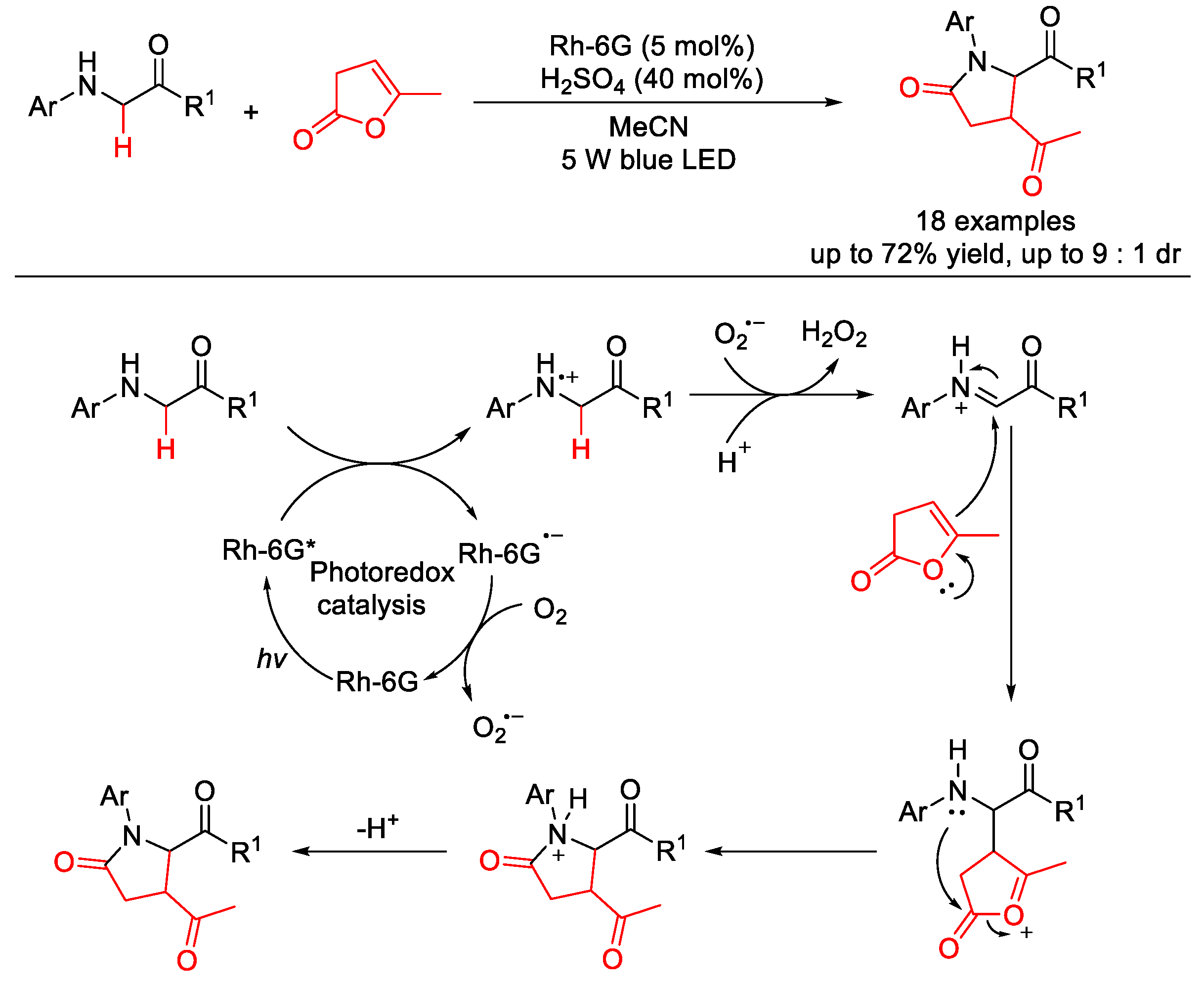
- Zhou, H.; Yang, X.; Li, S.; Zhu, Y.; Li, Y.; Zhang, Y. Visible light-induced aerobic oxidative cross-coupling of glycine esters with α-angelicalactone: A facile pathway to γ-lactams. Org. Biomol. Chem. 2018, 16, 6728–6734. [Google Scholar] [CrossRef] [PubMed]
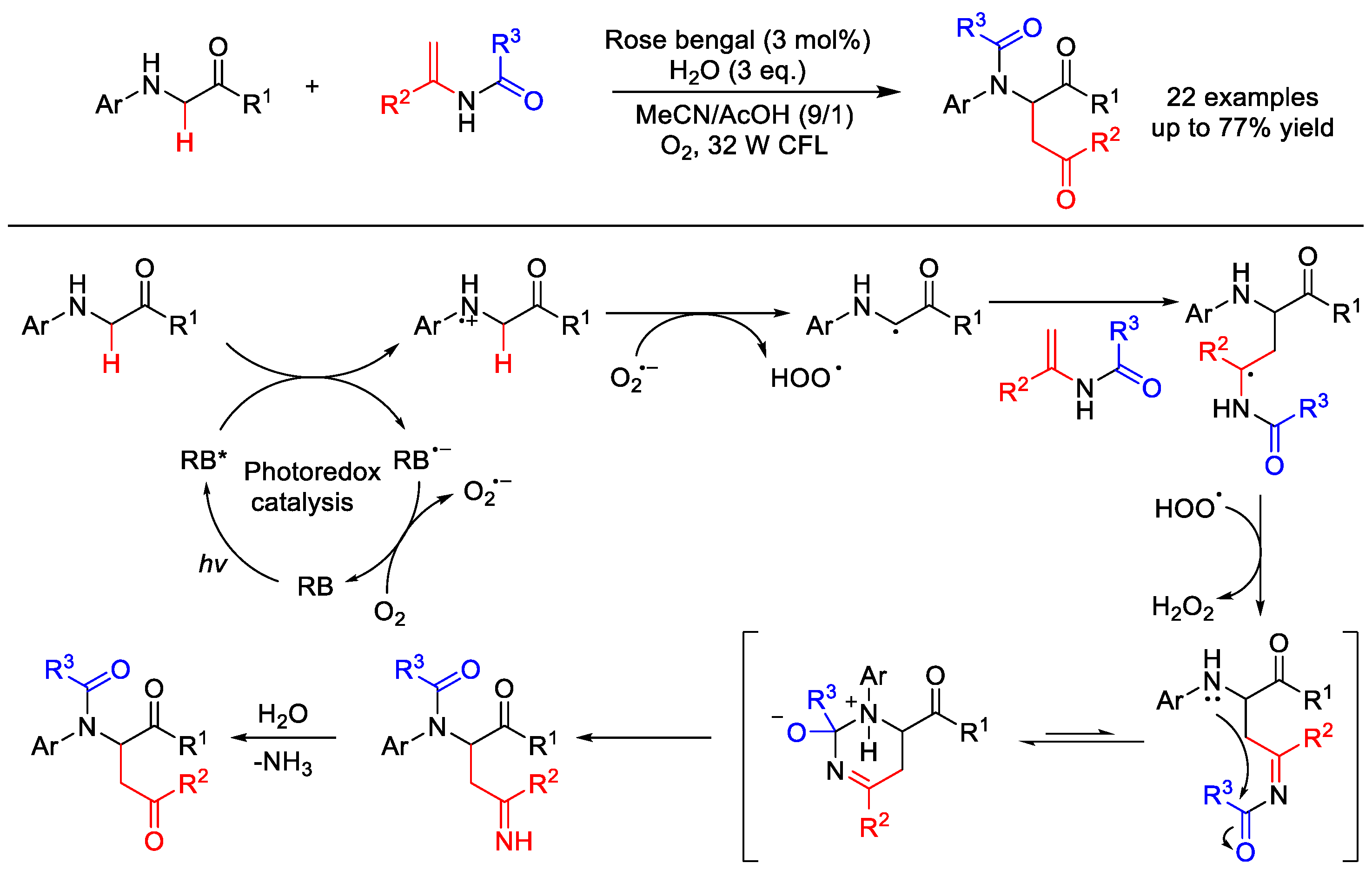
- Tang, L.; Li, X.M.; Matuska, J.H.; He, Y.-H.; Guan, Z. Intramolecular Transamidation of Secondary Amides via Visible-Light-Induced Tandem Reaction. Org. Lett. 2018, 20, 5618–5621. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, S.; Takao, G.; Kamijo, K.; Tsuno, T.; Ishiguro, K.; Murafuji, T. Alkylation of Nonacidic C(sp3)–H Bonds by Photoinduced Catalytic Michael-Type Radical Addition. Org. Lett. 2016, 18, 4912–4915. [Google Scholar] [CrossRef]
- Ashley, M.A.; Yamauchi, C.; Chu, J.C.K.; Otsuka, S.; Yorimitsu, H.; Rovis, T. Photoredox-Catalyzed Site-Selective α-C(sp3)–H Alkylation of Primary Amine Derivatives. Angew. Chem. Int. Ed. 2019, 58, 4002–4006. [Google Scholar] [CrossRef]
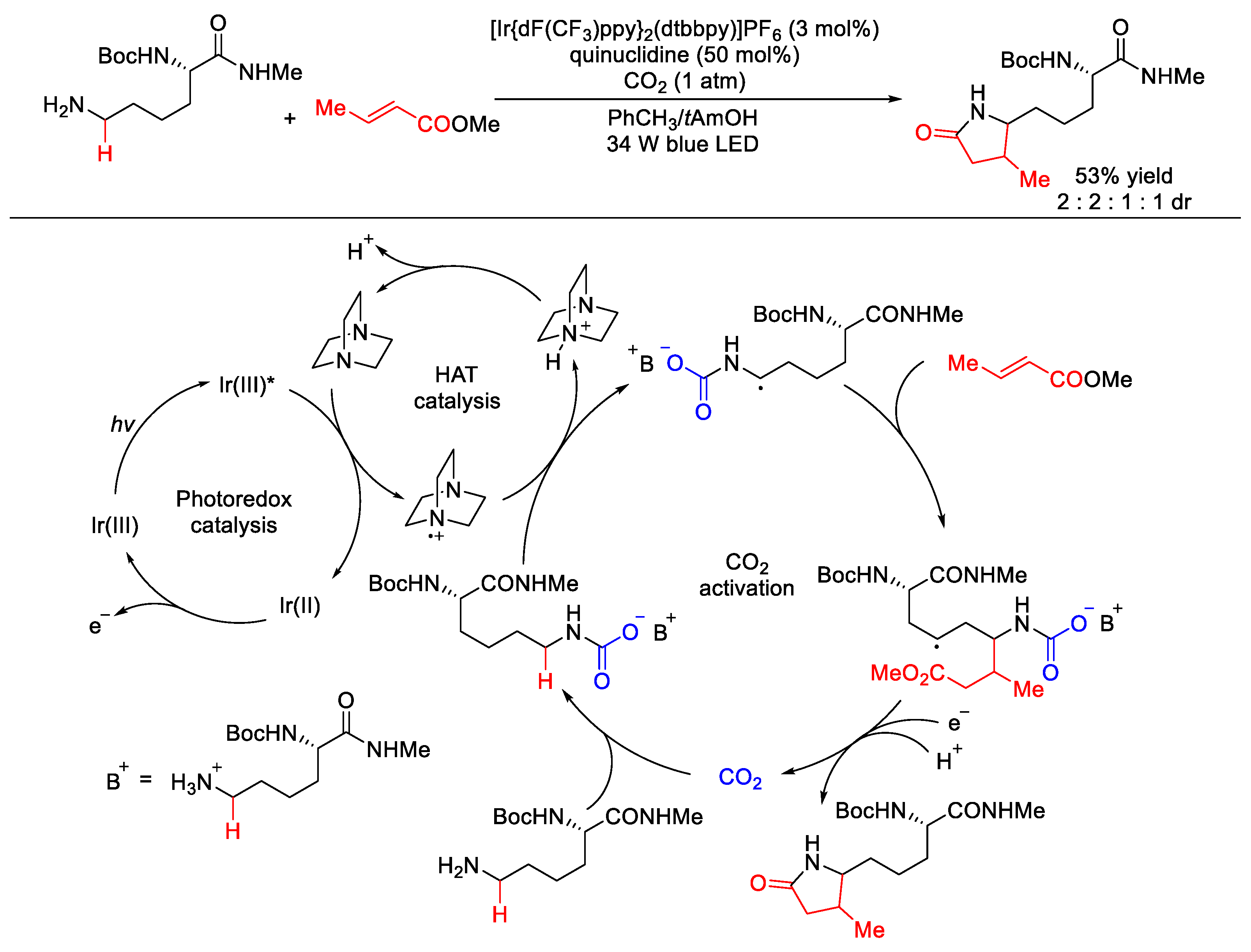
- Ye, J.; Kalvet, I.; Schoenebeck, F.; Rovis, T. Direct α-alkylation of primary aliphatic amines enabled by CO2 and electrostatics. Nat. Chem. 2018, 10, 1037–1041. [Google Scholar] [CrossRef]
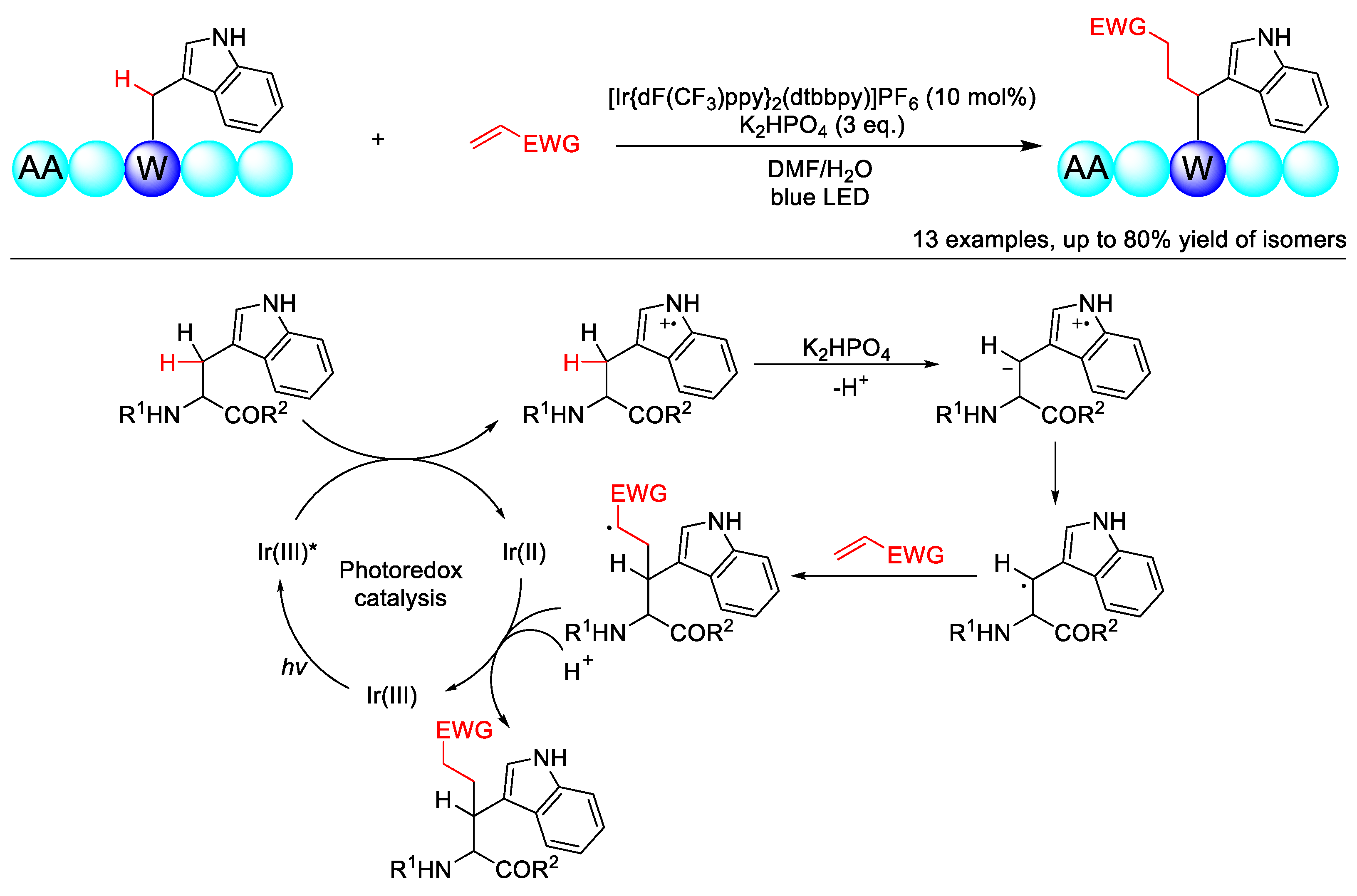
- Yu, Y.; Zhang, L.K.; Buevich, A.V.; Li, G.; Tang, H.; Vachal, P.; Colletti, S.L.; Shi, Z.-C. Chemoselective Peptide Modification via Photocatalytic Tryptophan β-Position Conjugation. J. Am. Chem. Soc. 2018, 140, 6797–6800. [Google Scholar] [CrossRef]
- Le, C.; Liang, Y.; Evans, R.W.; Li, X.; MacMillan, D.W.C. Selective sp3 C–H alkylation via polarity-match-based cross-coupling. Nature 2017, 547, 79–83. [Google Scholar] [CrossRef]
- Gao, X.-W.; Meng, Q.-Y.; Li, J.-X.; Zhong, J.-J.; Lei, T.; Li, X.-B.; Tung, C.-H.; Wu, L.-Z. Visible Light Catalysis Assisted Site-Specific Functionalization of Amino Acid Derivatives by C–H Bond Activation without Oxidant: Cross-Coupling Hydrogen Evolution Reaction. ACS Catal. 2015, 5, 2391–2396. [Google Scholar] [CrossRef]
- Furst, L.; Matsuura, B.S.; Narayanam, J.M.; Tucker, J.W.; Stephenson, C.R. Visible light-mediated intermolecular C–H functionalization of electron-rich heterocycles with malonates. Org. Lett. 2010, 12, 3104–3107. [Google Scholar] [CrossRef]
- Bottecchia, C.; Martín, R.; Abdiaj, I.; Crovini, E.; Alcazar, J.; Orduna, J.; Blesa, M.J.; Carrillo, J.R.; Prieto, P.; Noël, T. De novo Design of Organic Photocatalysts: Bithiophene Derivatives for the Visible-light Induced C–H Functionalization of Heteroarenes. Adv. Synth. Catal. 2019, 361, 945–950. [Google Scholar] [CrossRef]
- Chen, X.; Ye, F.; Luo, X.; Liu, X.; Zhao, J.; Wang, S.; Zhou, Q.; Chen, G.; Wang, P. Histidine-Specific Peptide Modification via Visible-Light-Promoted C–H Alkylation. J. Am. Chem. Soc. 2019, 141, 18230–18237. [Google Scholar] [CrossRef] [PubMed]
- Ciszewski, L.W.; Durka, J.; Gryko, D. Photocatalytic Alkylation of Pyrroles and Indoles with α-Diazo Esters. Org. Lett. 2019, 21, 7028–7032. [Google Scholar] [CrossRef]
