
Drugs Repurposing Using QSAR, Docking and Molecular Dynamics for Possible Inhibitors of the SARS-CoV-2 Mpro Protease

 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
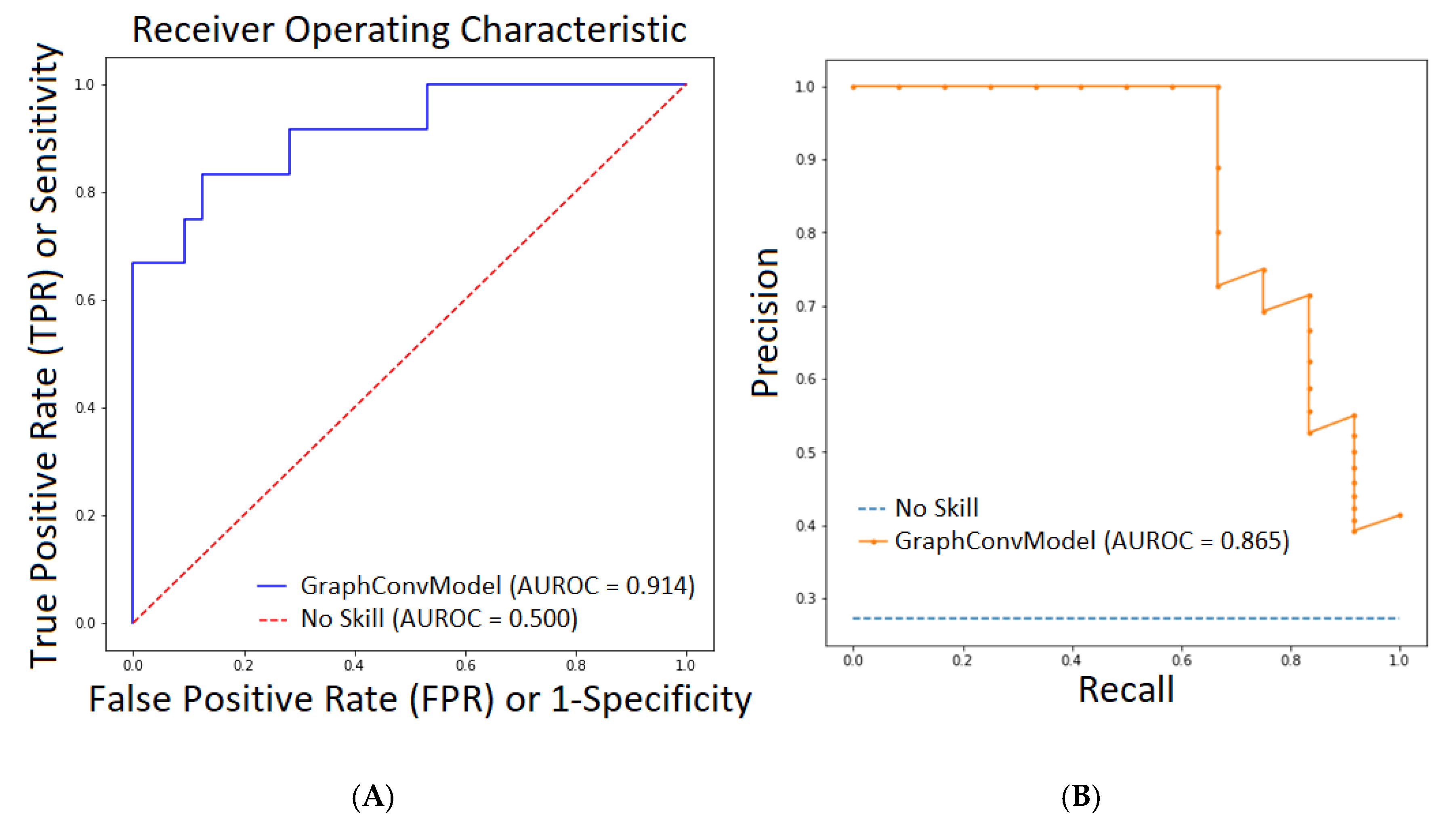
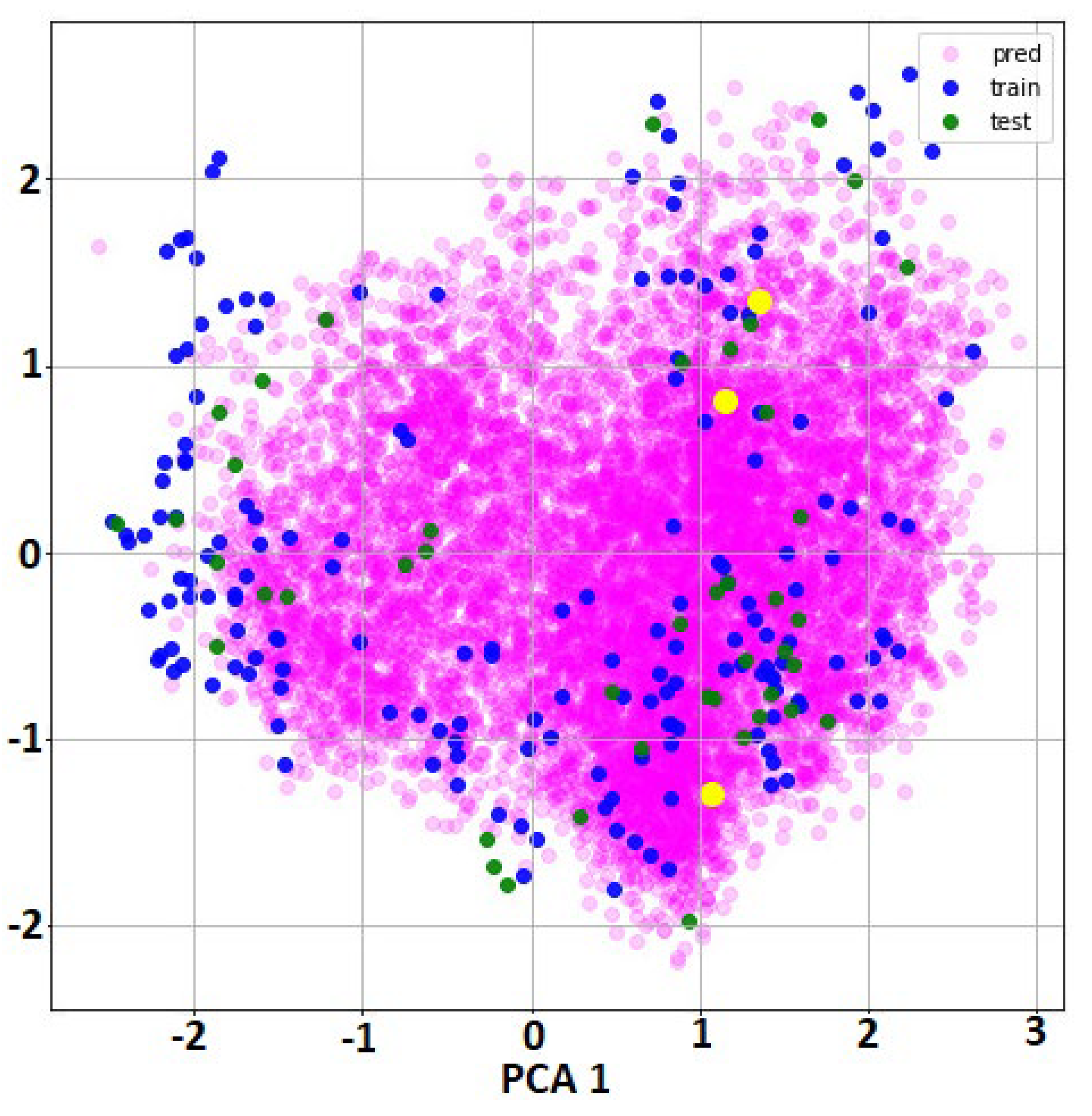
2.1. QSAR Modelling Results
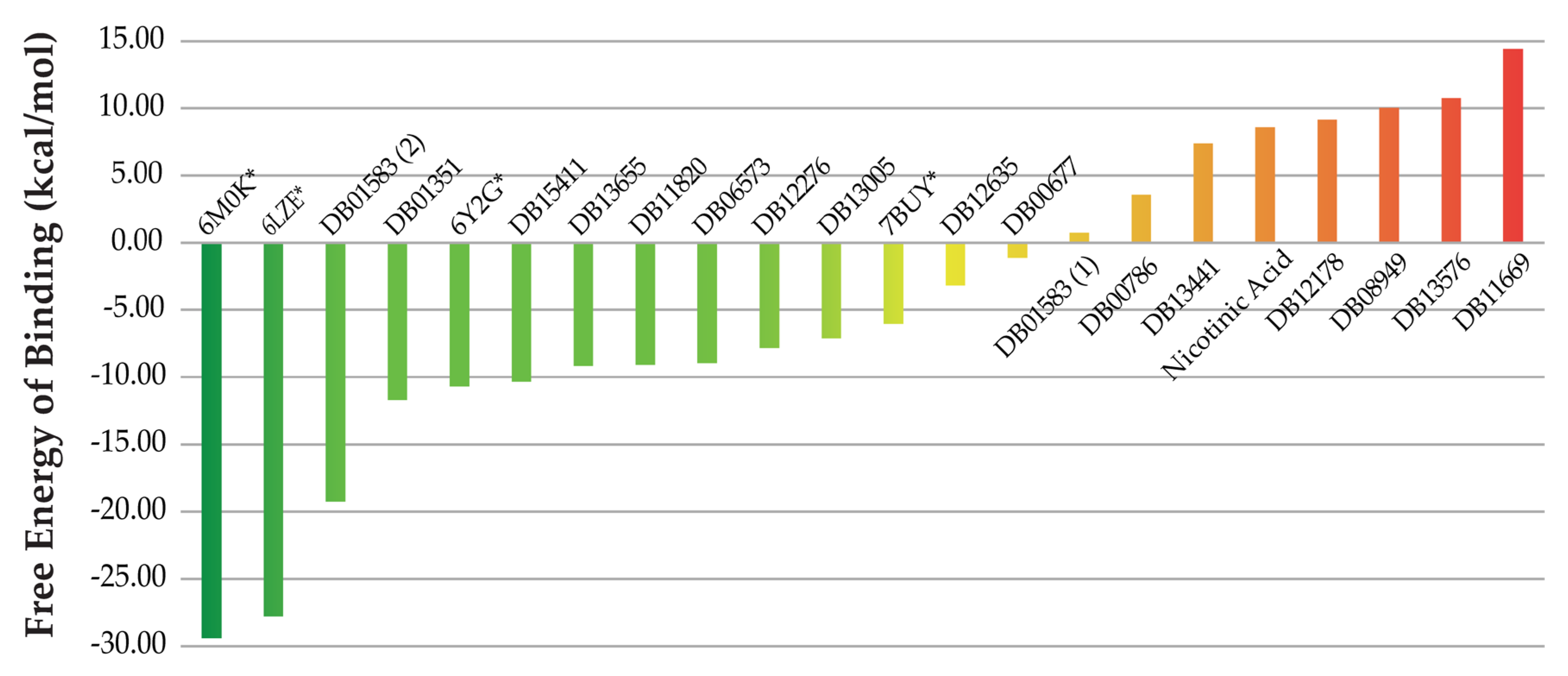
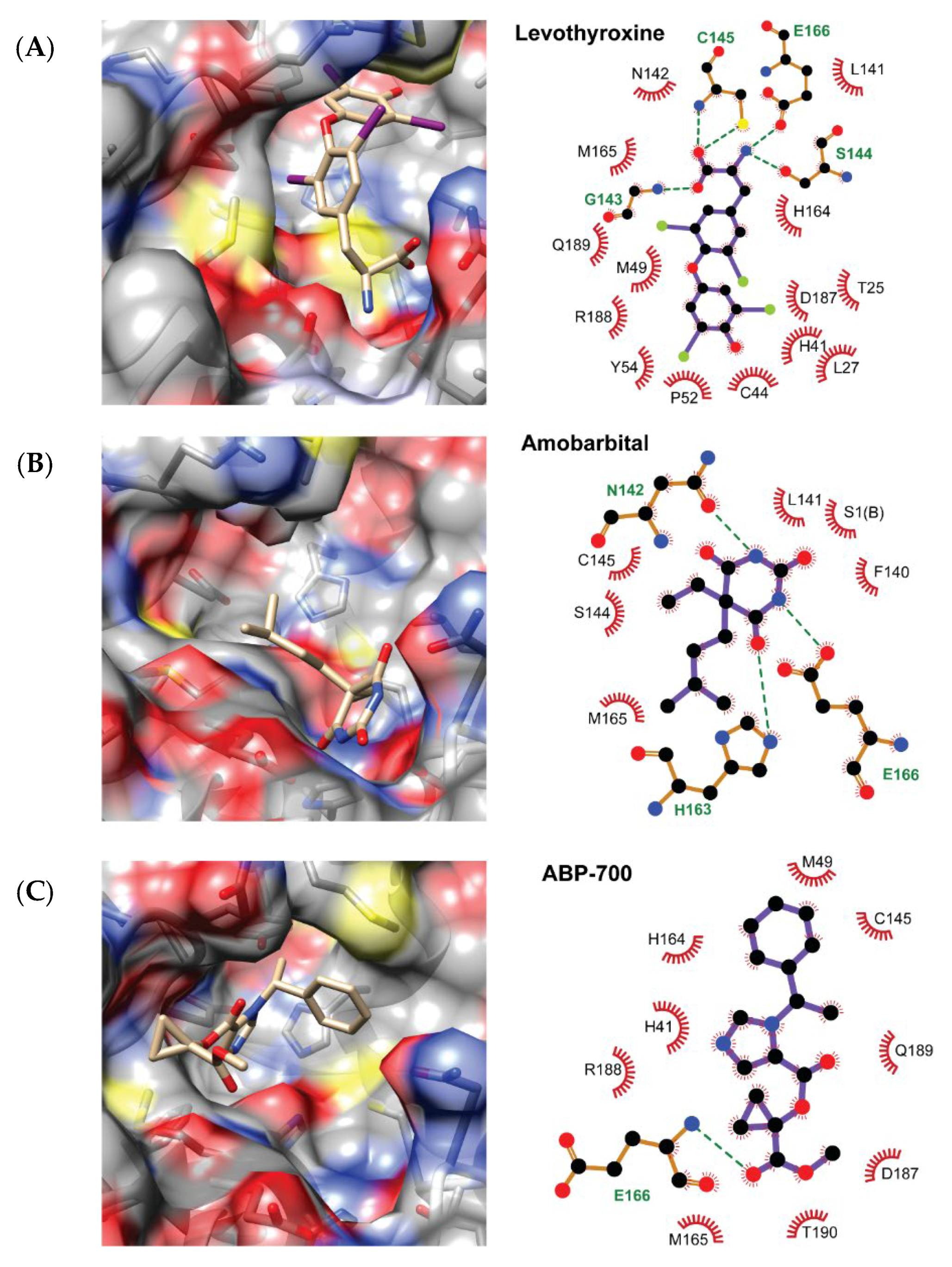
2.2. QSAR Virtual Screening, Docking and Molecular Dynamics
3. Discussion
4. Materials and Methods
4.1. QSAR Modelling
- -
- The entire dataset was available as antivirals_SMILES.csv in datasets folder of the repository (229 molecules as SMILES representation, antivirals_SMILES.csv in datasets folder).
