
Dimeric and Multimeric DNA Aptamers for Highly Effective Protein Recognition





Abstract
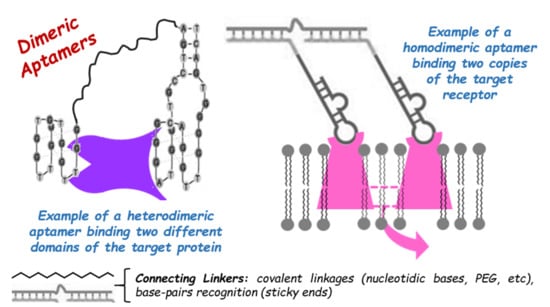
1. Introduction
2. Anti-Inflammatory Aptamers
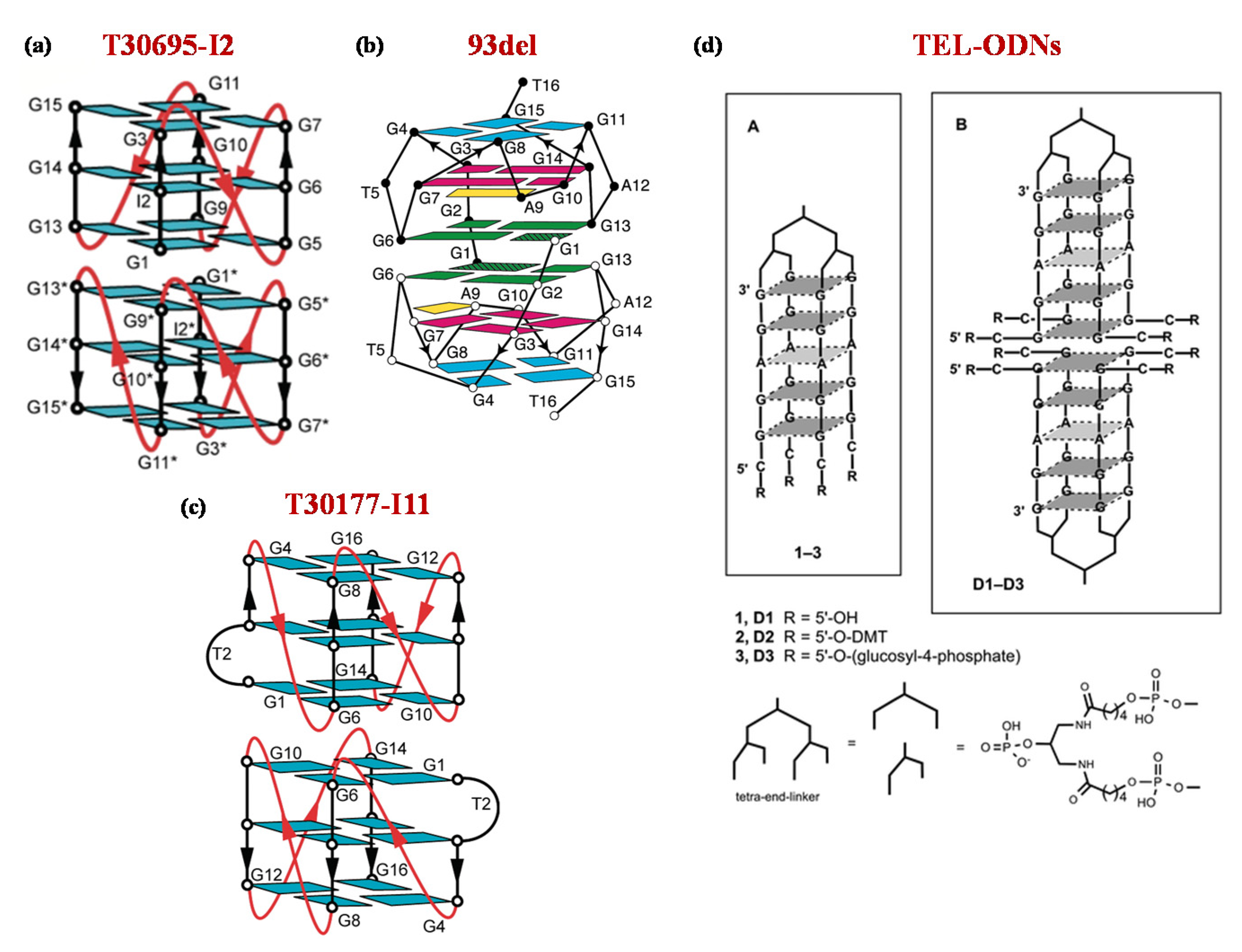
3. Antiviral Aptamers
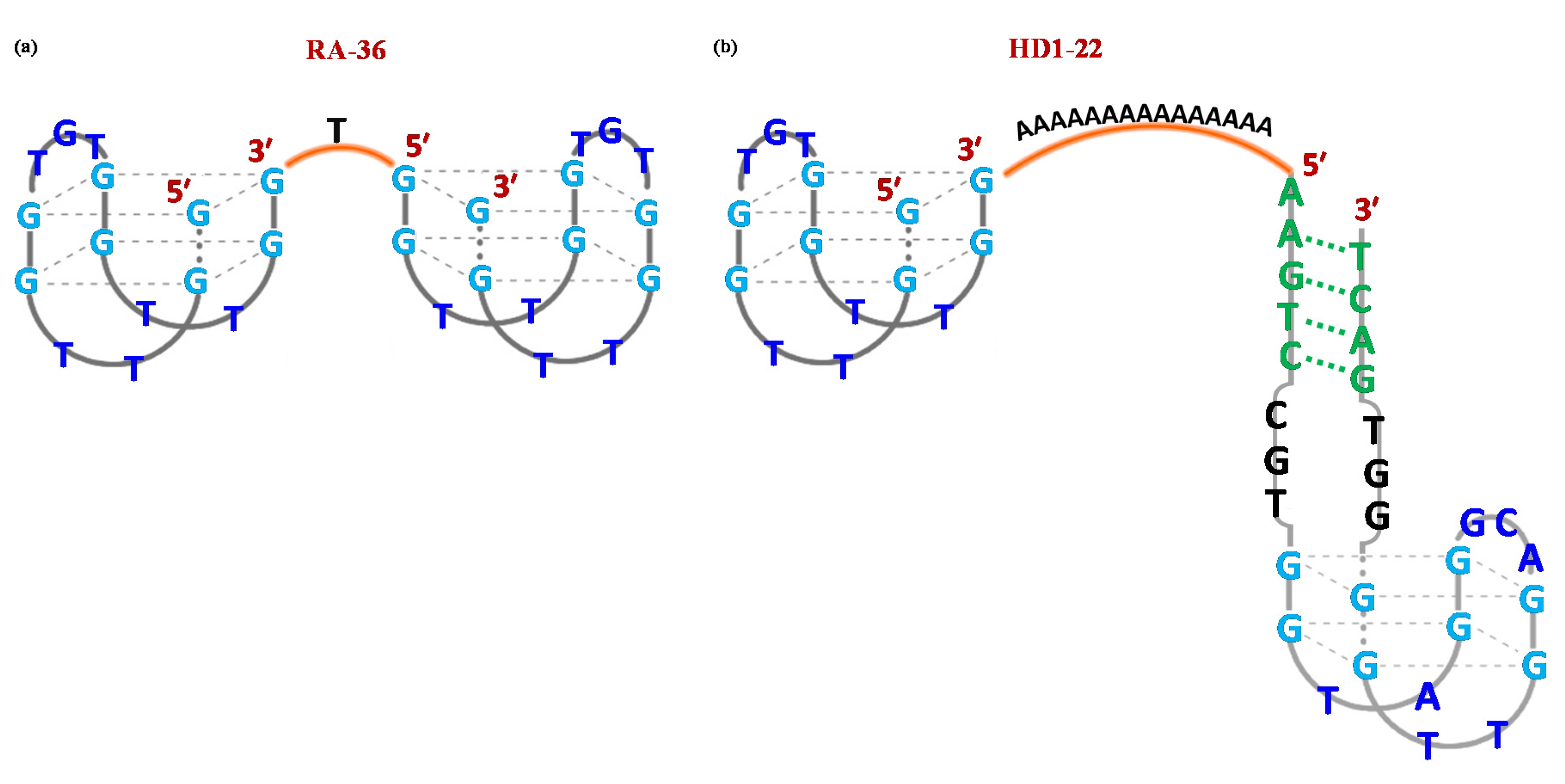
4. Anticoagulant Aptamers
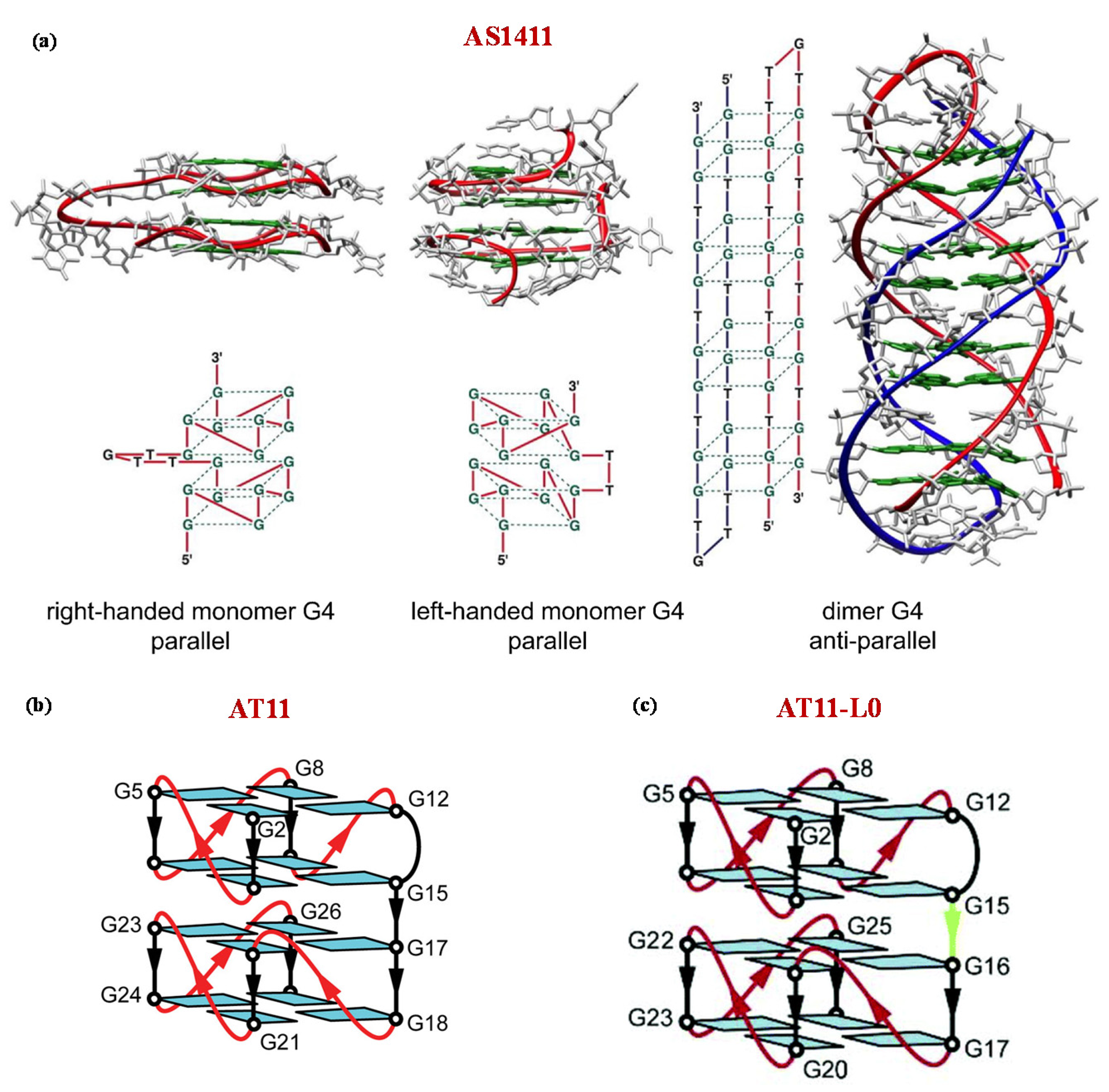
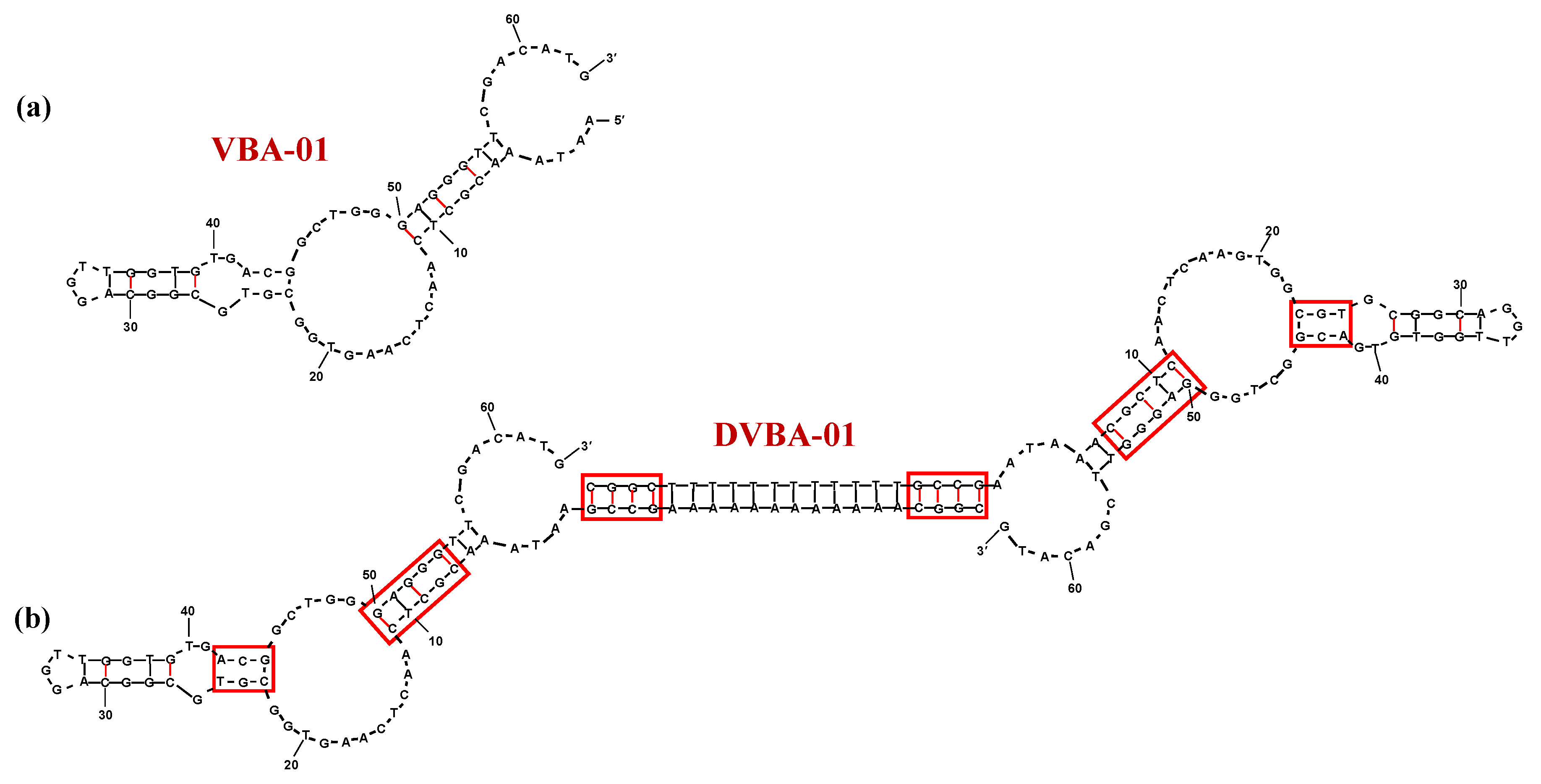
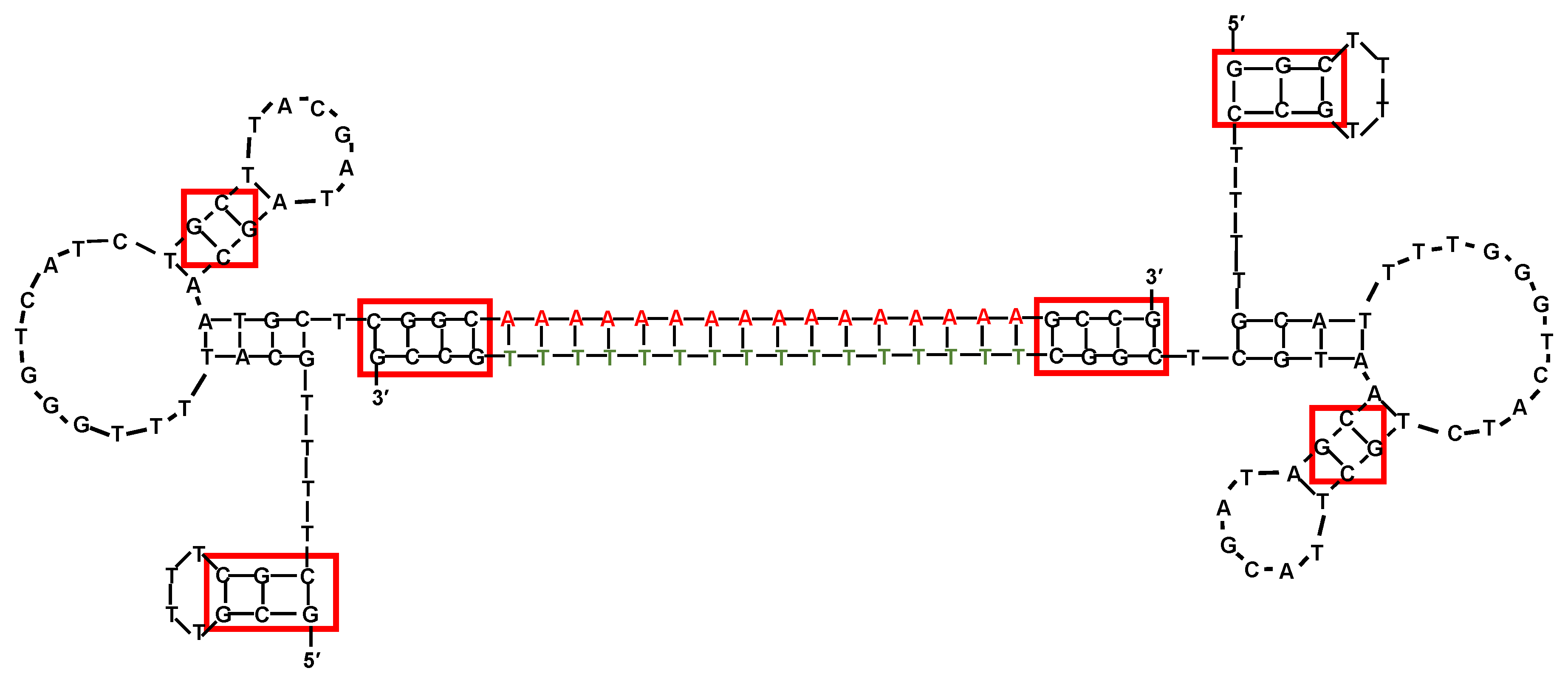
5. Anticancer Aptamers
5.1. Receptor Tyrosine Kinase PTK7
5.2. Nucleolin
5.3. Vitronectin
5.4. Prostate-Specific Membrane Antigen (PMSA)
5.5. Membrane-Bound Immunoglobulins M (mIgM)
5.6. T Cell Receptor Cluster of Differentiation 3 (TCR-CD3)
5.7. Vascular Endothelial Growth Factor (VEGF)
5.8. Hepatocyte Growth Factor Receptor (HGFR)
6. Conclusions
Funding
Conflicts of Interest
References
- Wang, R.E.; Wu, H.; Niu, Y.; Cai, J. Improving the stability of aptamers by chemical modification. Curr. Med. Chem. 2011, 18, 4126–4138. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liang, C.; Lv, Q.; Li, D.; Xu, X.; Liu, B.; Lu, A.; Zhang, G. Molecular selection, modification and development of therapeutic oligonucleotide aptamers. Int. J. Mol. Sci. 2016, 17, 358. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Yao, H.; Wang, L.; Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Chemical modifications of nucleic acid aptamers for therapeutic purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef] [PubMed]
- Röthlisberger, P.; Hollenstein, M. Aptamer chemistry. Adv. Drug Deliv. Rev. 2018, 134, 3–21. [Google Scholar] [CrossRef]
- Adachi, T.; Nakamura, Y. Aptamers: A review of their chemical properties and modifications for therapeutic application. Molecules 2019, 24, 4229. [Google Scholar] [CrossRef]
- Odeh, F.; Nsairat, H.; Alshaer, W.; Ismail, M.A.; Esawi, E.; Qaqish, B.; Al Bawab, A.; Ismail, S.I. Aptamers chemistry: Chemical modifications and conjugation strategies. Molecules 2020, 25, 3. [Google Scholar] [CrossRef]
- Parashar, A. Aptamers in therapeutics. J. Clin. Diagn. Res. 2016, 10, BE01–BE06. [Google Scholar] [CrossRef]
- Maier, K.E.; Levy, M. From selection hits to clinical leads: Progress in aptamer discovery. Mol. Ther. Methods Clin. Dev. 2016, 5, 16014–16023. [Google Scholar] [CrossRef]
- Nimjee, S.M.; White, R.R.; Becker, R.C.; Sullenger, B.A. Aptamers as therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 61–79. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef]
- Ismail, S.I.; Alshaer, W. Therapeutic aptamers in discovery, preclinical and clinical stages. Adv. Drug Deliv. Rev. 2018, 134, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Leslie, M.; Kameyama, H.; Volk, D.E.; Tanaka, T. Aptamer therapeutics in cancer: Current and future. Cancers 2018, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.H.; Elsherbiny, M.E.; Emara, M. Updates on aptamer research. Int. J. Mol. Sci. 2019, 20, 2511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lai, B.S.; Juhas, M. Recent advances in aptamer discovery and applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, C.; Larcher, L.M.; Barrero, R.A.; Veedu, R.N. Three decades of nucleic acid aptamer technologies: Lessons learned, progress and opportunities on aptamer development. Biotechnol. Adv. 2019, 37, 28–50. [Google Scholar] [CrossRef]
- Santosh, B.; Yadava, P.K. Nucleic acid aptamers: Research tools in disease diagnostics and therapeutics. Biomed. Res. Int. 2014, 2014, 50451–50464. [Google Scholar] [CrossRef]
- Ma, H.; Liu, J.; Ali, M.M.; Mahmood, M.A.I.; Labanieh, L.; Lu, M.; Iqbal, S.M.; Zhang, Q.; Zhao, W.; Wan, Y. Nucleic acid aptamers in cancer research, diagnosis and therapy. Chem. Soc. Rev. 2015, 44, 1240–1256. [Google Scholar] [CrossRef]
- Ku, T.H.; Zhang, T.; Luo, H.; Yen, T.M.; Chen, P.W.; Han, Y.; Lo, Y.H. Nucleic acid aptamers: An emerging tool for biotechnology and biomedical sensing. Sensors 2015, 15, 16281–16313. [Google Scholar] [CrossRef]
- Chandola, C.; Kalme, S.; Casteleijn, M.G.; Urtti, A.; Neerathilingam, M. Application of aptamers in diagnostics, drug-delivery and imaging. J. Biosci. 2016, 41, 535–561. [Google Scholar] [CrossRef]
- Musumeci, D.; Platella, C.; Riccardi, C.; Moccia, F.; Montesarchio, D. Fluorescence sensing using DNA aptamers in cancer research and clinical diagnostics. Cancers 2017, 9, 174. [Google Scholar] [CrossRef]
- Dhiman, A.; Kalra, P.; Bansal, V.; Bruno, J.G.; Sharma, T.K. Aptamer-based point-of-care diagnostic platforms. Sens. Actuators B Chem. 2017, 246, 535–553. [Google Scholar] [CrossRef]
- Hori, S.I.; Herrera, A.; Rossi, J.J.; Zhou, J. Current advances in aptamers for cancer diagnosis and therapy. Cancers 2018, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Kulabhusan, P.K.; Hussain, B.; Yüce, M. Current perspectives on aptamers as diagnostic tools and therapeutic agents. Pharmaceutics 2020, 12, 646. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature 1992, 355, 850–852. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX-A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef]
- Keefe, A.D.; Cload, S.T. SELEX with modified nucleotides. Curr. Opin. Chem. Biol. 2008, 12, 448–456. [Google Scholar] [CrossRef]
- Darmostuk, M.; Rimpelova, S.; Gbelcova, H.; Ruml, T. Current approaches in SELEX: An update to aptamer selection technology. Biotechnol. Adv. 2014, 33, 1141–1161. [Google Scholar] [CrossRef]
- Wu, Y.X.; Kwon, Y.J. Aptamers: The “evolution” of SELEX. Methods 2016, 106, 21–28. [Google Scholar] [CrossRef]
- Antipova, O.M.; Zavyalova, E.G.; Golovin, A.V.; Pavlova, G.V.; Kopylov, A.M.; Reshetnikov, R.V. Advances in the application of modified nucleotides in SELEX technology. Biochemistry 2018, 83, 1161–1172. [Google Scholar] [CrossRef]
- Bayat, P.; Nosrati, R.; Alibolandi, M.; Rafatpanah, H.; Abnous, K.; Khedri, M.; Ramezani, M. SELEX methods on the road to protein targeting with nucleic acid aptamers. Biochimie 2018, 154, 132–155. [Google Scholar] [CrossRef] [PubMed]
- Sola, M.; Menon, A.; Moreno, B.; Meraviglia-Crivelli, D.; Soldevilla, M.; Cartón-García, F.; Pastor, F. Aptamers against live targets: Is in vivo SELEX finally coming to edge? Mol. Ther. Nucleic Acids 2020, 21, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Vandghanooni, S.; Eskandani, M.; Barar, J.; Omidi, Y. Recent advances in aptamer-armed multimodal theranostic nanosystems for imaging and targeted therapy of cancer. Eur. J. Pharm. Sci. 2018, 117, 301–312. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Wen, N.; Xiao, D.; Yan, J.; Xiong, H.; Cai, S.; Liu, Z.; Liu, Y. Aptamer-based targeted drug delivery systems: Current potential and challenges. Curr. Med. Chem. 2020, 27, 2189–2219. [Google Scholar] [CrossRef] [PubMed]
- Broude, N.E. Stem-loop oligonucleotides: A robust tool for molecular biology and biotechnology. Trends Biotechnol. 2002, 20, 249–256. [Google Scholar] [CrossRef]
- Musumeci, D.; Riccardi, C.; Montesarchio, D. G-quadruplex forming oligonucleotides as anti-HIV agents. Molecules 2015, 20, 17511–17532. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S. Quadruplex nucleic acids as novel therapeutic targets. J. Med. Chem. 2016, 59, 5987–6011. [Google Scholar] [CrossRef]
- Platella, C.; Riccardi, C.; Montesarchio, D.; Roviello, G.N.; Musumeci, D. G-quadruplex-based aptamers against protein targets in therapy and diagnostics. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1429–1447. [Google Scholar] [CrossRef]