- Saget, T.; König, B. Photocatalytic Synthesis of Polycyclic Indolones. Chem. Eur. J. 2020, 26, 7004–7007. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, G.; Liu, L.; Dai, C.; Shi, D.-Q.; Zhao, Y. The protecting group enabledpara-selective C–H benzylation of anilidesviairon(II) catalysis: A convenient approach for the synthesis of triarylmethanes. Org. Chem. Front. 2020, 7, 1823–1827. [Google Scholar] [CrossRef]
- Xu, G.-Q.; Feng, Z.-T.; Xu, J.-T.; Wang, Z.-Y.; Qin, Y.; Xu, P.-F. Transition-Metal-Free Selective C–H Benzylation of Tertiary Arylamines by a Dearomatization-Aromatization Sequence. Chem. Eur. J. 2018, 24, 13778–13782. [Google Scholar] [CrossRef]
- Wessig, P. An efficient synthesis of bicyclic β-turn dipeptides via a photochemical key step. Tetrahedron Lett. 1999, 40, 5987–5988. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Heckroth, H. Reversible Diastereoselective Photocyclization of Phenylglyoxylamides of α-Amino Acid Methyl Esters to 3-Hydroxy β-Lactams. Synlett 2002, 2002, 0131–0133. [Google Scholar] [CrossRef]
- Ding, W.; Lu, L.Q.; Liu, J.; Liu, D.; Song, H.T.; Xiao, W.J. Visible Light Photocatalytic Radical-Radical Cross-Coupling Reactions of Amines and Carbonyls: A Route to 1,2-Amino Alcohols. J. Org. Chem. 2016, 81, 7237–7243. [Google Scholar] [CrossRef]
- Xia, Q.; Tian, H.; Dong, J.; Qu, Y.; Li, L.; Song, H.; Liu, Y.; Wang, Q. N-Arylamines Coupled with Aldehydes, Ketones, and Imines by Means of Photocatalytic Proton-Coupled Electron Transfer. Chem. Eur. J. 2018, 24, 9269–9273. [Google Scholar] [CrossRef]
- Deng, H.P.; Zhou, Q.; Wu, J. Microtubing-Reactor-Assisted Aliphatic C–H Functionalization with HCl as a Hydrogen-Atom-Transfer Catalyst Precursor in Conjunction with an Organic Photoredox Catalyst. Angew. Chem. Int. Ed. 2018, 57, 12661–12665. [Google Scholar] [CrossRef] [PubMed]
- Kandukuri, S.R.; Bahamonde, A.; Chatterjee, I.; Jurberg, I.D.; Escudero-Adán, E.C.; Melchiorre, P. X-ray characterization of an electron donor-acceptor complex that drives the photochemical alkylation of indoles. Angew. Chem. Int. Ed. 2015, 54, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Sevrin, M.J.; Furst, L.; Nguyen, J.D.; Collins III, J.L.; Stephenson, C.R.J. Lithium bis-catechol borate as an effective reductive quencher in photoredox catalysis. Tetrahedron 2018, 74, 3246–3252. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, M.I.; Horton, P.N.; Hursthouse, M.B.; Wood, M.E. α-Allylation of α-amino acids via 1,5-hydrogen atom transfer. Tetrahedron Lett. 2009, 50, 3400–3403. [Google Scholar] [CrossRef]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Kirk, K.L.; Nishida, M.; Fujii, S.; Kimoto, H. Photochemical trifluoromethylation of tyramine and L-tyrosine derivatives. J. Fluor. Chem. 1992, 59, 197–202. [Google Scholar] [CrossRef]
- Ichiishi, N.; Caldwell, J.P.; Lin, M.; Zhong, W.; Zhu, X.; Streckfuss, E.; Kim, H.Y.; Parish, C.A.; Krska, S.W. Protecting group free radical C–H trifluoromethylation of peptides. Chem. Sci. 2018, 9, 4168–4175. [Google Scholar] [CrossRef]