- -
- The external dataset used to predict anti-Mpro activity from drug repurposing was available as DB_SMILES4prediction.csv in the datasets folder of the repository (10,246 molecules with DB ID and SMILES formula).
- -
- For all molecules from the full dataset and external set for predictions, specific features were calculated using DeepChem function ConvMolFeaturizer, an implementation of the Duvenaud graph convolutions that computed a vector of 75 local descriptors for each atom in a molecule. Thus, each molecule was represented as an array with dimension number of atoms*75. As consequence, the initial input features were graph representations, not vector of values (as in classical QSAR). There was no possibility to cluster the molecules using this type of information.
- -
- We used 75 internal features for the convolutional graphs, batch size = 32 during 70 epochs and dropout = 0.05 as parameters for training with DeepChem function GraphConvModel. The optimization algorithm to find the best model was minimizing the error between the observed and predicted classes.
- -
- The dataset was randomly split (seed = 80) into 80–20% train-test subsets using DeepChem function SingletaskStratifiedSplitter that divides the dataset keeping the same ratio of classes across the training and test subsets. The result were 176 molecules in the training subset (train_subset.txt in datasets folder) and 44 in the test subset (test_subset.txt in datasets folder).
- -
- The training subset was used to build the best classifier using the two classes and the test subset was used to evaluate the model performance using AUROC as the performance metric. The training used a deterministic optimization and therefore it is possible to reproduce the same classifier. In addition, all the calculated features and the final model are available as files in a specific folder at the public repository (using specific DeepChem format).