- Kwok, C.K.; Merrick, C.J. G-Quadruplexes: Prediction, characterization, and biological application. Trends Biotechnol. 2017, 35, 997–1013. [Google Scholar] [CrossRef]
- Roxo, C.; Kotkowiak, W.; Pasternak, A. G-quadruplex forming aptamers characteristics, applications, and perspectives. Molecules 2019, 24, 3781. [Google Scholar] [CrossRef]
- Gatto, B.; Palumbo, M.; Sissi, C. Nucleic acid aptamers based on the G-quadruplex structure: Therapeutic and diagnostic potential. Curr. Med. Chem. 2009, 16, 1248–1265. [Google Scholar] [CrossRef]
- Collie, G.W.; Parkinson, G.N. The application of DNA and RNA G-quadruplexes to therapeutic medicines. Chem. Soc. Rev. 2011, 40, 5867–5892. [Google Scholar] [CrossRef]
- Dolinnaya, N.G.; Ogloblina, A.M.; Yakubovskaya, M.G. Structure, properties, and biological relevance of the DNA and RNA G-quadruplexes: Overview 50 years after their discovery. Biochemistry 2016, 81, 1602–1649. [Google Scholar] [CrossRef]
- Tucker, W.O.; Shum, K.T.; Tanner, J.A. G-quadruplex DNA aptamers and their ligands: Structure, function and application. Curr. Pharm. Des. 2012, 18, 2014–2026. [Google Scholar] [CrossRef]
- Bhattacharyya, D.; Arachchilage, G.M.; Basu, S. Metal cations in G-quadruplex folding and stability. Front. Chem. 2016, 4, 38. [Google Scholar] [CrossRef]
- Largy, E.; Marchand, A.; Amrane, S.; Gabelica, V.; Mergny, J.L. Quadruplex turncoats: Cation-dependent folding and stability of quadruplex-DNA double switches. J. Am. Chem. Soc. 2016, 138, 2780–2792. [Google Scholar] [CrossRef]
- Largy, E.; Mergny, J.L.; Gabelica, V. Role of alkali metal ions in G-quadruplex nucleic acid structure and stability. Met. Ions Life Sci. 2016, 16, 203–258. [Google Scholar] [CrossRef]
- Musumeci, D.; Montesarchio, D. Polyvalent nucleic acid aptamers and modulation of their activity: A focus on the thrombin binding aptamer. Pharmacol. Ther. 2012, 136, 202–215. [Google Scholar] [CrossRef]
- Vorobyeva, M.; Vorobjev, P.; Venyaminova, A. Multivalent aptamers: Versatile tools for diagnostic and therapeutic applications. Molecules 2016, 21, 1613. [Google Scholar] [CrossRef]
- Gao, S.; Zheng, X.; Jiao, B.; Wang, L. Post-SELEX optimization of aptamers. Anal. Bioanal. Chem. 2016, 408, 4567–4573. [Google Scholar] [CrossRef]
- Hasegawa, H.; Savory, N.; Abe, K.; Ikebukuro, K. Methods for improving aptamer binding affinity. Molecules 2016, 21, 421. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Nelson, R.M. Selectins. J. Clin. Investig. 1993, 91, 379–387. [Google Scholar] [CrossRef]
- Rosen, S.D.; Bertozzi, C.R. The selectins and their ligands. Curr. Opin. Cell Biol. 1994, 6, 663–673. [Google Scholar] [CrossRef]
- Rainer, T.H. L-selectin in health and disease. Resuscitation 2002, 52, 127–141. [Google Scholar] [CrossRef]
- Raffler, N.A.; Rivera-Nieves, J.; Ley, K. L-selectin in inflammation, infection and immunity. Drug Discov. Today Ther. Strateg. 2005, 2, 213–220. [Google Scholar] [CrossRef]
- Ivetic, A. A head-to-tail view of L-selectin and its impact on neutrophil behaviour. Cell Tissue Res. 2018, 371, 437–453. [Google Scholar] [CrossRef]
- Ivetic, A.; Green, H.L.H.; Hart, S.J. L-selectin: A major regulator of leukocyte adhesion, migration and signaling. Front. Immunol. 2019, 14, 1068. [Google Scholar] [CrossRef]
- Watson, S.R.; Chang, Y.F.; O’Connell, D.; Weigand, L.; Ringquist, S.; Parma, D.H. Anti-L-selectin aptamers: Binding characteristics, pharmacokinetic parameters, and activity against an intravascular target in vivo. Antisense Nucleic Acid Drug Dev. 2000, 10, 63–75. [Google Scholar] [CrossRef]
- Hicke, B.J.; Watson, S.R.; Koenig, A.; Lynott, C.K.; Bargatze, R.F.; Chang, Y.F.; Ringquist, S.; Moon-McDermott, L.; Jennings, S.; Fitzwater, T.; et al. DNA aptamers block L-selectin function in vivo: Inhibition of human lymphocyte trafficking in SCID mice. J. Clin. Investig. 1996, 98, 2688–2692. [Google Scholar] [CrossRef]
- Romig, T.S.; Bell, C.; Drolet, D.W. Aptamer affinity chromatography: Combinatorial chemistry applied to protein purification. J. Chromatogr. B Biomed. Sci. Appl. 1999, 731, 275–284. [Google Scholar] [CrossRef]
- Riese, S.B.; Buscher, K.; Enders, S.; Kuehne, C.; Tauber, R.; Dernedde, J. Structural requirements of mono- and multivalent L-selectin blocking aptamers for enhanced receptor inhibition in vitro and in vivo. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 901–908. [Google Scholar] [CrossRef]
- Chang, E.K.; Eckert, M.A.; Ali, M.M.; Riazifar, H.; Pone, E.J.; Liu, L.; Zhao, W. Facile supermolecular aptamer inhibitors of L-selectin. PLoS ONE 2015, 10, e0123034. [Google Scholar] [CrossRef]
- Ali, M.M.; Kang, D.-K.; Zhao, W.; Li, F.; Zhang, Z.; Zhang, K.; Le, X.C.; Ankrum, J.A. Rolling circle amplification: A versatile tool for chemical biology, materials science and medicine. Chem. Soc. Rev. 2014, 43, 3324–3341. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Bala, J.; Chinnapaiyan, S.; Dutta, R.K.; Unwalla, H. Aptamers in HIV research diagnosis and therapy. RNA Biol. 2018, 15, 327–337. [Google Scholar] [CrossRef]
- Ruggiero, E.; Richter, S.N. Survey and summary G-quadruplexes and G-quadruplex ligands: Targets and tools in antiviral therapy. Nucleic Acids Res. 2018, 46, 3270–3283. [Google Scholar] [CrossRef]
- Zou, X.; Wu, J.; Gu, J.; Shen, L.; Mao, L. Application of aptamers in virus detection and antiviral therapy. Front. Microbiol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- De Soultrait, V.R.; Lozach, P.Y.; Altmeyer, R.; Tarrago-Litvak, L.; Litvak, S.; Andréola, M.L. DNA aptamers derived from HIV-1 RNase H inhibitors are strong anti-integrase agents. J. Mol. Biol. 2002, 324, 195–203. [Google Scholar] [CrossRef]
- Andreola, M.-L. Closely related antiretroviral agents as inhibitors of two HIV-1 enzymes, ribonuclease H and integrase: “Killing two birds with one stone”. Curr. Pharm. Des. 2005, 10. [Google Scholar] [CrossRef]
- Métifiot, M.; Leon, O.; Tarrago-Litvak, L.; Litvak, S.; Andréola, M.L. Targeting HIV-1 integrase with aptamers selected against the purified RNase H domain of HIV-1 RT. Biochimie 2005, 87, 911–919. [Google Scholar] [CrossRef]
- Ojwang, J.O.; Buckheit, R.W.; Pommier, Y.; Mazumder, A.; De Vreese, K.; Este, J.A.; Reymen, D.; Pallansch, L.A.; Lackman-Smith, C.; Wallace, T.L.; et al. T30177, an oligonucleotide stabilized by an intramolecular guanosine octet, is a potent inhibitor of laboratory strains and clinical isolates of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 1995, 39, 2426–2435. [Google Scholar] [CrossRef]
- Mazumder, A.; Neamati, N.; Ojwang, J.O.; Sunder, S.; Rando, R.F.; Pommier, Y. Inhibition of the human immunodeficiency virus type 1 integrase by guanosine quartet structures. Biochemistry 1996, 35, 13762–13771. [Google Scholar] [CrossRef]
- Jing, N.; Rando, R.F.; Pommier, Y.; Hogan, M.E. Ion selective folding of loop domains in a potent anti-HIV oligonucleotide. Biochemistry 1997, 36, 12498–12505. [Google Scholar] [CrossRef]
- Jing, N.; Hogan, M.E. Structure-activity of tetrad-forming oligonucleotides as a potent anti-HIV therapeutic drug. J. Biol. Chem. 1998, 273, 34992–34999. [Google Scholar] [CrossRef]
- Do, N.Q.; Lim, K.W.; Teo, M.H.; Heddi, B.; Phan, A.T. Stacking of G-quadruplexes: NMR structure of a G-rich oligonucleotide with potential anti-HIV and anticancer activity. Nucleic Acids Res. 2011, 39, 9448–9457. [Google Scholar] [CrossRef]