- Baar, M.; Blechert, S. Graphitic carbon nitride polymer as a recyclable photoredox catalyst for fluoroalkylation of arenes. Chem. Eur. J. 2015, 21, 526–530. [Google Scholar] [CrossRef]
- Liu, P.; Liu, W.; Li, C.J. Catalyst-Free and Redox-Neutral Innate Trifluoromethylation and Alkylation of Aromatics Enabled by Light. J. Am. Chem. Soc. 2017, 139, 14315–14321. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Ito, Y.; Ide, T.; Masuda, S.; Hamashima, Y. Simple Photo-Induced Trifluoromethylation of Aromatic Rings. Synthesis 2018, 50, 2948–2953. [Google Scholar] [CrossRef]
- Ghosh, I.; Khamrai, J.; Savateev, A.; Shlapakov, N.; Antonietti, M.; König, B. Organic semiconductor photocatalyst can bifunctionalize arenes and heteroarenes. Science 2019, 365, 360–366. [Google Scholar] [CrossRef]
- Ding, B.; Weng, Y.; Liu, Y.; Song, C.; Yin, L.; Yuan, J.; Ren, Y.; Lei, A.; Chiang, C.-W. Selective Photoredox Trifluoromethylation of Tryptophan-Containing Peptides. Eur. J. Org. Chem. 2019, 2019, 7596–7605. [Google Scholar] [CrossRef]
- Qiu, Y.; Scheremetjew, A.; Finger, L.H.; Ackermann, L. Electrophotocatalytic Undirected C–H Trifluoromethylations of (Het)Arenes. Chem. Eur. J. 2020, 26, 3241–3246. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Li, G.X.; He, G.; Chen, G. Halogen-Bond-Promoted Photoactivation of Perfluoroalkyl Iodides: A Photochemical Protocol for Perfluoroalkylation Reactions. Org. Lett. 2017, 19, 1442–1445. [Google Scholar] [CrossRef]
- Lu, H.; Wang, D.-Y.; Zhang, A. Visible Light-Promoted Phosphine-Catalyzed Difluoroalkylation of Arenes and Heterocycles. J. Org. Chem. 2020, 85, 942–951. [Google Scholar] [CrossRef]
- Nebe, M.M.; Loeper, D.; Fürmeyer, F.; Opatz, T. Visible-Light Organophotoredox-Catalyzed Synthesis of Precursors for Horner-Type Olefinations. Eur. J. Org. Chem. 2018, 2018, 2471–2476. [Google Scholar] [CrossRef]
- Shi, C.; Jia, G. Chemistry of rhenium carbyne complexes. Coord. Chem. Rev. 2013, 257, 666–701. [Google Scholar] [CrossRef]
- Chalifoux, W.A.; Tykwinski, R.R. Synthesis of extended polyynes: Toward carbyne. Comptes Rendus Chimie 2009, 12, 341–358. [Google Scholar] [CrossRef]
- Wang, Z.; Herraiz, A.G.; Del Hoyo, A.M.; Suero, M.G. Generating carbyne equivalents with photoredox catalysis. Nature 2018, 554, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Liu, K.; Liu, C.; Lei, A. Olefinic C–H functionalization through radical alkenylation. Chem. Soc. Rev. 2015, 44, 1070–1082. [Google Scholar] [CrossRef]
- Feldman, K.S.; Ngernmeesri, P. Dragmacidin E synthesis studies. Preparation of a model cycloheptannelated indole fragment. Org. Lett. 2005, 7, 5449–5452. [Google Scholar] [CrossRef] [PubMed]
- Amaoka, Y.; Nagatomo, M.; Watanabe, M.; Tao, K.; Kamijo, S.; Inoue, M. Photochemically induced radical alkenylation of C(sp3)–H bonds. Chem. Sci. 2014, 5, 4339–4345. [Google Scholar] [CrossRef]
- Chen, H.; Guo, L.; Yu, S. Primary, Secondary, and Tertiary γ-C(sp3)-H Vinylation of Amides via Organic Photoredox-Catalyzed Hydrogen Atom Transfer. Org. Lett. 2018, 20, 6255–6259. [Google Scholar] [CrossRef] [PubMed]