4.2. Virtual Screening
4.3. Molecular Docking
4.4. Molecular Dynamics Simulations and Estimation of the Free Energies of Binding
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA J. Am. Med. Assoc. 2020, 323, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- López-Cortés, A.; Guevara-Ramírez, P.; Kyriakidis, N.C.; Barba-Ostria, C.; Cáceres, Á.L.; Guerrero, S.; Munteanu, C.R.; Tejera, E.; Ortiz-Prado, E.; Cevallos-Robalino, D.; et al. In Silico Analyses of Immune System Protein Interactome Network, Single-Cell RNA Sequencing of Human Tissues, and Artificial Neural Networks Reveal Potential Therapeutic Targets for Drug Repurposing Against COVID-19. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Altay, O.; Mohammadi, E.; Lam, S.; Turkez, H.; Boren, J.; Nielsen, J.; Uhlen, M.; Mardinoglu, A. Current Status of COVID-19 Therapies and Drug Repositioning Applications. iScience 2020, 23, 101303. [Google Scholar] [CrossRef]
- Oberfeld, B.; Achanta, A.; Carpenter, K.; Chen, P.; Gilette, N.M.; Langat, P.; Said, J.T.; Schiff, A.E.; Zhou, A.S.; Barczak, A.K.; et al. SnapShot: COVID-19. Cell 2020, 181, 954.e1. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Prado, E.; Simbaña-Rivera, K.; Gómez-Barreno, L.; Rubio-Neira, M.; Guaman, L.P.; Kyriakidis, N.C.; Muslin, C.; Jaramillo, A.M.G.; Barba-Ostria, C.; Cevallos-Robalino, D.; et al. Clinical, molecular, and epidemiological characterization of the SARS-CoV-2 virus and the Coronavirus Disease 2019 (COVID-19), a comprehensive literature review. Diagn. Microbiol. Infect. Dis. 2020, 98, 115094. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016.e19–1035.e19. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281.e6–292.e6. [Google Scholar] [CrossRef]
- Cao, Y.; Li, L.; Feng, Z.; Wan, S.; Huang, P.; Sun, X.; Wen, F.; Huang, X.; Ning, G.; Wang, W. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020, 6, 1–4. [Google Scholar]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894.e9–904.e9. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Mü, M.A.; Drosten, C.; Pö, S.; Krü, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor Article SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 1–10. [Google Scholar] [CrossRef]
- Zhao, Q.; Weber, E.; Yang, H. Recent Developments on Coronavirus Main Protease/3C Like Protease Inhibitors. Recent Pat. Antiinfect. Drug Discov. 2013, 8, 150–156. [Google Scholar] [CrossRef]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) Structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Donchenko, A.P.; Blinov, V.M.; Koonin, E.V. Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases. A distinct protein superfamily with a common structural fold. FEBS Lett. 1989, 243, 103–114. [Google Scholar] [CrossRef]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of Wide-Spectrum Inhibitors Targeting Coronavirus Main Proteases. PLoS Biol. 2005, 3, e324. [Google Scholar] [CrossRef]
- Khan, S.A.; Zia, K.; Ashraf, S.; Uddin, R.; Ul-Haq, Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Guy, R.K.; DiPaola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020. [Google Scholar] [CrossRef]
- Tsuji, M. Potential anti-SARS-CoV-2 drug candidates identified through virtual screening of the ChEMBL database for compounds that target the main coronavirus protease. FEBS Open Bio. 2020, 10, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6. [Google Scholar] [CrossRef]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.; Ikram, S.; Ahmad, F.; Rehman, I.U.; Mushtaq, M. SARS-CoV-2 RNA Dependent RNA polymerase (RdRp)—A drug repurposing study. Heliyon 2020. [Google Scholar] [CrossRef]