- Phan, A.T.; Kuryavyi, V.; Ma, J.B.; Faure, A.; Andréola, M.L.; Patel, D.J. An interlocked dimeric parallel-stranded DNA quadruplex: A potent inhibitor of HIV-1 integrase. Proc. Natl. Acad. Sci. USA 2005, 102, 634–639. [Google Scholar] [CrossRef]
- Mukundan, V.T.; Do, N.Q.; Phan, A.T. HIV-1 integrase inhibitor T30177 forms a stacked dimeric G-quadruplex structure containing bulges. Nucleic Acids Res. 2011, 39, 8984–8991. [Google Scholar] [CrossRef]
- Nici, F.; Oliviero, G.; Falanga, A.P.; D’Errico, S.; Marzano, M.; Musumeci, D.; Montesarchio, D.; Noppen, S.; Pannecouque, C.; Piccialli, G.; et al. Anti-HIV activity of new higher order G-quadruplex aptamers obtained from tetra-end-linked oligonucleotides. Org. Biomol. Chem. 2018, 16, 2349–2355. [Google Scholar] [CrossRef]
- Borbone, N.; Amato, J.; Oliviero, G.; D’Atri, V.; Gabelica, V.; De Pauw, E.; Piccialli, G.; Mayol, L. d(CGGTGGT) forms an octameric parallel G-quadruplex via stacking of unusual G(:C):G(:C):G(:C):G(:C) octads. Nucleic Acids Res. 2011, 39, 7848–7857. [Google Scholar] [CrossRef]
- D’Atri, V.; Borbone, N.; Amato, J.; Gabelica, V.; D’Errico, S.; Piccialli, G.; Mayol, L.; Oliviero, G. DNA-based nanostructures: The effect of the base sequence on octamer formation from d(XGGYGGT) tetramolecular G-quadruplexes. Biochimie 2014, 99, 119–128. [Google Scholar] [CrossRef]
- D’Atri, V.; Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Mayol, L.; Piccialli, V.; Haider, S.; Hoorelbeke, B.; Balzarini, J.; et al. New anti-HIV aptamers based on tetra-end-linked DNA G-quadruplexes: Effect of the base sequence on anti-HIV activity. Chem. Commun. 2012, 48, 9516–9518. [Google Scholar] [CrossRef]
- D’Onofrio, J.; Petraccone, L.; Erra, E.; Martino, L.; Di Fabio, G.; De Napoli, L.; Giancola, C.; Montesarchio, D. 5′-modified G-quadruplex forming oligonucleotides endowed with anti-HIV activity: Synthesis and biophysical properties. Bioconjugate Chem. 2007, 18, 1194–1204. [Google Scholar] [CrossRef]
- Huntington, J.A. Molecular recognition mechanisms of thrombin. J. Thromb. Haemost. 2005, 3, 1861–1872. [Google Scholar] [CrossRef]
- Wolberg, A.S. Thrombin generation and fibrin clot structure. Blood Rev. 2007, 21, 131–142. [Google Scholar] [CrossRef]
- Di Cera, E. Thrombin. Mol. Aspects Med. 2008, 29, 203–254. [Google Scholar] [CrossRef]
- Licari, L.G.; Kovacic, J.P. Thrombin physiology and pathophysiology. J. Vet. Emerg. Crit. Care 2009, 19, 11–22. [Google Scholar] [CrossRef]
- Mazepa, M.; Hoffman, M.; Monroe, D. Superactivated platelets: Thrombus regulators, thrombin generators, and potential clinical targets. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1747–1752. [Google Scholar] [CrossRef]
- Posma, J.J.N.; Posthuma, J.J.; Spronk, H.M.H. Coagulation and non-coagulation effects of thrombin. J. Thromb. Haemost. 2016, 14, 1908–1916. [Google Scholar] [CrossRef]
- Huntington, J.A.; Baglin, T.P. Targeting thrombin rational drug design from natural mechanisms. Trends Pharmacol. Sci. 2003, 24, 589–595. [Google Scholar] [CrossRef]
- Hirsh, J. Current anticoagulant therapy unmet clinical needs. Thromb. Res. 2003, 109, S1–S8. [Google Scholar] [CrossRef]
- Gómez Outes, A.; Suárez Gea, M.L.; Pozo Hernández, C.; Lecumberri, R.; Rocha, E.; Vargas Castrillón, E. New parenteral anticoagulants in development. Ther. Adv. Cardiovasc. Dis. 2011, 5, 33–59. [Google Scholar] [CrossRef]
- Zavyalova, E.G.; Ustinov, N.; Golovin, A.; Pavlova, G.; Kopylov, A. G-quadruplex aptamers to human thrombin versus other direct thrombin inhibitors: The focus on mechanism of action and drug efficiency as anticoagulants. Curr. Med. Chem. 2016, 23, 2230–2244. [Google Scholar] [CrossRef]
- Becker, R.C.; Povsic, T.; Cohen, M.G.; Rusconi, C.P.; Sullenger, B. Nucleic acid aptamers as antithrombotic agents: Opportunities in extracellular therapeutics. Thromb. Haemost. 2010, 103, 586–595. [Google Scholar] [CrossRef]
- Nimjee, S.M.; Povsic, T.J.; Sullenger, B.A.; Becker, R.C. Translation and clinical development of antithrombotic aptamers. Nucleic Acid Ther. 2016, 26, 147–155. [Google Scholar] [CrossRef]
- Ponce, A.T.; Hong, K.L. A mini-review: Clinical development and potential of aptamers for thrombotic events treatment and monitoring. Biomedicines 2019, 7, 55. [Google Scholar] [CrossRef]
- Riccardi, C.; Napolitano, E.; Platella, C.; Musumeci, D.; Montesarchio, D. G-quadruplex-based aptamers targeting human thrombin: Discovery, chemical modifications and antithrombotic effects. Pharmacol. Ther. 2020, 107649. [Google Scholar] [CrossRef]
- Tasset, D.M.; Kubik, M.F.; Steiner, W. Oligonucleotide inhibitors of human thrombin that bind distinct epitopes. J. Mol. Biol. 1997, 272, 688–698. [Google Scholar] [CrossRef]
- Marson, G.; Palumbo, M.; Sissi, C. Folding versus charge: Understanding selective target recognition by the thrombin aptamers. Curr. Pharm. Des. 2012, 18, 2027–2035. [Google Scholar] [CrossRef]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Macaya, R.F.; Schultze, P.; Smith, F.W.; Roe, J.A.; Feigon, J. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl. Acad. Sci. USA 1993, 90, 3745–3749. [Google Scholar] [CrossRef]
- Wang, K.Y.; Bolton, P.H.; McCurdy, S.; Shea, R.G.; Swaminathan, S. A DNA aptamer which binds to and inhibits thrombin exhibits a new structural motif for DNA. Biochemistry 1993, 32, 1899–1904. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.Y.; Bolton, P.H.; Krawczyk, S.H.; Bischofberger, N.; Swaminathan, S. The tertiary structure of a DNA aptamer which binds to and inhibits thrombin determines activity. Biochemistry 1993, 32, 11285–11292. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, K.; Padmanabhan, K.P.; Ferrara, J.D.; Sadler, J.E.; Tulinsky, A. The structure of α-thrombin inhibited by a 15-mer single-stranded DNA aptamer. J. Biol. Chem. 1993, 268, 17651–17654. [Google Scholar] [CrossRef] [PubMed]
- Schultze, P.; Macaya, R.F.; Feigon, J. Three-dimensional solution structure of the thrombin-binding DNA aptamer d(GGTTGGTGTGGTTGG). J. Mol. Biol. 1994, 235, 1532–1547. [Google Scholar] [CrossRef]
- Padmanabhan, K.; Tulinsky, A. An ambiguous structure of a DNA 15-mer thrombin complex. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996, 52, 272–282. [Google Scholar] [CrossRef]
- Kelly, J.A.; Feigon, J.; Yeates, T.O. Reconciliation of the X-ray and NMR structures of the thrombin-binding aptamer d(GGTTGGTGTGGTTGG). J. Mol. Biol. 1996, 256, 417–422. [Google Scholar] [CrossRef]
- Zavyalova, E.G.; Golovin, A.; Reshetnikov, R.; Mudrik, N.; Panteleyev, D.; Pavlova, G.; Kopylov, A. Novel modular DNA aptamer for human thrombin with high anticoagulant activity. Curr. Med. Chem. 2011, 18, 3343–3350. [Google Scholar] [CrossRef]
- Poniková, S.; Tlučková, K.; Antalík, M.; Víglaský, V.; Hianik, T. The circular dichroism and differential scanning calorimetry study of the properties of DNA aptamer dimers. Biophys. Chem. 2011, 155, 29–35. [Google Scholar] [CrossRef]
- Amato, T.; Virgilio, A.; Pirone, L.; Vellecco, V.; Bucci, M.; Pedone, E.; Esposito, V.; Galeone, A. Investigating the properties of TBA variants with twin thrombin binding domains. Sci. Rep. 2019, 9, 9184. [Google Scholar] [CrossRef]
- Müller, J.; Wulffen, B.; Pötzsch, B.; Mayer, G. Multidomain targeting generates a high-affinity thrombin-inhibiting bivalent aptamer. ChemBioChem 2007, 8, 2223–2226. [Google Scholar] [CrossRef]