- Hoshikawa, T.; Kamijo, S.; Inoue, M. Photochemically induced radical alkynylation of C(sp3)-H bonds. Org. Biomol. Chem. 2013, 11, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Morcillo, S.P.; Dauncey, E.M.; Kim, J.H.; Douglas, J.J.; Sheikh, N.S.; Leonori, D. Photoinduced Remote Functionalization of Amides and Amines Using Electrophilic Nitrogen Radicals. Angew. Chem. Int. Ed. 2018, 57, 12945–12949. [Google Scholar] [CrossRef]
- Wu, X.F. Acylation of (Hetero)Arenes through C–H Activation with Aroyl Surrogates. Chem. Eur. J. 2015, 21, 12252–12265. [Google Scholar] [CrossRef]
- Santiago, C.; Sotomayor, N.; Lete, E. Pd(II)-Catalyzed C–H Acylation of (Hetero)arenes—Recent Advances. Molecules 2020, 25, 3247. [Google Scholar] [CrossRef]
- Takechi, H.; Tateuchi, S.; Machida, M.; Nishibata, Y.; Aoe, K.; Sato, Y.; Kanaoka, Y. Photoreactions of succinimides with an N-acyl group in the side chain. Synthesis and stereochemistry of tricyclic pyrrolo[1,2-α]pyrazine ring systems. Chem. Pharm. Bull. 1986, 34, 3142–3152. [Google Scholar] [CrossRef]
- Kamijo, S.; Takao, G.; Kamijo, K.; Hirota, M.; Tao, K.; Murafuji, T. Photo-induced Substitutive Introduction of the Aldoxime Functional Group to Carbon Chains: A Formal Formylation of Non-Acidic C(sp3)–H Bonds. Angew. Chem. Int. Ed. 2016, 55, 9695–9699. [Google Scholar] [CrossRef] [PubMed]
- Manna, M.K.; Bairy, G.; Jana, R. Dual visible-light photoredox and palladium(II) catalysis for dehydrogenative C2-acylation of indoles at room temperature. Org. Biomol. Chem. 2017, 15, 5899–5903. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, S.; Hoshikawa, T.; Inoue, M. Photochemically induced radical transformation of C(sp3)–H bonds to C(sp3)-CN bonds. Org. Lett. 2011, 13, 5928–5931. [Google Scholar] [CrossRef] [PubMed]
- Akula, P.S.; Hong, B.-C.; Lee, G.-H. Visible-light-induced C(sp3)–H activation for a C–C bond forming reaction of 3,4-dihydroquinoxalin-2(1H)-one with nucleophiles using oxygen with a photoredox catalyst or under catalyst-free conditions. RSC Adv. 2018, 8, 19580–19584. [Google Scholar] [CrossRef]
- Wakaki, T.; Sakai, K.; Enomoto, T.; Kondo, M.; Masaoka, S.; Oisaki, K.; Kanai, M. C(sp3)–H Cyanation Promoted by Visible-Light Photoredox/Phosphate Hybrid Catalysis. Chem. Eur. J. 2018, 24, 8051–8055. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Lessi, M.; Manzini, C.; Marianetti, G.; Bellina, F. Transition Metal-Free Direct C–H (Hetero)arylation of Heteroarenes: A Sustainable Methodology to Access (Hetero)aryl-Substituted Heteroarenes. Adv. Synth. Catal. 2015, 357, 3777–3814. [Google Scholar] [CrossRef]
- Nareddy, P.; Jordan, F.; Szostak, M. Recent Developments in Ruthenium-Catalyzed C–H Arylation: Array of Mechanistic Manifolds. ACS Catal. 2017, 7, 5721–5745. [Google Scholar] [CrossRef]
- Zhu, S.; Rueping, M. Merging visible-light photoredox and Lewis acid catalysis for the functionalization and arylation of glycine derivatives and peptides. Chem. Commun. 2012, 48, 11960–11962. [Google Scholar] [CrossRef]
- Dong, J.; Xia, Q.; Lv, X.; Yan, C.; Song, H.; Liu, Y.; Wang, Q. Photoredox-Mediated Direct Cross-Dehydrogenative Coupling of Heteroarenes and Amines. Org. Lett. 2018, 20, 5661–5665. [Google Scholar] [CrossRef]