- Dyall, J.; Coleman, C.M.; Hart, B.J.; Venkataraman, T.; Holbrook, M.R.; Kindrachuk, J.; Johnson, R.F.; Olinger, G.G.; Jahrling, P.B.; Laidlaw, M.; et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob. Agents Chemother. 2014, 58, 4885–4893. [Google Scholar] [CrossRef]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. Unravelling lead antiviral phytochemicals for the inhibition of SARS-CoV-2 Mpro enzyme through in silico approach. Life Sci. 2020, 255, 117831. [Google Scholar] [CrossRef] [PubMed]
- Nukoolkarn, V.; Lee, V.S.; Malaisree, M.; Aruksakulwong, O.; Hannongbua, S. Molecular dynamic simulations analysis of ritronavir and lopinavir as SARS-CoV 3CLpro inhibitors. J. Theor. Biol. 2008, 254, 861–867. [Google Scholar] [CrossRef]
- Cournia, Z.; Allen, B.K.; Beuming, T.; Pearlman, D.A.; Radak, B.K.; Sherman, W. Rigorous Free Energy Simulations in Virtual Screening. J. Chem. Inf. Model. 2020, 60, 4153–4169. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Jin, Z.; Zhao, Y.; Sun, Y.; Zhang, B.; Wang, H.; Wu, Y.; Zhu, Y.; Zhu, C.; Hu, T.; Du, X.; et al. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020, 27, 529–532. [Google Scholar] [CrossRef]
- Bellera, C.L.; Balcazar, D.E.; Alberca, L.; Labriola, C.A.; Talevi, A.; Carrillo, C. Identification of Levothyroxine Antichagasic Activity Through Computer-Aided Drug Repurposing. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef]
- Kim, M.; Kim, Y.B. In Silico Synergistic Drug Repurposing for Combating Novel Coronavirus (COVID-19) Outbreaks. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Gysi, D.M.; Valle, Í.D.; Zitnik, M.; Ameli, A.; Gan, X.; Varol, O.; Sanchez, H.; Baron, R.M.; Ghiassian, D.; Loscalzo, J.; et al. Network Medicine Framework for Identifying Drug Repurposing Opportunities for COVID-19. arXiv 2020, arXiv:2004.07229. [Google Scholar]
- Sugandh, K.; Pratima, K.; Geetanjali, A.; Preethy, V.; Shaheerah, K.; Gulam Hussain, S.; Anshuman, D. Identification of Drugs Targeting Multiple Viral and Human Proteins Using Computational Analysis for Repurposing Against COVID-19. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Do Av Sá, L.G.; Da Silva, C.R.; S Campos, R.D.; De A Neto, J.B.; Sampaio, L.S.; Do Nascimento, F.B.; Barroso, F.D.; Da Silva, L.J.; Queiroz, H.A.; Cândido, T.M.; et al. Synergistic anticandidal activity of etomidate and azoles against clinical fluconazole-resistant isolates. Future Microbiol. 2019, 14, 1477–1488. [Google Scholar] [CrossRef]
- Pizzorno, A.; Terrier, O.; de Lamballerie, C.N.; Julien, T.; Padey, B.; Traversier, A.; Roche, M.; Hamelin, M.E.; Rhéaume, C.; Croze, S.; et al. Repurposing of drugs as novel influenza inhibitors from clinical gene expression infection signatures. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; Aksoydan, B.; Dogan, B.; Sahin, K.; Shahraki, A.; Birgül-İyison, N. Screening of Clinically Approved and Investigation Drugs as Potential Inhibitors of SARS-CoV-2 Main Protease and Spike Receptor-Binding Domain Bound with ACE2 COVID19 Target Proteins: A Virtual Drug Repurposing Study. ChemRxiv 2020, 1–31. [Google Scholar] [CrossRef]
- Sangjae, S.; Jung Woo, P.; Dosik, A.; Junwon, Y.; Hyojung, P.; Soonwook, H. Supercomputer-aided Drug Repositioning at Scale: Virtual Screening for SARS-CoV-2 Protease Inhibitor. ChemRxiv 2020, 2. [Google Scholar] [CrossRef]
- Eleftheriou, P.; Amanatidou, D.; Petrou, A.; Geronikaki, A. In Silico Evaluation of the Effectivity of Approved Protease Inhibitors against the Main Protease of the Novel SARS-CoV-2 Virus. Molecules 2020, 25, 2529. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Duvenaud, C.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Gómez-Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R.P.; Duvenaud, D.; Aguilera-Iparraguirre Rafael Gómez-Bombarelli, J. Convolutional Networks on Graphs for Learning Molecular Fingerprints; Neural Information Processing Systems Foundation, Inc.: Long Beach, CA, USA, 2015. [Google Scholar]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning; Springer Series in Statistics; Springer: New York, NY, USA, 2009; ISBN 978-0-387-84857-0. [Google Scholar]