- Müller, J.; Freitag, D.; Mayer, G.; Pötzsch, B. Anticoagulant characteristics of HD1-22, a bivalent aptamer that specifically inhibits thrombin and prothrombinase. J. Thromb. Haemost. 2008, 6, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Taira, K.I.; Sode, K.; Ikebukuro, K. Improvement of aptamer affinity by dimerization. Sensors 2008, 8, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Heyduk, T. Bivalent ligands with long nanometer-scale flexible linkers. Biochemistry 2009, 48, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Hughes, Q.W.; Le, B.T.; Gilmore, G.; Baker, R.I.; Veedu, R.N. Construction of a bivalent thrombin binding aptamer and its antidote with improved properties. Molecules 2017, 22, 1770. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.M.; Xiao, Y.; Soh, H.T. Selection is more intelligent than design: Improving the affinity of a bivalent ligand through directed evolution. Nucleic Acids Res. 2012, 40, 11777–11783. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Cao, Z.; Tan, W. Molecular assembly for high-performance bivalent nucleic acid inhibitor. Proc. Natl. Acad. Sci. USA 2008, 105, 5664–5669. [Google Scholar] [CrossRef]
- Di Giusto, D.A.; King, G.C. Construction, stability, and activity of multivalent circular anticoagulant aptamers. J. Biol. Chem. 2004, 279, 46483–46489. [Google Scholar] [CrossRef]
- Di Giusto, D.A.; Knox, S.M.; Lai, Y.; Tyrelle, G.D.; Aung, M.T.; King, G.C. Multitasking by multivalent circular DNA aptamers. ChemBioChem 2006, 7, 535–544. [Google Scholar] [CrossRef]
- Hsiao, K.Y.; Sun, H.S.; Tsai, S.J. Circular RNA—New member of noncoding RNA with novel functions. Exp. Biol. Med. 2017, 242, 1136–1141. [Google Scholar] [CrossRef]
- Li, J.; Mohammed-Elsabagh, M.; Paczkowski, F.; Li, Y. Circular nucleic acids: Discovery, functions and applications. ChemBioChem 2020, 22, 1547–1566. [Google Scholar] [CrossRef]
- Riccardi, C.; Meyer, A.; Vasseur, J.J.; Russo Krauss, I.; Paduano, L.; Oliva, R.; Petraccone, L.; Morvan, F.; Montesarchio, D. Stability is not everything: The case of the cyclization of the thrombin binding aptamer. ChemBioChem 2019, 20, 1789–1794. [Google Scholar] [CrossRef]
- Riccardi, C.; Meyer, A.; Vasseur, J.J.; Russo Krauss, I.; Paduano, L.; Morvan, F.; Montesarchio, D. Fine-tuning the properties of the thrombin binding aptamer through cyclization: Effect of the 5′-3′ connecting linker on the aptamer stability and anticoagulant activity. Bioorg. Chem. 2020, 94, 103379. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Meyer, A.; Vasseur, J.J.; Cavasso, D.; Russo Krauss, I.; Paduano, L.; Morvan, F.; Montesarchio, D. Design, synthesis and characterization of cyclic NU172 analogues: A biophysical and biological insight. Int. J. Mol. Sci. 2020, 21, 3860. [Google Scholar] [CrossRef] [PubMed]
- Russo Krauss, I.; Merlino, A.; Giancola, C.; Randazzo, A.; Mazzarella, L.; Sica, F. Thrombin-aptamer recognition: A revealed ambiguity. Nucleic Acids Res. 2011, 39, 7858–7867. [Google Scholar] [CrossRef] [PubMed]
- Russo Krauss, I.; Merlino, A.; Randazzo, A.; Novellino, E.; Mazzarella, L.; Sica, F. High-resolution structures of two complexes between thrombin and thrombin-binding aptamer shed light on the role of cations in the aptamer inhibitory activity. Nucleic Acids Res. 2012, 40, 8119–8128. [Google Scholar] [CrossRef]
- Yigit, M.V.; Mazumdar, D.; Lu, Y. MRI detection of thrombin with aptamer functionalized superparamagnetic iron oxide nanoparticles. Bioconjugate Chem. 2008, 19, 412–417. [Google Scholar] [CrossRef]
- Musumeci, D.; Oliviero, G.; Roviello, G.N.; Bucci, E.M.; Piccialli, G. G-quadruplex-forming oligonucleotide conjugated to magnetic nanoparticles: Synthesis, characterization, and enzymatic stability assays. Bioconjugate Chem. 2012, 23, 382–391. [Google Scholar] [CrossRef]
- Yu, J.; Yang, L.; Liang, X.; Dong, T.; Liu, H. Bare magnetic nanoparticles as fluorescence quenchers for detection of thrombin. Analyst 2015, 140, 4114–4120. [Google Scholar] [CrossRef]
- Shiang, Y.C.; Huang, C.C.; Wang, T.H.; Chien, C.W.; Chang, H.T. Aptamer-conjugated nanoparticles efficiently control the activity of thrombin. Adv. Funct. Mater. 2010, 20, 3175–3182. [Google Scholar] [CrossRef]
- Shiang, Y.C.; Hsu, C.L.; Huang, C.C.; Chang, H.T. Gold nanoparticles presenting hybridized self-assembled aptamers that exhibit enhanced inhibition of thrombin. Angew. Chem. Int. Ed. Eng. 2011, 50, 7660–7665. [Google Scholar] [CrossRef]
- Hsu, C.L.; Chang, H.T.; Chen, C.T.; Wei, S.C.; Shiang, Y.C.; Huang, C.C. Highly efficient control of thrombin activity by multivalent nanoparticles. Chem. A Eur. J. 2011, 17, 10994–11000. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Wei, S.C.; Jian, J.W.; Chang, H.T.; Chen, W.H.; Huang, C.C. Highly flexible and stable aptamer-caged nanoparticles for control of thrombin activity. RSC Adv. 2012, 2, 1577–1584. [Google Scholar] [CrossRef]
- Huang, S.S.; Wei, S.C.; Chang, H.T.; Lin, H.J.; Huang, C.C. Gold nanoparticles modified with self-assembled hybrid monolayer of triblock aptamers as a photoreversible anticoagulant. J. Control. Release 2016, 221, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cui, Y.; He, Q.; Yang, Y.; Fei, J.; Li, J. Selective recognition of co-assembled thrombin aptamer and docetaxel on mesoporous silica nanoparticles against tumor cell proliferation. Chemistry 2011, 17, 13170–13174. [Google Scholar] [CrossRef]
- Babu, E.; Mareeswaran, P.M.; Rajagopal, S. Highly sensitive optical biosensor for thrombin based on structure switching aptamer-luminescent silica nanoparticles. J. Fluoresc. 2013, 23, 137–146. [Google Scholar] [CrossRef]
- Yue, Q.; Shen, T.; Wang, L.; Xu, S.; Li, H.; Xue, Q.; Zhang, Y.; Gu, X.; Zhang, S.; Liu, J. A convenient sandwich assay of thrombin in biological media using nanoparticle-enhanced fluorescence polarization. Biosens. Bioelectron. 2014, 56, 231–236. [Google Scholar] [CrossRef]
- Riccardi, C.; Russo Krauss, I.; Musumeci, D.; Morvan, F.; Meyer, A.; Vasseur, J.J.; Paduano, L.; Montesarchio, D. Fluorescent thrombin binding aptamer-tagged nanoparticles for an efficient and reversible control of thrombin activity. ACS Appl. Mater. Interfaces 2017, 9, 35574–35587. [Google Scholar] [CrossRef]
- Lai, P.X.; Mao, J.Y.; Unnikrishnan, B.; Chu, H.W.; Wu, C.W.; Chang, H.T.; Huang, C.C. Self-assembled, bivalent aptamers on graphene oxide as an efficient anticoagulant. Biomater. Sci. 2018, 6, 1882–1891. [Google Scholar] [CrossRef]
- Lin, T.-X.; Lai, P.-X.; Mao, J.-Y.; Chu, H.-W.; Unnikrishnan, B.; Anand, A.; Huang, C.-C. Supramolecular aptamers on graphene oxide for efficient inhibition of thrombin activity. Front. Chem. 2019, 7. [Google Scholar] [CrossRef]
- Ireson, C.R.; Kelland, L.R. Discovery and development of anticancer aptamers. Mol. Cancer Ther. 2006, 5, 2957–2962. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, H.J.; Heo, K. Therapeutic aptamers: Developmental potential as anticancer drugs. BMB Rep. 2015, 48, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.O.; Kim, G.; Hwang, J.; Han, K.H.; Kwak, M.; Lee, P.C.W. Nucleic acid nanotechnology for cancer treatment. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188377. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Xiang, J. Aptamers, the nucleic acid antibodies, in cancer therapy. Int. J. Mol. Sci. 2020, 21, 2793. [Google Scholar] [CrossRef] [PubMed]