- Chen, H.; Fan, W.; Yuan, X.-A.; Yu, S. Site-selective remote C(sp3)–H heteroarylation of amides via organic photoredox catalysis. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Shaw, M.H.; Shurtleff, V.W.; Terrett, J.A.; Cuthbertson, J.D.; MacMillan, D.W. Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science 2016, 352, 1304–1308. [Google Scholar] [CrossRef] [PubMed]
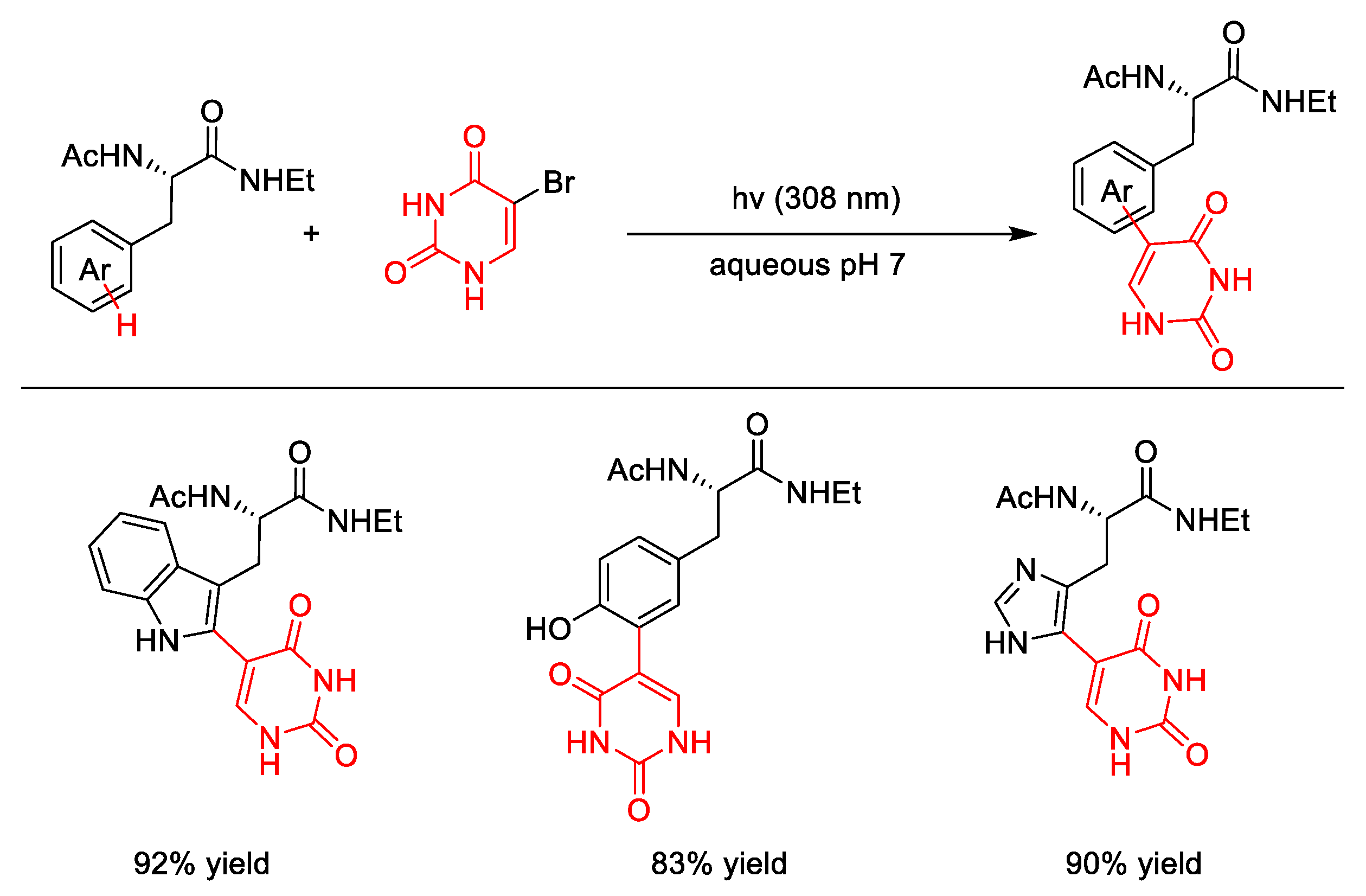
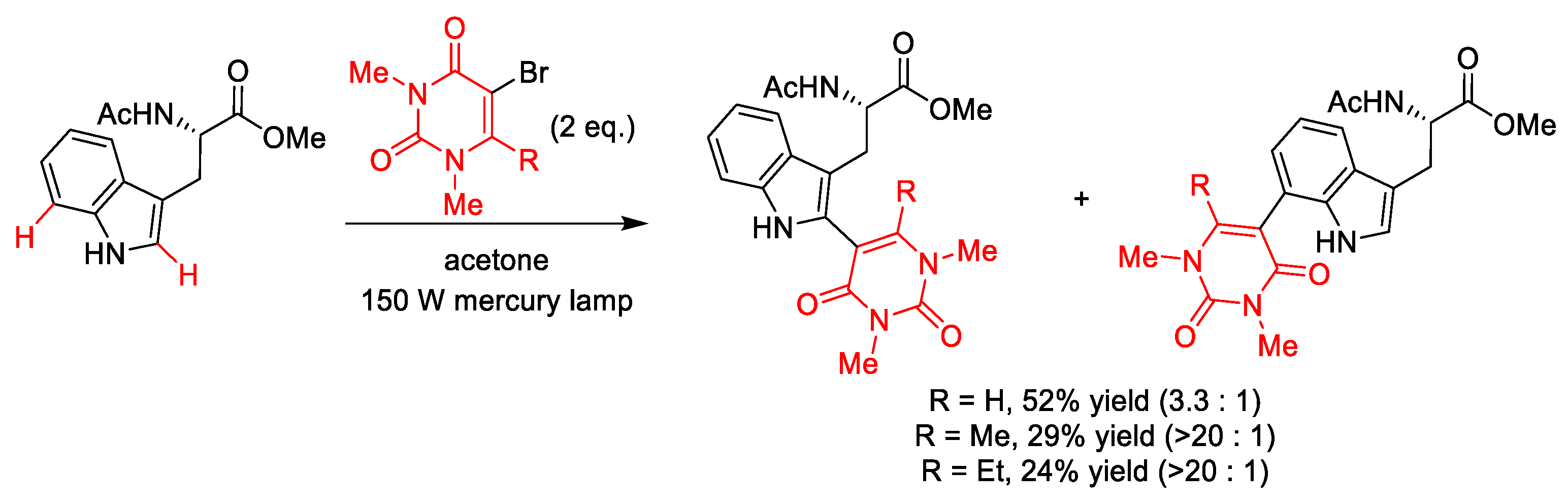
- Dietz, T.M.; Koch, T.H. Photochemical coupling of 5-bromouracil to tryptophan, tyrosine and histidine, peptide-like derivatives in aqueous fluid solution. Photochem. Photobiol. 1987, 46, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Celewicz, L. Photochemical coupling of 5-bromo-1,3-dimethyluracil and its 6-alkyl derivatives to 3-methylindole and Nα-acetyl-l-tryptophan methyl ester. J. Photoch. Photobio. B 1989, 3, 565–574. [Google Scholar] [CrossRef]
- Brase, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic azides: An exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef]
- Hili, R.; Yudin, A.K. Making carbon-nitrogen bonds in biological and chemical synthesis. Nat. Chem. Biol. 2006, 2, 284–287. [Google Scholar] [CrossRef]
- Wang, Y.; Li, G.X.; Yang, G.; He, G.; Chen, G. A visible-light-promoted radical reaction system for azidation and halogenation of tertiary aliphatic C–H bonds. Chem. Sci. 2016, 7, 2679–2683. [Google Scholar] [CrossRef]
- Bosnidou, A.E.; Muñiz, K. Intermolecular Radical C(sp3)–H Amination under Iodine Catalysis. Angew. Chem. Int. Ed. 2019, 58, 7485–7489. [Google Scholar] [CrossRef]
- Stateman, L.M.; Wappes, E.A.; Nakafuku, K.M.; Edwards, K.M.; Nagib, D.A. Catalytic β C–H amination via an imidate radical relay. Chem. Sci. 2019, 10, 2693–2699. [Google Scholar] [CrossRef]
- Kim, H.; Kim, T.; Lee, D.G.; Roh, S.W.; Lee, C. Nitrogen-centered radical-mediated C–H imidation of arenes and heteroarenes via visible light induced photocatalysis. Chem. Commun. 2014, 50, 9273–9276. [Google Scholar] [CrossRef]
- Ruffoni, A.; Juliá, F.; Svejstrup, T.D.; McMillan, A.J.; Douglas, J.J.; Leonori, D. Practical and regioselective amination of arenes using alkyl amines. Nat. Chem. 2019, 11, 426–433. [Google Scholar] [CrossRef]
- Kuribara, T.; Nakajima, M.; Nemoto, T. Visible-Light-Induced Metal-/Photocatalyst-Free C–H Bond Imidation of Arenes. Org. Lett. 2020, 22, 2235–2239. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, K.L.; Scharf, D.H.; Litomska, A.; Hertweck, C. Enzymatic Carbon-Sulfur Bond Formation in Natural Product Biosynthesis. Chem. Rev. 2017, 117, 5521–5577. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Prieto, M.A.; Miatello, R.M. Organosulfur compounds and cardiovascular disease. Mol. Aspects Med. 2010, 31, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Takimiya, K.; Osaka, I.; Mori, T.; Nakano, M. Organic semiconductors based on [1]benzothieno[3,2-b][1]benzothiophene substructure. Acc. Chem. Res. 2014, 47, 1493–1502. [Google Scholar] [CrossRef]