- Davis, J.; Goadrich, M. The relationship between precision-recall and ROC curves. In ACM International Conference Proceeding Series; ACM Press: New York, NY, USA, 2006; Volume 148, pp. 233–240. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the protein databank and cambridge structural database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Lopes, S.P.; Castillo, Y.P.; Monteiro, M.L.; de Menezes, R.R.; Almeida, R.N.; Martins, A.M.C.; Sousa, D.P. Trypanocidal Mechanism of Action and in silico Studies of p-Coumaric Acid Derivatives. Int. J. Mol. Sci. 2019, 20, 5916. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Turkez, H.; Nóbrega, F.R.; Ozdemir, O.; Bezerra Filho, C.D.; Almeida, R.N.; Tejera, E.; Perez-Castillo, Y.; Sousa, D.P. NFBTA: A Potent Cytotoxic Agent against Glioblastoma. Molecules 2019, 24, 2411. [Google Scholar] [CrossRef]
- Perez-Castillo, Y.; Lima, T.C.; Ferreira, A.R.; Silva, C.R.; Campos, R.S.; Neto, J.B.A.; Magalhães, H.I.F.; Cavalcanti, B.C.; Júnior, H.V.N.; de Sousa, D.P. Bioactivity and Molecular Docking Studies of Derivatives from Cinnamic and Benzoic Acids. Biomed Res. Int. 2020, 2020, 6345429. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro TAD, V.W.D.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; Gohlke, H.; et al. AMBER 2018 Reference Manuel; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metrics | Train | Test |
|---|---|---|
| Accuracy | 0.977 | 0.841 |
| Precision | 0.970 | 0.830 |
| AUROC | 0.998 | 0.914 |
| PRC-AUC | 0.995 | 0.865 |
| Name | Probability | DrugBank ID | Name | Probability | DrugBank ID |
|---|---|---|---|---|---|
| Inositol nicotinate | 0.999 | DB08949 | Aluminium nicotinate | 0.992 | DB13576 |
| Telinavir | 0.998 | DB12178 | Amobarbital | 0.991 | DB01351 |
| Ortataxel | 0.998 | DB11669 | ABP-700 | 0.991 | DB15411 |
| Niceritrol | 0.997 | DB13441 | Rebastinib | 0.988 | DB13005 |
| Rebimastat | 0.996 | DB06573 | Bismuth subcitrate potassium | 0.987 | DB09275 |
| Apomine | 0.994 | DB12276 | Drometrizole trisiloxane | 0.987 | DB11585 |
| Mecobalamin | 0.994 | DB03614 | Aleplasinin | 0.985 | DB12635 |
| Nikethamide | 0.993 | DB13655 | Liotrix | 0.984 | DB01583 |
| Hydroxocobalamin | 0.993 | DB00200 | Nifurtimox | 0.983 | DB11820 |
| Marimastat | 0.992 | DB00786 | Isoflurophate | 0.982 | DB00677 |
Sample Availability: All scripts, datasets, molecular features, the best classifier and the prediction results can be accessed as a free GitHub repository at https://github.com/muntisa/Anticoronavirals-Classifier-using-DeepChem. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tejera, E.; Munteanu, C.R.; López-Cortés, A.; Cabrera-Andrade, A.; Pérez-Castillo, Y. Drugs Repurposing Using QSAR, Docking and Molecular Dynamics for Possible Inhibitors of the SARS-CoV-2 Mpro Protease. Molecules 2020, 25, 5172. https://doi.org/10.3390/molecules25215172
Tejera E, Munteanu CR, López-Cortés A, Cabrera-Andrade A, Pérez-Castillo Y. Drugs Repurposing Using QSAR, Docking and Molecular Dynamics for Possible Inhibitors of the SARS-CoV-2 Mpro Protease. Molecules. 2020; 25(21):5172. https://doi.org/10.3390/molecules25215172
Chicago/Turabian StyleTejera, Eduardo, Cristian R. Munteanu, Andrés López-Cortés, Alejandro Cabrera-Andrade, and Yunierkis Pérez-Castillo. 2020. "Drugs Repurposing Using QSAR, Docking and Molecular Dynamics for Possible Inhibitors of the SARS-CoV-2 Mpro Protease" Molecules 25, no. 21: 5172. https://doi.org/10.3390/molecules25215172
APA StyleTejera, E., Munteanu, C. R., López-Cortés, A., Cabrera-Andrade, A., & Pérez-Castillo, Y. (2020). Drugs Repurposing Using QSAR, Docking and Molecular Dynamics for Possible Inhibitors of the SARS-CoV-2 Mpro Protease. Molecules, 25(21), 5172. https://doi.org/10.3390/molecules25215172