- Barsan, V.; Ramakrishna, S.; Davis, K.L. Immunotherapy for the treatment of acute lymphoblastic leukemia. Curr. Oncol. Rep. 2020, 22, 11. [Google Scholar] [CrossRef]
- Whitehead, T.P.; Metayer, C.; Wiemels, J.L.; Singer, A.W.; Miller, M.D. Childhood leukemia and primary prevention. Curr. Probl. Pediatr. Adolesc. Health Care 2016, 46, 317–352. [Google Scholar] [CrossRef]
- Shangguan, D.; Li, Y.; Tang, Z.; Cao, Z.C.; Chen, H.W.; Mallikaratchy, P.; Sefah, K.; Yang, C.J.; Tan, W. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc. Natl. Acad. Sci. USA 2006, 103, 11838–11843. [Google Scholar] [CrossRef]
- Zhang, Z.; Ali, M.M.; Eckert, M.A.; Kang, D.K.; Chen, Y.Y.; Sender, L.S.; Fruman, D.A.; Zhao, W. A polyvalent aptamer system for targeted drug delivery. Biomaterials 2013, 34, 9728–9735. [Google Scholar] [CrossRef]
- Kuai, H.; Zhao, Z.; Mo, L.; Liu, H.; Hu, X.; Fu, T.; Zhang, X.; Tan, W. Circular bivalent aptamers enable in vivo stability and recognition. J. Am. Chem. Soc. 2017, 139, 9128–9131. [Google Scholar] [CrossRef]
- Fujiki, H.; Watanabe, T.; Suganuma, M. Cell-surface nucleolin acts as a central mediator for carcinogenic, anti-carcinogenic, and disease-related ligands. J. Cancer Res. Clin. Oncol. 2014, 140, 689–699. [Google Scholar] [CrossRef]
- Koutsioumpa, M.; Papadimitriou, E. Cell surface nucleolin as a target for anti-cancer therapies. Recent Pat. Anticancer Drug Discov. 2014, 9, 137–152. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Bambury, R.M.; Van Allen, E.M.; Drabkin, H.A.; Lara, P.N.J.; Harzstark, A.L.; Wagle, N.; Figlin, R.A.; Smith, G.W.; Garraway, L.A.; et al. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Investig. New Drugs 2014, 32, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Kahlon, J.B.; Thomas, S.D.; Trent, J.O.; Miller, D.M. Antiproliferative activity of G-rich oligonucleotides correlates with protein binding. J. Biol. Chem. 1999, 274, 26369–26377. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Reyes-reyes, E.M.; Malik, M.T.; Murphy, E.M.; Toole, M.G.O.; Trent, J.O. G-quadruplex oligonucleotide AS1411 as a cancer-targeting agent: Uses and mechanisms. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1414–1428. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Zeng, X.; Wu, J.; Zhu, X.; Yu, X.; Zhang, X.; Zhang, J.; Liu, G.; Mei, L. Polydopamine-based surface modification of novel nanoparticle-aptamer bioconjugates for in vivo breast cancer targeting and enhanced therapeutic effects. Theranostics 2016, 6, 470–484. [Google Scholar] [CrossRef]
- Xu, G.; Yu, X.; Zhang, J.; Sheng, Y.; Liu, G.; Tao, W.; Mei, L. Robust aptamer–polydopamine-functionalized M-PLGA–TPGS nanoparticles for targeted delivery of docetaxel and enhanced cervical cancer therapy. Int. J. Nanomed. 2016, 11, 2953–2965. [Google Scholar] [CrossRef]
- Chen, D.; Li, B.; Cai, S.; Wang, P.; Peng, S.; Sheng, Y.; He, Y.; Gu, Y.; Chen, H. Dual targeting luminescent gold nanoclusters for tumor imaging and deep tissue therapy. Biomaterials 2016, 100, 1–16. [Google Scholar] [CrossRef]
- Riccardi, C.; Fàbrega, C.; Grijalvo, S.; Vitiello, G.; D’Errico, G.; Eritja, R.; Montesarchio, D. AS1411-decorated niosomes as effective nanocarriers for Ru(III)-based drugs in anticancer strategies. J. Mater. Chem. B 2018, 6, 5368–5384. [Google Scholar] [CrossRef]
- Yang, S.; Ren, Z.; Chen, M.; Wang, Y.; You, B.; Chen, W.; Qu, C.; Liu, Y.; Zhang, X. Nucleolin targeting AS1411-aptamer-modified graft polymeric micelle with dual pH/redox sensitivity designed to enhance tumor therapy through the codelivery of doxorubicin/TLR4 siRNA and suppression of invasion. Mol. Pharm. 2018, 15, 314–325. [Google Scholar] [CrossRef]
- Chen, W.H.; Yang Sung, S.; Fadeev, M.; Cecconello, A.; Nechushtai, R.; Willner, I. Targeted VEGF-triggered release of an anti-cancer drug from aptamer-functionalized metal–organic framework nanoparticles. Nanoscale 2018, 25. [Google Scholar] [CrossRef]
- Dailey, M.M.; Clarke Miller, M.; Bates, P.J.; Lane, A.N.; Trent, J.O. Resolution and characterization of the structural polymorphism of a single quadruplex-forming sequence. Nucleic Acids Res. 2010, 38, 4877–4888. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Lee, Y.B.; Lee, J.H.; Lee, D.H.; Cho, E.J.; Yu, S.J.; Kim, Y.J.; Kim, J.I.; Im, J.H.; Lee, J.H.; et al. Modified AS1411 aptamer suppresses hepatocellular carcinoma by up-regulating galectin-14. PLoS ONE 2016, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Sun, L.; Li, K.; Yang, X.; Cai, B.; Zhang, Y.; Zhu, Y.; Ma, Y.; Guan, Z.; Wu, Y.; et al. The bioactivity of D-/L-isonucleoside- and 2′-deoxyinosine-incorporated aptamer AS1411s including DNA replication/microRNA expression. Mol. Ther. Nucleic Acids 2017, 9, 218–229. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D.; Russo Krauss, I.; Piccolo, M.; Irace, C.; Paduano, L.; Montesarchio, D. Exploring the conformational behaviour and aggregation properties of lipid-conjugated AS1411 aptamers. Int. J. Biol. Macromol. 2018, 118, 1384–1399. [Google Scholar] [CrossRef] [PubMed]
- Do, N.Q.; Chung, W.J.; Truong, T.H.A.; Heddi, B.; Phan, A.T. G-quadruplex structure of an anti-proliferative DNA sequence. Nucleic Acids Res. 2017, 45, 7487–7493. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.; Lopes-Nunes, J.; Lopes, A.C.; Cabral Campello, M.P.; Paulo, A.; Queiroz, J.A.; Cruz, C. Aptamer-guided acridine derivatives for cervical cancer. Org. Biomol. Chem. 2019, 17, 2992–3002. [Google Scholar] [CrossRef]
- Seiffert, D.; Crain, K.; Wagner, N.V.; Loskutoff, D.J. Vitronectin gene expression in vivo. Evidence for extrahepatic synthesis and acute phase regulation. J. Biol. Chem. 1994, 269, 19836–19842. [Google Scholar] [CrossRef]
- Zhuang, P.; Chen, A.I.; Peterson, C.B. Native and multimeric vitronectin exhibit similar affinity for heparin: Differences in heparin binding properties induced upon denaturation are due to self-association into a multivalent form. J. Biol. Chem. 1997, 272, 6858–6867. [Google Scholar] [CrossRef]
- Aaboe, M.; Offersen, B.V.; Christensen, A.; Andreasen, P.A. Vitronectin in human breast carcinomas. Biochim. Biophys. Acta Mol. Basis Dis. 2003, 1638, 72–82. [Google Scholar] [CrossRef]
- Kadowaki, M.; Sangai, T.; Nagashima, T.; Sakakibara, M.; Yoshitomi, H.; Takano, S.; Sogawa, K.; Umemura, H.; Fushimi, K.; Nakatani, Y.; et al. Identification of vitronectin as a novel serum marker for early breast cancer detection using a new proteomic approach. J. Cancer Res. Clin. Oncol. 2011, 137, 1105–1115. [Google Scholar] [CrossRef]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Akram, M.; Iqbal, M.; Daniyal, M.; Khan, A.U. Awareness and current knowledge of breast cancer. Biol. Res. 2017, 50, 33. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, M.; Misso, G.; Ferraro, M.G.; Riccardi, C.; Capuozzo, A.; Zarone, M.R.; Maione, F.; Trifuoggi, M.; Stiuso, P.; D’Errico, G.; et al. Exploring cellular uptake, accumulation and mechanism of action of a cationic Ru-based nanosystem in human preclinical models of breast cancer. Sci. Rep. 2019, 9, 7006. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.R.; Gagliano, J.; Xiao, J.; Libby, K.; Saito, S.; Yu, G.; Cubicciotti, R.; Macosko, J.; Colyer, C.L.; Guthold, M.; et al. Combining capillary electrophoresis and next-generation sequencing for aptamer selection. Anal. Bioanal. Chem. 2015, 407, 1527–1532. [Google Scholar] [CrossRef]