- Kazemi, M.; Shiri, L.; Kohzadi, H. Recent Advances in Aryl Alkyl and Dialkyl Sulfide Synthesis. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 978–1003. [Google Scholar] [CrossRef]
- Na, C.G.; Alexanian, E.J. A General Approach to Site-Specific, Intramolecular C–H Functionalization Using Dithiocarbamates. Angew. Chem. Int. Ed. 2018, 57, 13106–13109. [Google Scholar] [CrossRef]
- Bala, V.; Gupta, G.; Sharma, V.L. Chemical and medicinal versatility of dithiocarbamates: An overview. Mini Rev. Med. Chem. 2014, 14, 1021–1032. [Google Scholar] [CrossRef]
- Oliveira, J.W.F.; Rocha, H.A.O.; De Medeiros, W.M.T.Q.; Silva, M.S. Application of Dithiocarbamates as Potential New Antitrypanosomatids-Drugs: Approach Chemistry, Functional and Biological. Molecules 2019, 24, 2806. [Google Scholar] [CrossRef]
- Dinh, A.N.; Nguyen, A.D.; Aceves, E.M.; Albright, S.T.; Cedano, M.R.; Smith, D.K.; Gustafson, J.L. Photocatalytic Oxidative C–H Thiolation: Synthesis of Benzothiazoles and Sulfenylated Indoles. Synlett 2019, 30, 1648–1655. [Google Scholar] [CrossRef]
- Gaich, T.; Baran, P.S. Aiming for the ideal synthesis. J. Org. Chem. 2010, 75, 4657–4673. [Google Scholar] [CrossRef]
- Chen, K.; Baran, P.S. Total synthesis of eudesmane terpenes by site-selective C–H oxidations. Nature 2009, 459, 824–828. [Google Scholar] [CrossRef] [PubMed]
- White, M.C. Chemistry. Adding aliphatic C–H bond oxidations to synthesis. Science 2012, 335, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Long, T.E. Asymmetric synthesis of monocyclic β-lactams from l-cysteine using photochemistry. Tetrahedron Lett. 2011, 52, 5051–5054. [Google Scholar] [CrossRef]
- Li, G.X.; Morales-Rivera, C.A.; Gao, F.; Wang, Y.; He, G.; Liu, P.; Chen, G. A unified photoredox-catalysis strategy for C(sp3)–H hydroxylation and amidation using hypervalent iodine. Chem. Sci. 2017, 8, 7180–7185. [Google Scholar] [CrossRef] [PubMed]
- Richers, J.; Heilmann, M.; Drees, M.; Tiefenbacher, K. Synthesis of Lactones via C–H Functionalization of Nonactivated C(sp3)–H Bonds. Org. Lett. 2016, 18, 6472–6475. [Google Scholar] [CrossRef]
- Hollister, K.A.; Conner, E.S.; Spell, M.L.; Deveaux, K.; Maneval, L.; Beal, M.W.; Ragains, J.R. Remote Hydroxylation through Radical Translocation and Polar Crossover. Angew. Chem. Int. Ed. 2015, 54, 7837–7841. [Google Scholar] [CrossRef]
- Voica, A.F.; Mendoza, A.; Gutekunst, W.R.; Fraga, J.O.; Baran, P.S. Guided desaturation of unactivated aliphatics. Nat. Chem. 2012, 4, 629–635. [Google Scholar] [CrossRef]
- Chen, X.; Kopecky, D.J.; Mihalic, J.; Jeffries, S.; Min, X.; Heath, J.; Deignan, J.; Lai, S.; Fu, Z.; Guimaraes, C.; et al. Structure-guided design, synthesis, and evaluation of guanine-derived inhibitors of the eIF4E mRNA-cap interaction. J. Med. Chem. 2012, 55, 3837–3851. [Google Scholar] [CrossRef]
- Surry, D.S.; Buchwald, S.L. Biaryl phosphane ligands in palladium-catalyzed amination. Angew. Chem. Int. Ed. 2008, 47, 6338–6361. [Google Scholar] [CrossRef]
- Onouchi, H.; Miyagawa, T.; Furuko, A.; Maeda, K.; Yashima, E. Enantioselective esterification of prochiral phosphonate pendants of a polyphenylacetylene assisted by macromolecular helicity: Storage of a dynamic macromolecular helicity memory. J. Am. Chem. Soc. 2005, 127, 2960–2965. [Google Scholar] [CrossRef]
- Demmer, C.S.; Krogsgaard-Larsen, N.; Bunch, L. Review on modern advances of chemical methods for the introduction of a phosphonic acid group. Chem. Rev. 2011, 111, 7981–8006. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.; Peng, L.; Wang, G.; Wang, F.; Luo, Y.; Lei, A. Visible light mediated aerobic radical C–H phosphorization toward arylphosphonates. Org. Chem. Front. 2016, 3, 749–752. [Google Scholar] [CrossRef]
- Shaikh, R.S.; Ghosh, I.; König, B. Direct C–H Phosphonylation of Electron-Rich Arenes and Heteroarenes by Visible-Light Photoredox Catalysis. Chem. Eur. J. 2017, 23, 12120–12124. [Google Scholar] [CrossRef]
- Kirk, K.L. Fluorination in Medicinal Chemistry: Methods, Strategies, and Recent Developments. Org. Process Res. Dev. 2008, 12, 305–321. [Google Scholar] [CrossRef]
- Shinada, N.K.; De Brevern, A.G.; Schmidtke, P. Halogens in Protein-Ligand Binding Mechanism: A Structural Perspective. J. Med. Chem. 2019, 62, 9341–9356. [Google Scholar] [CrossRef] [PubMed]
- Kee, C.W.; Chin, K.F.; Wong, M.W.; Tan, C.H. Selective fluorination of alkyl C–H bonds via photocatalysis. Chem. Commun. 2014, 50, 8211–8214. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.B.; Zhu, C.; Chen, C. Visible light-promoted metal-free sp3-C–H fluorination. Chem. Commun. 2014, 50, 11701–11704. [Google Scholar] [CrossRef]
- Egami, H.; Masuda, S.; Kawato, Y.; Hamashima, Y. Photofluorination of Aliphatic C–H Bonds Promoted by the Phthalimide Group. Org. Lett. 2018, 20, 1367–1370. [Google Scholar] [CrossRef]
- Xia, J.B.; Zhu, C.; Chen, C. Visible light-promoted metal-free C–H activation: Diarylketone-catalyzed selective benzylic mono- and difluorination. J. Am. Chem. Soc. 2013, 135, 17494–17500. [Google Scholar] [CrossRef]
- Bloom, S.; McCann, M.; Lectka, T. Photocatalyzed benzylic fluorination: Shedding “light” on the involvement of electron transfer. Org. Lett. 2014, 16, 6338–6341. [Google Scholar] [CrossRef]
- Bume, D.D.; Pitts, C.R.; Jokhai, R.T.; Lectka, T. Direct, visible light-sensitized benzylic C–H fluorination of peptides using dibenzosuberenone: Selectivity for phenylalanine-like residues. Tetrahedron 2016, 72, 6031–6036. [Google Scholar] [CrossRef]
- Halperin, S.D.; Fan, H.; Chang, S.; Martin, R.E.; Britton, R. A convenient photocatalytic fluorination of unactivated C–H bonds. Angew. Chem. Int. Ed. 2014, 53, 4690–4693. [Google Scholar] [CrossRef] [PubMed]
- Nukuna, B.N.; Goshe, M.B.; Anderson, V.E. Sites of hydroxyl radical reaction with amino acids identified by 2H NMR detection of induced 1H/2H exchange. J. Am. Chem. Soc. 2001, 123, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Watts, Z.I.; Easton, C.J. Peculiar stability of amino acids and peptides from a radical perspective. J. Am. Chem. Soc. 2009, 131, 11323–11325. [Google Scholar] [CrossRef] [PubMed]