- Stuart, C.H.; Riley, K.R.; Boyacioglu, O.; Herpai, D.M.; Debinski, W.; Qasem, S.; Marini, F.C.; Colyer, C.L.; Gmeiner, W.H. Selection of a novel aptamer against vitronectin using capillary electrophoresis and next generation sequencing. Mol. Ther. Nucleic Acids 2016, 5, e386. [Google Scholar] [CrossRef]
- Boyacioglu, O.; Stuart, C.H.; Kulik, G.; Gmeiner, W.H. Dimeric DNA aptamer complexes for high-capacity-targeted drug delivery using ph-sensitive covalent linkages. Mol. Ther. Nucleic Acids 2013, 2, e107. [Google Scholar] [CrossRef]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.W.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- Schülke, N.; Varlamova, O.A.; Donovan, G.P.; Ma, D.; Gardner, J.P.; Morrissey, D.M.; Arrigale, R.R.; Zhan, C.; Chodera, A.J.; Surowitz, K.G.; et al. The homodimer of prostate-specific membrane antigen is a functional target for cancer therapy. Proc. Natl. Acad. Sci. USA 2003, 100, 12590–12595. [Google Scholar] [CrossRef]
- Aggarwal, S.; Singh, P.; Topaloglu, O.; Isaacs, J.T.; Demneade, S.R. A dimeric peptide that binds selectively to prostate-specific membrane antigen and inhibits its enzymatic activity. Cancer Res. 2006, 66, 9171–9177. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.A.; Glaser, S.L.; Dorfman, R.F.; Bracci, P.M.; Eberle, E.; Holly, E.A. Expert review of non-Hodgkin’s lymphomas in a population-based cancer registry: Reliability of diagnosis and subtype classifications. Cancer Epidemiol. Biomark. Prev. 2004, 13, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.O.; Gascoyne, R.D.; Lunning, M.A.; Cavalli, F. Non-Hodgkin lymphoma. Lancet 2017, 390, 298–310. [Google Scholar] [CrossRef]
- Tang, Z.; Shangguan, D.; Wang, K.; Shi, H.; Sefah, K.; Mallikratchy, P.; Chen, H.W.; Li, Y.; Tan, W. Selection of aptamers for molecular recognition and characterization of cancer cells. Anal. Chem. 2007, 79, 4900–4907. [Google Scholar] [CrossRef]
- Mallikaratchy, P.R.; Ruggiero, A.; Gardner, J.R.; Kuryavyi, V.; Maguire, W.F.; Heaney, M.L.; McDevitt, M.R.; Patel, D.J.; Scheinberg, D.A. A multivalent DNA aptamer specific for the B-cell receptor on human lymphoma and leukemia. Nucleic Acids Res. 2011, 39, 2458–2469. [Google Scholar] [CrossRef]
- Mallikaratchy, P. Evolution of complex target SELEX to identify aptamers against mammalian cell-surface antigens. Molecules 2017, 22, 215. [Google Scholar] [CrossRef]
- Zumrut, H.E.; Ara, M.N.; Fraile, M.; Maio, G.; Mallikaratchy, P. Ligand-guided selection of target-specific aptamers: A screening technology for identifying specific aptamers against cell-surface proteins. Nucleic Acid Ther. 2016, 26, 190–198. [Google Scholar] [CrossRef]
- Zumrut, H.E.; Mallikaratchy, P.R. Ligand Guided Selection () of artificial nucleic acid ligands against cell surface targets. ACS Appl. Bio Mater. 2020, 3, 2545–2552. [Google Scholar] [CrossRef]
- Zümrüt, H.E.; Batool, S.; Van, N.; George, S.; Bhandari, S.; Mallikaratchy, P. Structural optimization of an aptamer generated from Ligand-Guided Selection (LIGS) resulted in high affinity variant toward mIgM expressed on Burkitt’s lymphoma cell lines. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1825–1832. [Google Scholar] [CrossRef]
- Moccia, F.; Platella, C.; Musumeci, D.; Batool, S.; Zumrut, H.; Bradshaw, J.; Mallikaratchy, P.; Montesarchio, D. The role of G-quadruplex structures of LIGS-generated aptamers R1.2 and R1.3 in IgM specific recognition. Int. J. Biol. Macromol. 2019, 133, 839–849. [Google Scholar] [CrossRef]
- Batool, S.; Argyropoulos, K.V.; Azad, R.; Okeoma, P.; Zumrut, H.; Bhandari, S.; Dekhang, R.; Mallikaratchy, P.R. Dimerization of an aptamer generated from ligand-guided selection (LIGS) yields a high affinity scaffold against B-cells. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Kuhns, M.S.; Badgandi, H.B. Piecing together the family portrait of TCR-CD3 complexes. Immunol. Rev. 2012, 250, 120–143. [Google Scholar] [CrossRef] [PubMed]
- Inthagard, J.; Edwards, J.; Roseweir, A.K. Immunotherapy: Enhancing the efficacy of this promising therapeutic in multiple cancers. Clin. Sci. 2019, 133, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Zumrut, H.E.; Batool, S.; Argyropoulos, K.V.; Williams, N.; Azad, R.; Mallikaratchy, P.R. Integrating ligand-receptor interactions and in vitro evolution for streamlined discovery of artificial nucleic acid ligands. Mol. Ther. Nucleic Acids 2019, 17, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Freage, L.; Jamal, D.; Williams, N.B.; Mallikaratchy, P.R. A homodimeric aptamer variant generated from ligand-guided selection activates T-cell receptor cluster of differentiation three complex. Mol. Ther. Nucleic Acids 2020, 22, 167–178. [Google Scholar] [CrossRef]
- Senger, D.R.; Van De Water, L.; Brown, L.F.; Nagy, J.A.; Yeo, K.T.; Yeo, T.K.; Berse, B.; Jackman, R.W.; Dvorak, A.M.; Dvorak, H.F. Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Rev. 1993, 12, 303–324. [Google Scholar] [CrossRef]
- Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef]
- Guo, P.; Fang, Q.; Tao, H.Q.; Schafer, C.A.; Fenton, B.M.; Ding, I.; Hu, B.; Cheng, S.Y. Overexpression of vascular endothelial growth factor by MCF-7 breast cancer cells promotes estrogen-independent tumor growth in vivo. Cancer Res. 2003, 63, 4684–4691. [Google Scholar]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Holmes, D.I.R.; Zachary, I. The vascular endothelial growth factor (VEGF) family: Angiogenic factors in health and disease. Genome Biol. 2005, 6, 209. [Google Scholar] [CrossRef][Green Version]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) Signaling in Tumour Vascularization: Potential and Challenges. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Prabhu, K.; Krishnankutty, R.; Kuttikrishnan, S.; Tsakou, M.; Alali, F.Q.; Dermime, S.; Mohammad, R.M.; Uddin, S. Vascular endothelial growth factor (VEGF) signaling in tumour vascularization: Potential and challenges. Curr. Vasc. Pharmacol. 2017, 15, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Ema, M. Roles of VEGF-A signalling in development, regeneration, and tumours. J. Biochem. 2014, 156, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in signaling and disease: Beyond discovery and development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.L.; Rak, J.W.; Klement, G.; Kerbel, R.S. Vascular endothelial growth factor isoform expression as a determinant of blood vessel patterning in human melanoma xenografts. Cancer Res. 2002, 62, 1838–1846. [Google Scholar]
- Catena, R.; Muniz-Medina, V.; Moralejo, B.; Javierre, B.; Best, C.J.M.; Emmert-Buck, M.R.; Green, J.E.; Baker, C.C.; Calvo, A. Increased expression of VEGF121/VEGF165-189 ratio results in a significant enhancement of human prostate tumor angiogenesis. Int. J. Cancer 2007, 120, 2096–2109. [Google Scholar] [CrossRef]
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef]
- Meadows, K.L.; Hurwitz, H.I. Anti-VEGF therapies in the clinic. Cold Spring Harb. Perspect. Med. 2012, 2, a006577. [Google Scholar] [CrossRef]
- Riccardi, C.; Napolitano, E.; Platella, C.; Musumeci, D.; Melone, M.A.B.; Montesarchio, D. Anti-VEGF DNA-based aptamers in cancer therapeutics and diagnostics. Med. Res. Rev. 2020. [Google Scholar] [CrossRef]
- Ikebukuro, K.; Hasegawa, H.; Sode, K. Selection and characterization of DNA aptamers against VEGF165 with aptamer blotting method and its application. Nucleic Acids Symp. Ser. 2007, 51, 399–400. [Google Scholar] [CrossRef]