- Halperin, S.D.; Kwon, D.; Holmes, M.; Regalado, E.L.; Campeau, L.C.; DiRocco, D.A.; Britton, R. Development of a Direct Photocatalytic C–H Fluorination for the Preparative Synthesis of Odanacatib. Org. Lett. 2015, 17, 5200–5203. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Nodwell, M.B.; Yang, H.; Malik, N.; Merkens, H.; Benard, F.; Martin, R.E.; Schaffer, P.; Britton, R. Site-Selective, Late-Stage C–H 18F-Fluorination on Unprotected Peptides for Positron Emission Tomography Imaging. Angew. Chem. Int. Ed. 2018, 57, 12733–12736. [Google Scholar] [CrossRef]
- Short, M.A.; Shehata, M.F.; Sanders, M.A.; Roizen, J.L. Sulfamides direct radical-mediated chlorination of aliphatic C–H bonds. Chem. Sci. 2020, 11, 217–223. [Google Scholar] [CrossRef]
































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
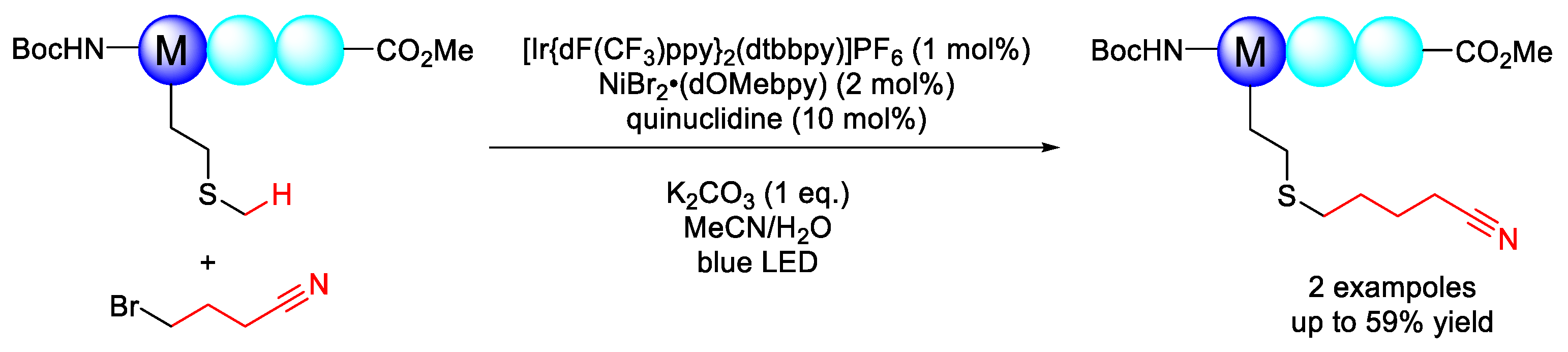{kind=link}
{kind=link}
{kind=link}
{kind=link}
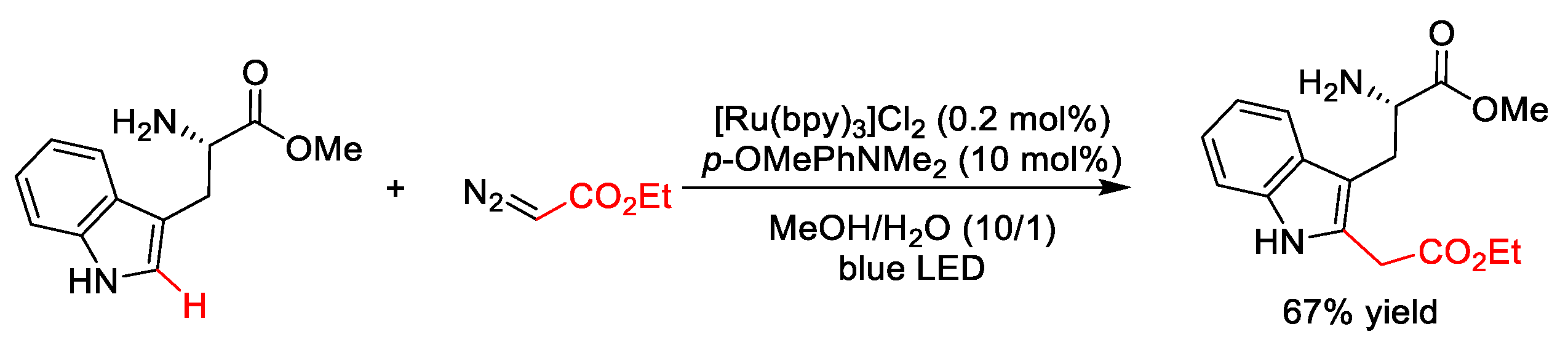{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
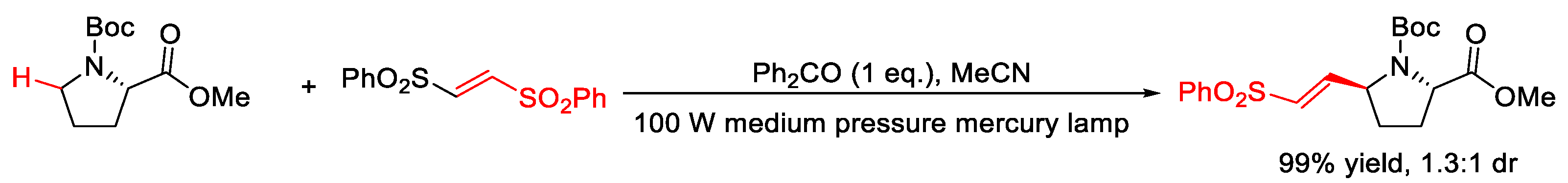{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
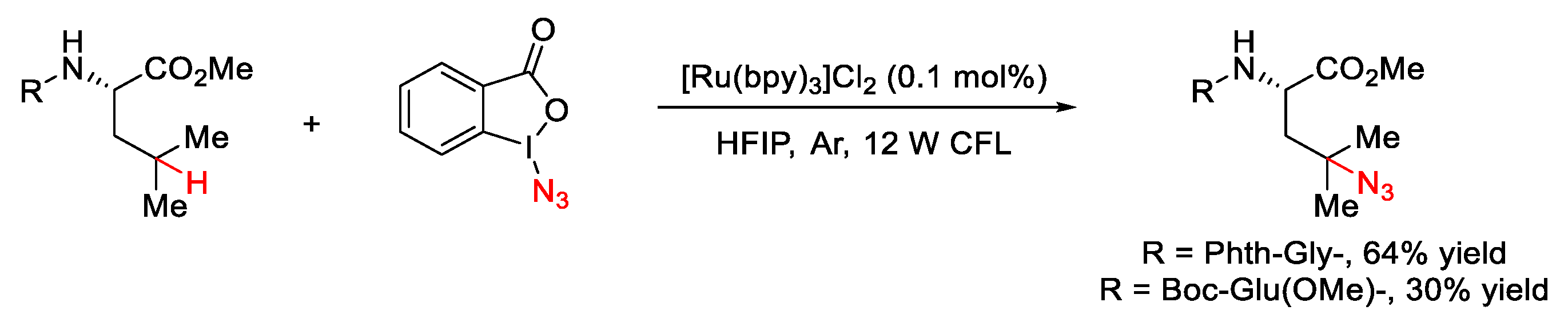{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
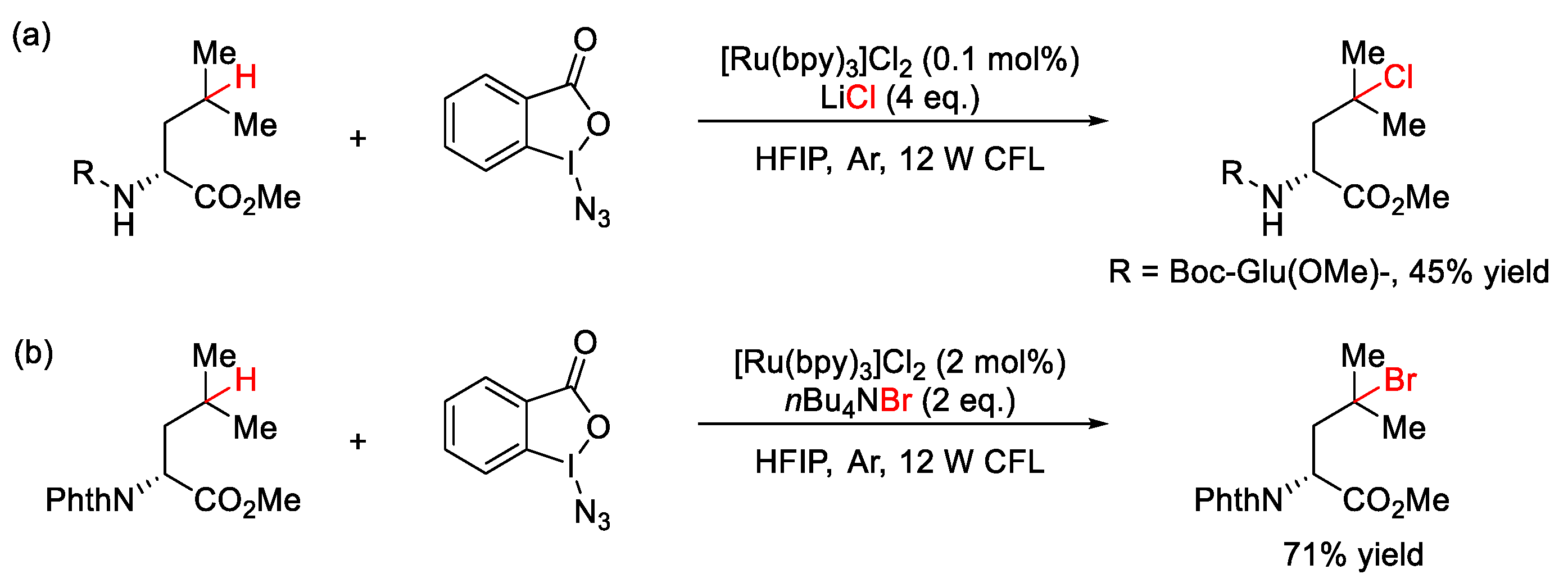{kind=link}
{kind=link}
 | |||||
| Entry | Substrates | Products | Conditions | Yields | Ref. |
| 1 |  |  | AQN (2 mol%) O2 free MeCN 11 W CFL | 34% | [125] |
| 2 | acetophenone (20 mol%) MeCN 19 W CFL | 85% | [126] | ||
| 3 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 (5 mol%) Cs2CO3 (1 eq.) MeCN/H2O blue LED | 5 examples up to 56% | [77] | ||
| 4 | MeCN LED (365 nm) | 45% | [127] | ||
| 5 |  |  | 9-fluorenone (5 mol%) MeCN 11 W CFL | 52% 1:1 dr | [128] |
| 6 | TCB (10 mol%) MeCN hv (302 nm) | 62% 1:1 dr | [129] | ||
| 7 | MeCN LED (365 nm) | 43% 1:1 dr | [127] | ||
| 8 |  |  | MeCN hv (365 nm) | 70% | [127] |
| 9 |  |  | Ir[dF(CF3)ppy]2(dtbbpy)PF6 (5 mol%) Cs2CO3 (1 eq.) MeCN/H2O blue LED | 39% 1.5:1 dr | [77] |
| 10 |  |  | Ir[dF(CF3)ppy]2(dtbbpy)PF6 (5 mol%) Cs2CO3 (1 eq.) MeCN/H2O blue LED | 25% | [77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Z.; Liu, X.; Liu, C.; Zhang, Y.; Rao, Y. Recent Advances in Rapid Synthesis of Non-proteinogenic Amino Acids from Proteinogenic Amino Acids Derivatives via Direct Photo-Mediated C–H Functionalization. Molecules 2020, 25, 5270. https://doi.org/10.3390/molecules25225270
Yuan Z, Liu X, Liu C, Zhang Y, Rao Y. Recent Advances in Rapid Synthesis of Non-proteinogenic Amino Acids from Proteinogenic Amino Acids Derivatives via Direct Photo-Mediated C–H Functionalization. Molecules. 2020; 25(22):5270. https://doi.org/10.3390/molecules25225270
Chicago/Turabian StyleYuan, Zhenbo, Xuanzhong Liu, Changmei Liu, Yan Zhang, and Yijian Rao. 2020. "Recent Advances in Rapid Synthesis of Non-proteinogenic Amino Acids from Proteinogenic Amino Acids Derivatives via Direct Photo-Mediated C–H Functionalization" Molecules 25, no. 22: 5270. https://doi.org/10.3390/molecules25225270
APA StyleYuan, Z., Liu, X., Liu, C., Zhang, Y., & Rao, Y. (2020). Recent Advances in Rapid Synthesis of Non-proteinogenic Amino Acids from Proteinogenic Amino Acids Derivatives via Direct Photo-Mediated C–H Functionalization. Molecules, 25(22), 5270. https://doi.org/10.3390/molecules25225270