- Hasegawa, H.; Sode, K.; Ikebukuro, K. Selection of DNA aptamers against VEGF165 using a protein competitor and the aptamer blotting method. Biotechnol. Lett. 2008, 30, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Yung, L.Y.L. Probing high affinity sequences of DNA aptamer against VEGF165. PLoS ONE 2012, 7, e31196. [Google Scholar] [CrossRef] [PubMed]
- Manochehry, S.; Mcconnell, E.M.; Li, Y. Unraveling determinants of affinity enhancement in dimeric aptamers for a dimeric protein. Sci. Rep. 2019, 9, 17824. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, T.; Abe, K.; Savory, N.; Tsukakoshi, K.; Yoshida, W.; Ferri, S.; Sode, K.; Ikebukuro, K. Improvement of the VEGF binding ability of DNA aptamers through in silico maturation and multimerization strategy. J. Biotechnol. 2015, 212, 99–105. [Google Scholar] [CrossRef]
- Manochehry, S.; Gu, J.; McConnell, E.; Salena, B.; Li, Y. In vitro selection of new DNA aptamers for human vascular endothelial growth factor 165. ChemBioChem 2020, 21, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.; Janjic, N. High-Affinity Oligonucleotide Ligands to Vascular Endothelial Growth Factor (VEGF). U.S. Patent 7,153,948, 26 December 2006. [Google Scholar]
- Potty, A.S.R.; Kourentzi, K.; Fang, H.; Jackson, G.W.; Zhang, X.; Legge, G.B.; Willson, R.C. Biophysical characterization of DNA aptamer interactions with vascular endothelial growth factor. Biopolymers 2009, 91, 145–156. [Google Scholar] [CrossRef]
- Nonaka, Y.; Sode, K.; Ikebukuro, K. Screening and improvement of an anti-VEGF DNA aptamer. Molecules 2010, 15, 215–225. [Google Scholar] [CrossRef]
- Edwards, S.L.; Poongavanam, V.; Kanwar, J.R.; Roy, K.; Hillman, K.M.; Prasad, N.; Leth-Larsen, R.; Petersen, M.; Marušič, M.; Plavec, J.; et al. Targeting VEGF with LNA-stabilized G-rich oligonucleotide for efficient breast cancer inhibition. Chem. Commun. 2015, 51, 9499–9502. [Google Scholar] [CrossRef]
- Marušič, M.; Veedu, R.N.; Wengel, J.; Plavec, J. G-rich VEGF aptamer with locked and unlocked nucleic acid modifications exhibits a unique G-quadruplex fold. Nucleic Acids Res. 2013, 41, 9524–9536. [Google Scholar] [CrossRef]
- Nonaka, Y.; Yoshida, W.; Abe, K.; Ferri, S.; Schulze, H.; Bachmann, T.T.; Ikebukuro, K. Affinity improvement of a VEGF aptamer by in silico maturation for a sensitive VEGF-detection system. Anal. Chem. 2013, 85, 1132–1137. [Google Scholar] [CrossRef]
- Moccia, F.; Riccardi, C.; Musumeci, D.; Leone, S.; Oliva, R.; Petraccone, L.; Montesarchio, D. Insights into the G-rich VEGF-binding aptamer V7t1: When two G-quadruplexes are better than one! Nucleic Acids Res. 2019, 47, 8318–8331. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Roviello, G.N.; Rigione, G.; Capasso, D.; Di Gaetano, S.; Riccardi, C.; Roviello, V.; Montesarchio, D. Benzodifuran derivatives as potential antiproliferative agents: Possible correlation between their bioactivity and aggregation properties. ChemPlusChem 2017, 82, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Vicidomini, C.; Cioffi, F.; Broersen, K.; Roviello, V.; Riccardi, C.; Montesarchio, D.; Capasso, D.; Gaetano, S.D.; Roviello, G.N. Benzodifurans for biomedical applications: BZ4, a selective anti-proliferative and anti-amyloid lead compound. Future Med. Chem. 2019, 11, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Musumeci, D.; Platella, C.; Gaglione, R.; Arciello, A.; Montesarchio, D. Tuning the polymorphism of the anti-VEGF G-rich V7t1 aptamer by covalent dimeric constructs. Int. J. Mol. Sci. 2020, 21, 1963. [Google Scholar] [CrossRef] [PubMed]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.L.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef]
- Gentile, A.; Trusolino, L.; Comoglio, P.M. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008, 27, 85–94. [Google Scholar] [CrossRef]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into μ-opioid receptor activation. Nature 2015, 90, 972–981. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef]
- Graveel, C.R.; Tolbert, D.; Vande Woude, G.F. MET: A critical player in tumorigenesis and therapeutic target. Cold Spring Harb. Perspect. Biol. 2013, 5, a009209. [Google Scholar] [CrossRef]
- Ueki, R.; Sando, S. A DNA aptamer to c-Met inhibits cancer cell migration. Chem. Commun. 2014, 50, 13131–13134. [Google Scholar] [CrossRef] [PubMed]
- Ueki, R.; Ueki, A.; Kanda, N.; Sando, S. Oligonucleotide-Based Mimetics of Hepatocyte Growth Factor. Angew. Chem. Int. Ed. 2016, 55, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. Eng. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Fasting, C.; Schalley, C.A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.W.; Haag, R. Multivalency as a chemical organization and action principle. Angew. Chem. Int. Ed. Eng. 2012, 51, 10472–10498. [Google Scholar] [CrossRef]
- Yokobayashi, Y. Aptamer-based and aptazyme-based riboswitches in mammalian cells. Curr. Opin. Chem. Biol. 2019, 52, 72–78. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti-inflammatory aptamers | |||||||
| Name | Sequence (5′→3′) | Features | Ref. | ||||
| LD201 | CAAGGTAACCAGTACAAGGTGCTAAACGTAATGGCTTCG | Kd: 1.8 nM | [59] | ||||
| LD174 | CATTCACCATGGCCCCTTCCTACGTATGTTCTGCGGGTG | Kd: 5.5 nM | |||||
| LD196 | TGGCGGTACGGGCCGTGCACCCACTTACCTGGGAAGTGA | Kd: 3.1 nM | |||||
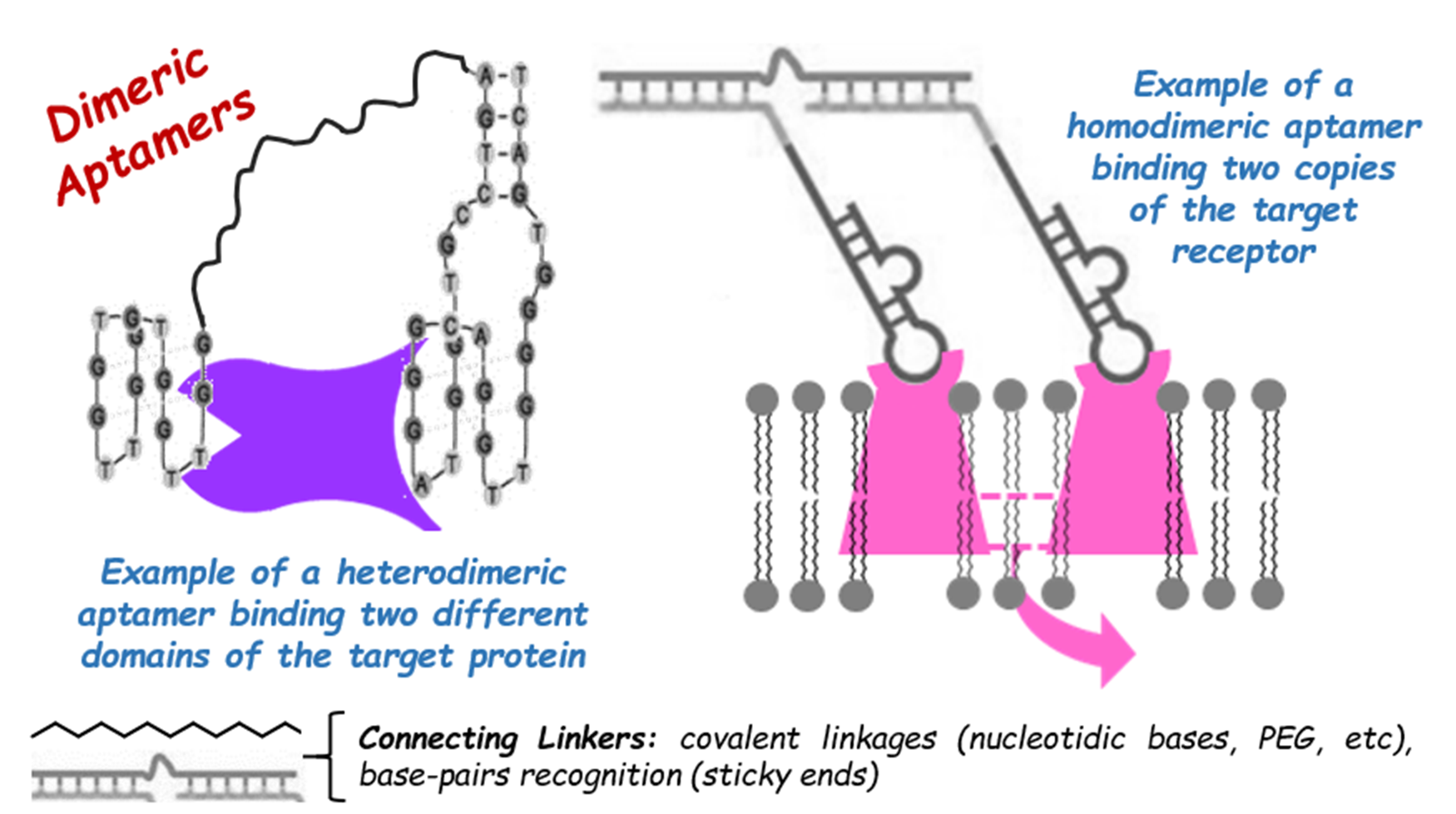
| LD201* | GCGGTAACCAGTACAAGGTGCTAAACGTAATGGCGC | IC50: 8 nM | Tm: 48.7 °C | [61] | |||
| mΔ1 | GCCAGTACAAGGTGCTAAACGTAATGGC | IC50: 10.6 nM | Tm: 55.7 °C | ||||
| dΔ1-A3 | mΔ1-AAA-mΔ1 | IC50: 12 nM; No enhancement vs. mΔ1 | |||||
| dΔ1-A9 | mΔ1-AAAAAAAAA-mΔ1 | IC50: 0.3 nM; ca. 35-fold enhancement vs. mΔ1 | |||||
| dΔ1-A15 | mΔ1-AAAAAAAAAAAAAAA-mΔ1 | IC50: 1.6 nM; ca. 7-fold enhancement vs. mΔ1 | |||||
| dΔ1-A20 | mΔ1-AAAAAAAAAAAAAAAAAAAA-mΔ1 | IC50: 1.6 nM; ca. 7-fold enhancement vs. mΔ1 | |||||
| tΔ1-A9 | mΔ1-AAAAAAAAA-mΔ1-AAAAAAAAA-mΔ1 | IC50: 0.8 nM; ca. 13-fold enhancement vs. mΔ1 | |||||
| Antiviral aptamers | |||||||
| Name | Sequence (5′→3′) | Features | Ref. | ||||
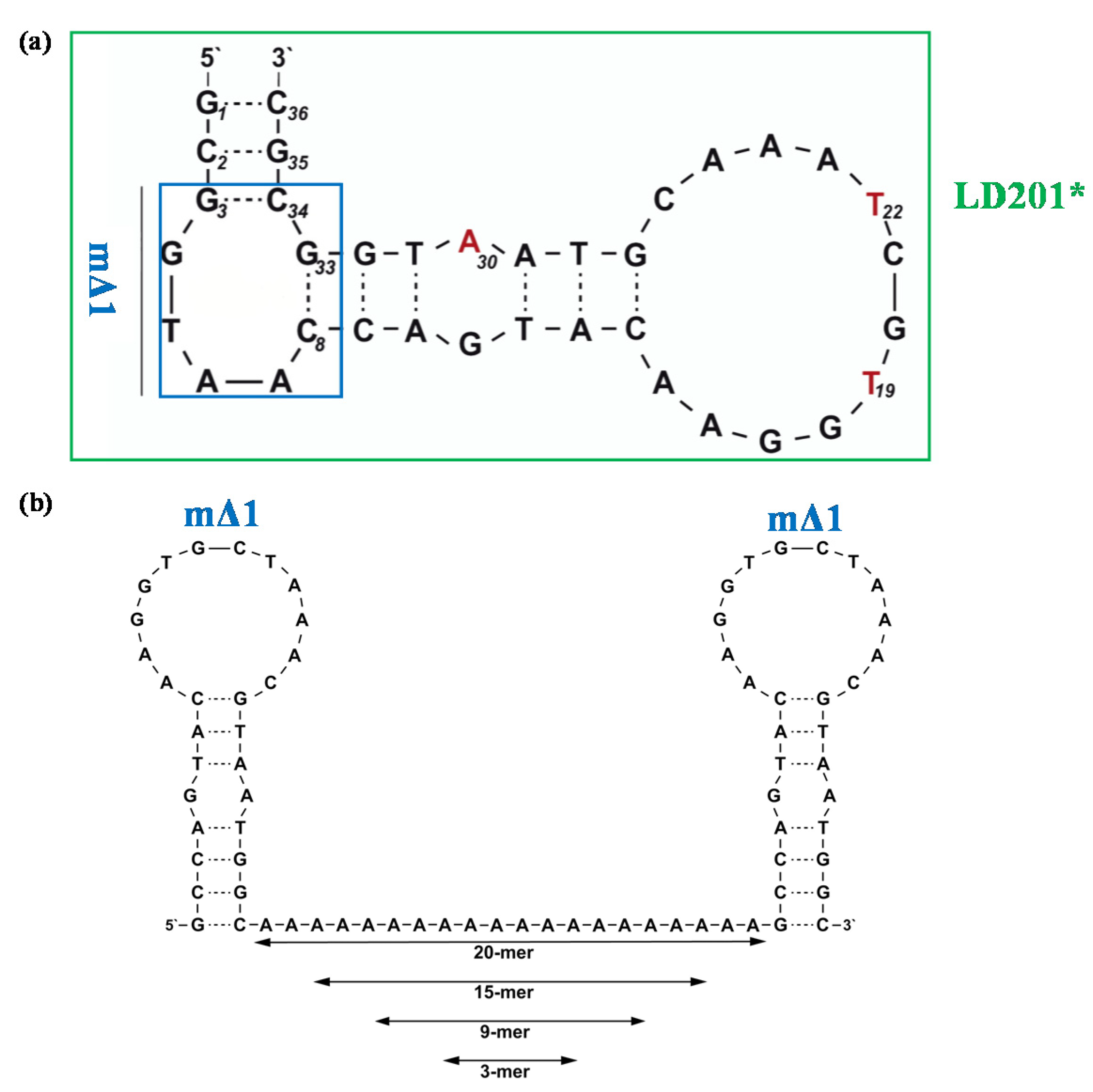
| 1 | OH-(CGGAGG)4-TEL | EC50: > 5 | [78] | ||||
| 2 | O-DMT-(CGGAGG)4-TEL | EC50: 0.54 | |||||
| 3 | O-pGlc-(CGGAGG)4-TEL | EC50: > 5 | |||||
| Anticoagulant aptamers | |||||||
| Name | Sequence (5′→3′) | Features | Ref. | ||||
| TBA15 | GGTTGGTGTGGTTGG | Tm: 33 °C | PT: 25.7 s; | [109] | |||
| AA | 5′-TBA-A-3′-3′-A-TBA-5′ | Tm: 37 °C | PT: 32.6 s; ca. 1.3-fold enhancement vs. TBA | ||||
| TT | 5′-TBA-T-3′-3′-T-TBA-5′ | Tm: 33 °C | PT: 29.6 s; ca. 1.2-fold enhancement vs. TBA | ||||
| gly | 5′-TBA-3′-gly-3′-TBA-5′ | Tm: 35 °C | No enhancement vs. TBA | ||||
| AglyA | 5′-TBA-A-3′-gly-3′-A-TBA-5′ | Tm: 35 °C | PT: 30.5 s; ca. 1.2-fold enhancement vs. TBA | ||||
| TglyT | 5′-TBA-T-3′-gly-3′-T-TBA-5′ | Tm: 29 °C | No activity | ||||
| TBA | GGTTGGTGTGGTTGG | Kd: 7.10 nM | [111] | ||||
| TBA29 | AGTCCGTGTAGGGCAGGTTGGGGTGACT | Kd: 2.40 nM | |||||
| HD1-22 | TBA15-A15-TBA29 | Kd: 0.65 nM; ca. 11-fold enhancement vs. TBA15 and 4-fold vs. TBA29 | |||||
| Linker 0 | TBA15-TBA29 | Kd: 0.14 nM; ca. 144-fold enhancement vs. TBA15 and 25-fold vs. TBA29 | [112] | ||||
| Linker 5 | TBA-T5-TBA29 | Kd: 0.06 nM; ca. 337-fold enhancement vs. TBA15 and 58-fold vs. TBA29 | |||||
| Linker 10 | TBA-T10-TBA29 | Kd: 0.74 nM; ca. 27-fold enhancement vs. TBA15 and 5-fold vs. TBA29 | |||||
| Linker 20 | TBA-T20-TBA29 | Kd: 0.35 nM; ca. 58-fold enhancement vs. TBA15 and 10-fold vs. TBA29 | |||||
| A1(12nm)A2 | TBA29-(spacer 18)5-TBA15 | Kd: 0.3 nM; ca. 13-fold enhancement vs. TBA15 and 10-fold vs. TBA29 | [113] | ||||
| A1(24nm)A2 | TBA29-(spacer 18)10-TBA15 | Kd: 0.06 nM; ca. 67-fold enhancement vs. TBA15 and 48-fold vs. TBA29 | |||||
| A2(24nm)A1 | TBA15-(spacer 18)10-TBA29 | Kd: 0.03 nM; ca. 133-fold enhancement vs. TBA15 and 97-fold vs. TBA29 | |||||
| RNV216A | TBA15/iT | TCT: 27 s; | [114] | ||||
| RNV219 | TBA29/ iT | TCT: 19 s; | |||||
| RNV220 | RNV216A-TEG-RNV219 | TCT: 40 s; ca. 1.5-fold enhancement vs. RNV216A and 2-fold vs. RNV219 | |||||
| RNV220-T | RNV216A-TTTT-RNV219 | TCT:30 s; ca. 1.1-fold enhancement vs. RNV216A and 1.6-fold vs. RNV219 | |||||
| TBV-08 | AGCAGCACAGAGGTCAGATG-TBA15-TGAGACCTTGCATGCGACTTGGTGAGCACGTGAGA-TBA29-CCTATGCGTGCTACCGTGAA | Kd: 8.1 pM; ca. 308-fold enhancement vs. TBA15 and 185-fold vs. TBA29 | [115] | ||||
| 16T | TBA15-T16-TBA29 | Kd: 120 pM; ca. 21-fold enhancement vs. TBA15 and 13-fold vs. TBA29 | |||||
| Bi-4S | TBA27-(S)4-TBA15 | 2903 cps/sec; No enhancement | [116] | ||||
| Bi-6S | TBA27-(S)6-TBA15 | 97 cps/sec; ca. 11-fold enhancement vs. TBA15 | |||||
| Bi-8S | TBA27-(S)8-TBA15 | 63 cps/sec; ca. 17-fold enhancement vs. TBA15 | |||||
| Bi-10S | TBA27-(S)10-TBA15 | 346 cps/sec; ca. 3-fold enhancement vs. TBA15 | |||||
| Anticancer aptamers | |||||||
| Name | Sequence (5′→3′) | Features | Ref. | ||||
| VBA-01 | AATAAACGCTCAACTCAAGTGGCGTGCGGCAGGTTGGTGTGACGGCTGGGAGGGTTCGACATG | Kd: 405 nM | [177] | ||||
| DVBA-01 | VBA-01-duplexDNA-VBA-01 | Kd: 485 nM: no enhancement | |||||
| DVBA/Dox | DVBA-01 + Dox | Kd: 28 nM; ca. 14-fold enhancement vs. VBA-01 | |||||
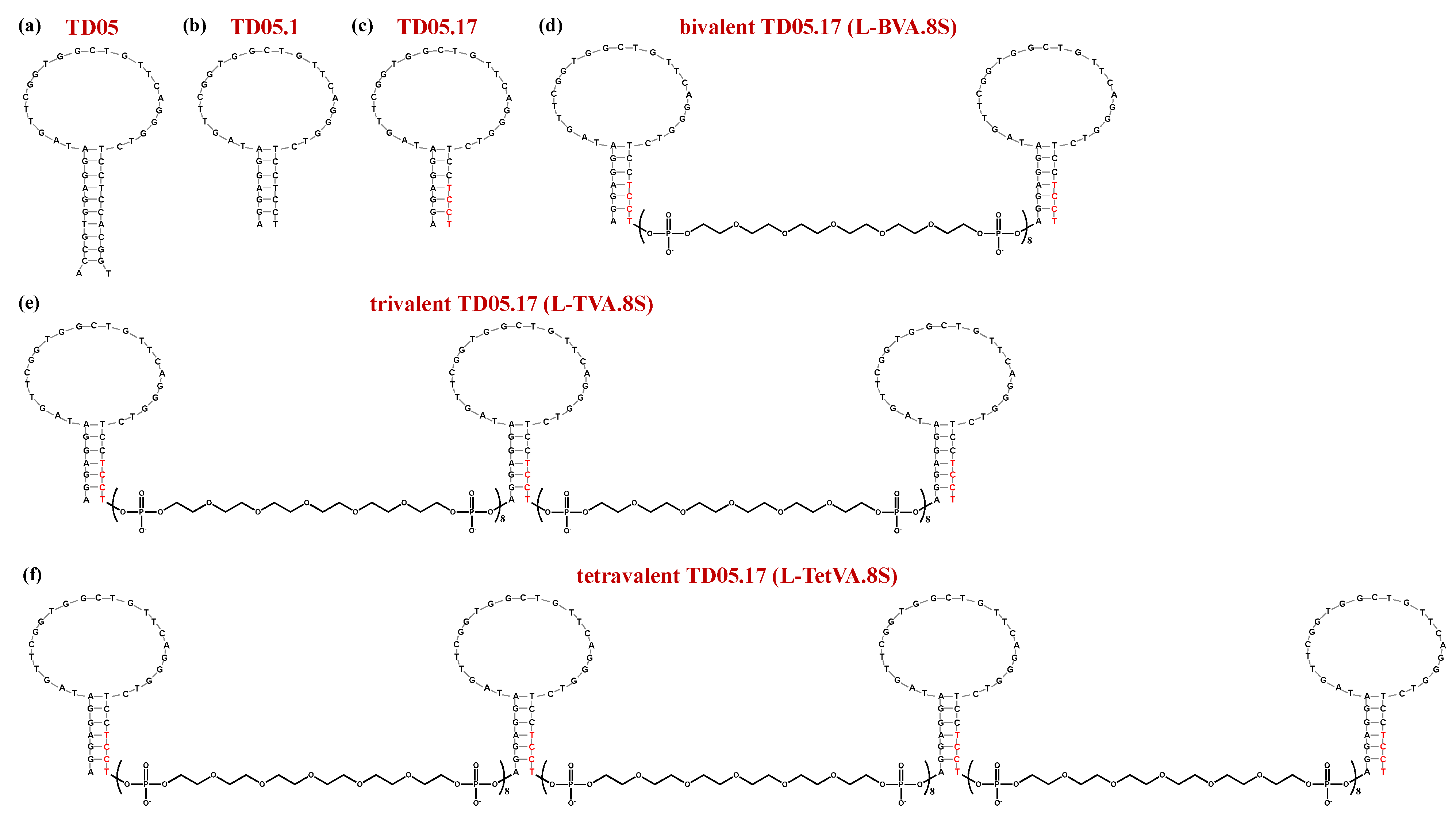
| TD05 | ACCGTGGAGGATAGTTCGGTGGCTGTTCAGGGTCTCCTCCACGGT | Kd: 359 nM at 4 °C | [185] | ||||
| TD05.1 | AGGAGGATAGTTCGGTGGCTGTTCAGGGTCTCCTCCT | Kd: 53 nM at 4 °C; Kd > 10,000 nM at 37 °C | |||||
| TD05.17 | AGGAGGATAGTTCGGTGGCTGTTCAGGGTCTCCTCCT | Kd: 43 nM at 4 °C: Kd > 10,000 nM at 37 °C | |||||
| TVA.8S | TD05.1-(sp18)8-TD05.1-(sp18)8-TD05.1 | Kd: 490 nM at 37 °C; ca. 20-fold enhancement vs. TD05.1 | |||||
| TetVA.8S | TD05.1-(sp18)8-TD05.1-(sp18)8-TD05.1-(sp18)8-TD05.1 | Kd: 425 nM at 37 °C; ca. 24-fold enhancement vs. TD05.1 | |||||
| L-BVA.8S | sp18-TD05.17-(sp18)8-TD05.17-sp18 | Kd: 6222 nM at 37 °C; ca. 1.6-fold enhancement vs. TD05.17 | |||||
| L-TVA.8S | sp18-TD05.17-(sp18)8-TD05.17-(sp18)8-TD05.17-sp18 | Kd: 256 nM at 37 °C; ca. 40-fold enhancement vs. TD05.17 | |||||
| L-TetVA.8S | sp18-TD05.17-(sp18)8-TD05.17-(sp18)8-TD05.17-(sp18)8-TD05.17-sp18 | Kd: 272 nM at 37 °C; ca. 36-fold enhancement vs. TD05.17 | |||||
| R1.2 | CACTGGGTGGGGTTAGCGGGCGATTTAGGGATCTTGAGTGGT | Kd: 35.5 nM at 4 °C; Kd: 65.6 nM at 37 °C | [189] | ||||
| R1.3 | CACTGGGTGGGGTTAGCGGGCGATTTAGGGATCTT | Kd: 134 nM at 4 °C | |||||
| DR1.2_3S | R1.2-(spacer)3-R1.2 | Kd: 55.8 nM at 4 °C: no enhancement Kd: 11.4 nM at 37 °C; ca. 6-fold enhancement vs. R1.2 | [191] | ||||
| DR1.2_5S | R1.2-(spacer)5-R1.2 | Kd: 31.7 nM at 4 °C; ca. 1.1-fold enhancement vs. R1.2 Kd: 20.8 nM at 37 °C; ca. 3.1-fold enhancement vs. R1.2 | |||||
| DR1.2_7S | R1.2-(spacer)7-R1.2 | Kd: 23.9 nM at 4 °C; ca. 1.5-fold enhancement vs. R1.2 Kd: 48.6 nM at 37 °C; ca. 1.3-fold enhancement vs. R1.2 | |||||
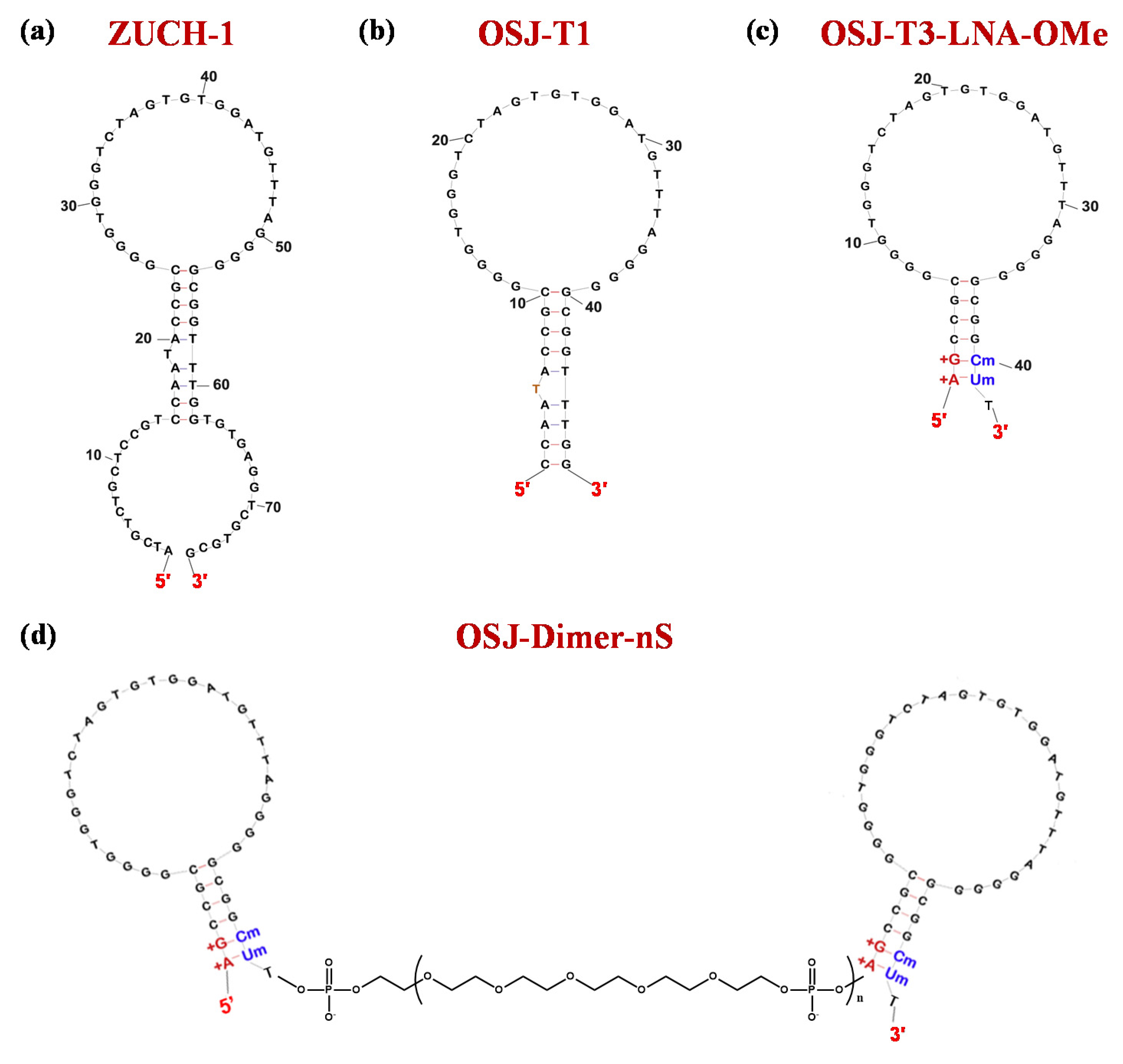
| ZUCH-1 | ATCGTCTGCTCCGTCCAATACCGCGGGGTGGGTCTAGTGTGGATGTTTAGGGGGCGGTTTGGTGTGAGGTCGCGCG | Kd: 3 nM | [194] | ||||
| OSJ-T1 | CCAATACCGCGGGGTGGGTCTAGTGTGGATGTTTAGGGGGCGGTTTGG | Kd: 2.3 nM | [195] | ||||
| OSJ-T2 | CCAATACCGCGGGGTGGGTCTAGTGTGGATGTTTAGGGGGCGGTATTGG | Kd: 2.7 nM | |||||
| OSJ-T3 | GCCGCGGGGTGGGTCTAGTGTGGATGTTTAGGGGGCGGC | Kd: 2.1 nM | |||||
| OSJ-T3-LNA-OMe | AGCCGCGGGGTGGGTCTAGTGTGGATGTTTAGGGGGCGGCU | Kd: 1.7 nM | |||||
| OSJ-dimer-2S | OSJ-T3-LNA-OMe-(S)2-OSJ-T3-LNA-OMe | Kd: 0.5 nM; ca. 3.4-fold enhancement vs. OSJ-T3-LNA-OMe | |||||
| OSJ-dimer-4S | OSJ-T3-LNA-OMe-(S)4-OSJ-T3-LNA-OMe | Kd: 0.3 nM; ca. 5.7-fold enhancement vs. OSJ-T3-LNA-OMe | |||||
| OSJ-dimer-6S | OSJ-T3-LNA-OMe-(S)6-OSJ-T3-LNA-OMe | Kd: 0.4 nM; ca. 4.3-fold enhancement vs. OSJ-T3-LNA-OMe | |||||
| OSJ-dimer-8S | OSJ-T3-LNA-OMe-(S)8-OSJ-T3-LNA-OMe | Kd: 1.7 nM: no enhancement | |||||
| VEa5 | ATACCAGTCTATTCAATTGGGCCCGTCCGTATGGTGGGTGTGCTGGCAGATAGTATGTGCAATCA | Kd: 130 nM | [210,211] | ||||
| del5-1 | ATACCAGTCTATTCAATTGGGCCCGTCCGTATGGTGGGTGTGCTGGCCAG | Kd: 476 nM | [112] | ||||
| SL2-B | CAATTGGGCCCGTCCGTATGGTGGGT | Kd: 37.9 nM | [213] | ||||
| VEa5 homodimer | VEa5-VEa5 | Kd: 6.2 nM; ca. 21-fold enhancement vs. VEa5 | [112] | ||||
| del5-1 homodimer | del5-1-del5-1 | Kd: 17.2 nM; ca. 28-fold enhancement vs. del5-1 | [112] | ||||
| SL2-B homodimer | SL2-B-SL2-B | Kd: 14 nM; ca. 2.7-fold enhancement vs. SL2-B | [213] | ||||
| 2G19 | CTGGCCAGGTACCAAAAGATGATCTTGGGCCCGTCCGAATGGTGGGTGTTCTGGCCAG | Kd: 52 nM | [214] | ||||
| 2G19 homodimer | 2G19-2G19 | Kd: 2.0 nM; ca. 26-fold enhancement vs. 2G19 | |||||
| bivalent SL5 | SL5-SL5 | 1.9 nM; ca. 27-fold enhancement vs. 2G19 | |||||
| trivalent SL5 | SL5-SL5-SL5 | 0.37 nM; ca. 141-fold enhancement vs. 2G19 | |||||
| H4 | TTACGTCAAGGTGTCACTCCCTAGGGGTCCAGGCGAAGCTTAGTAGGGGTGTCCCCTCCCAGAAGCATCTCTTTGGCGTG | Kd: 4 nM | [215] | ||||
| H4 homodimer | H4-T100-H4 | Kd: 1.4 nM; ca. 2.9-fold enhancement vs. H4 | |||||
| +5′G+3′C | GCCCGTCTTCCAGACAAGAGTGCAGGG C | Kd: 9.9 nM | [213] | ||||
| +5′G+3′C homodimer | +5′G+3′C-T20-+5′G+3′C | Kd: 5.5 nM; ca. 1.8-fold enhancement vs. +5′G+3′C | |||||
| +5′G+3′C homodimer | +5′G+3′C-T60-+5′G+3′C | Kd: 7.0 nM; ca. 1.4-fold enhancement vs. +5′G+3′C | |||||
| Vap7 | ATACCAGTCTATTCAATTGCACTCTGTGGGGGTGGACGGGCCGGGTAGATAGTATGTGCAATC | Kd: 20 nM vs. VEGF165; Kd: 1.0 nM vs. VEGF121 | [218] | ||||
| V7t1 | TGTGGGGGTGGACGGGCCGGGTAGA | Kd: 1.4 nM vs. VEGF165; Kd: 1.1 nM vs. VEGF121 | |||||
| 3R02 | TGTGGGGGTGGACTGGGTGGGTACC | Kd: 0.3 nM vs. VEGF165 | [221] | ||||
| 3R02 homodimer | 3R02-T10-3R02 | Kd: 0.03 nM vs. VEGF165; ca. 10-fold enhancement vs. 3R02 | |||||
| del5-1/V7t1 heterodimer | del5-1-V7t1 | Kd: 0.47 nM; ca. 1000-fold enhancement vs. del5-1 and 3-fold vs. V7t1 | [218] | ||||
| CLN0003_SL1 (SL1) | ATCAGGCTGGATGGTAGCTCGGTCGGGGTGGGTGGGTTGGCAAGTCTGAT | Kd: 123 nM | [232] | ||||
| ss-0 | ATCAGGCTGGATGGTAGCTCGGTCGGGGTGGGTGGGTTGGCAAGTCTGAT−CGTGTCACGGATGGTAGCTCGGTCGGGGTGGGTGGGTTGGCAGTGACACG | n.d. | [233] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riccardi, C.; Napolitano, E.; Musumeci, D.; Montesarchio, D. Dimeric and Multimeric DNA Aptamers for Highly Effective Protein Recognition. Molecules 2020, 25, 5227. https://doi.org/10.3390/molecules25225227
Riccardi C, Napolitano E, Musumeci D, Montesarchio D. Dimeric and Multimeric DNA Aptamers for Highly Effective Protein Recognition. Molecules. 2020; 25(22):5227. https://doi.org/10.3390/molecules25225227
Chicago/Turabian StyleRiccardi, Claudia, Ettore Napolitano, Domenica Musumeci, and Daniela Montesarchio. 2020. "Dimeric and Multimeric DNA Aptamers for Highly Effective Protein Recognition" Molecules 25, no. 22: 5227. https://doi.org/10.3390/molecules25225227
APA StyleRiccardi, C., Napolitano, E., Musumeci, D., & Montesarchio, D. (2020). Dimeric and Multimeric DNA Aptamers for Highly Effective Protein Recognition. Molecules, 25(22), 5227. https://doi.org/10.3390/molecules25225227