
Interactive Field Effect of Atomic Bonding Forces on the Equivalent Elastic Modulus Estimation of Micro-Level Single-Crystal Copper by Utilizing Atomistic-Continuum Finite Element Simulation


Abstract
:1. Introduction
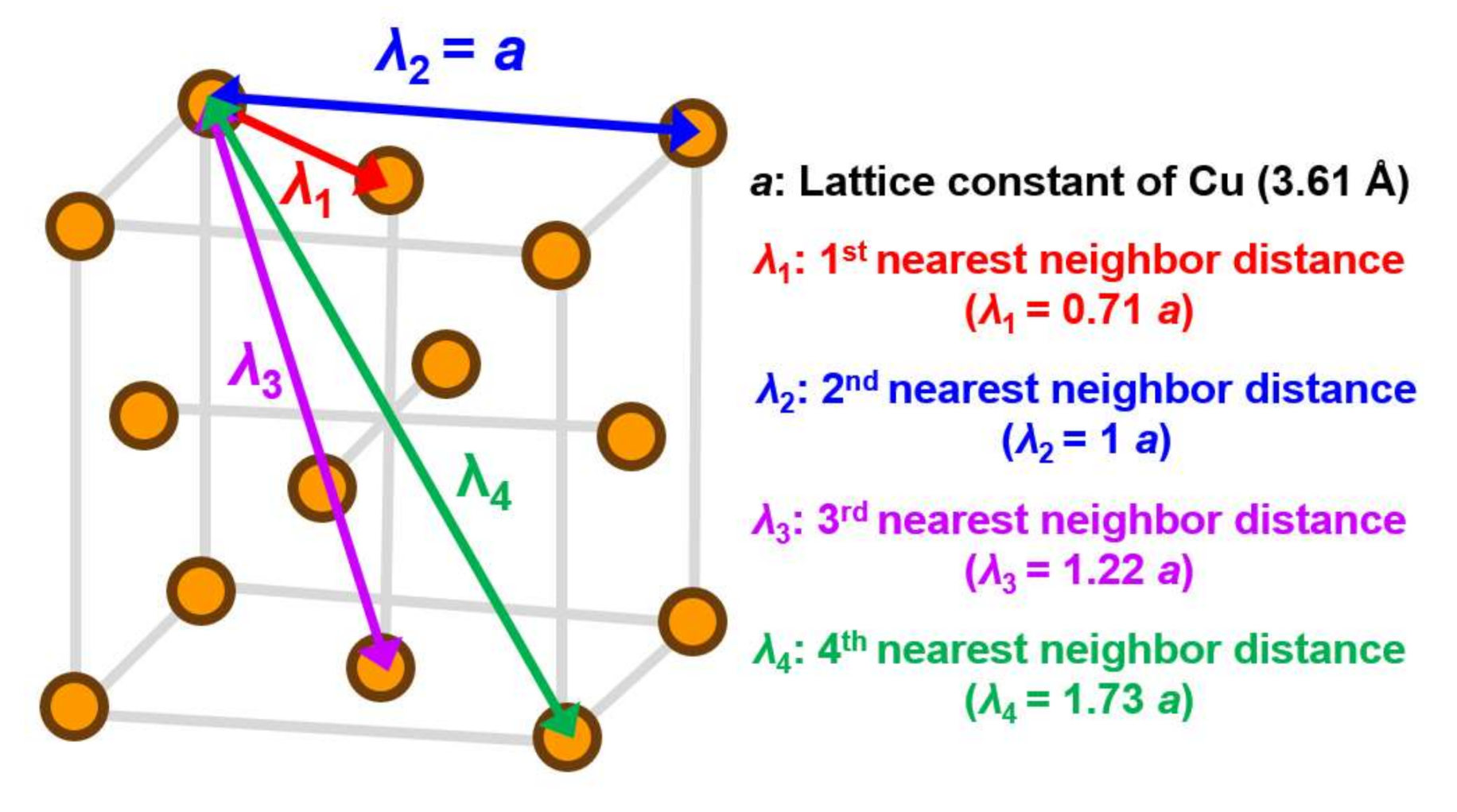
2. Potential Function of Cu Metal Atoms
2.1. Morse Potential
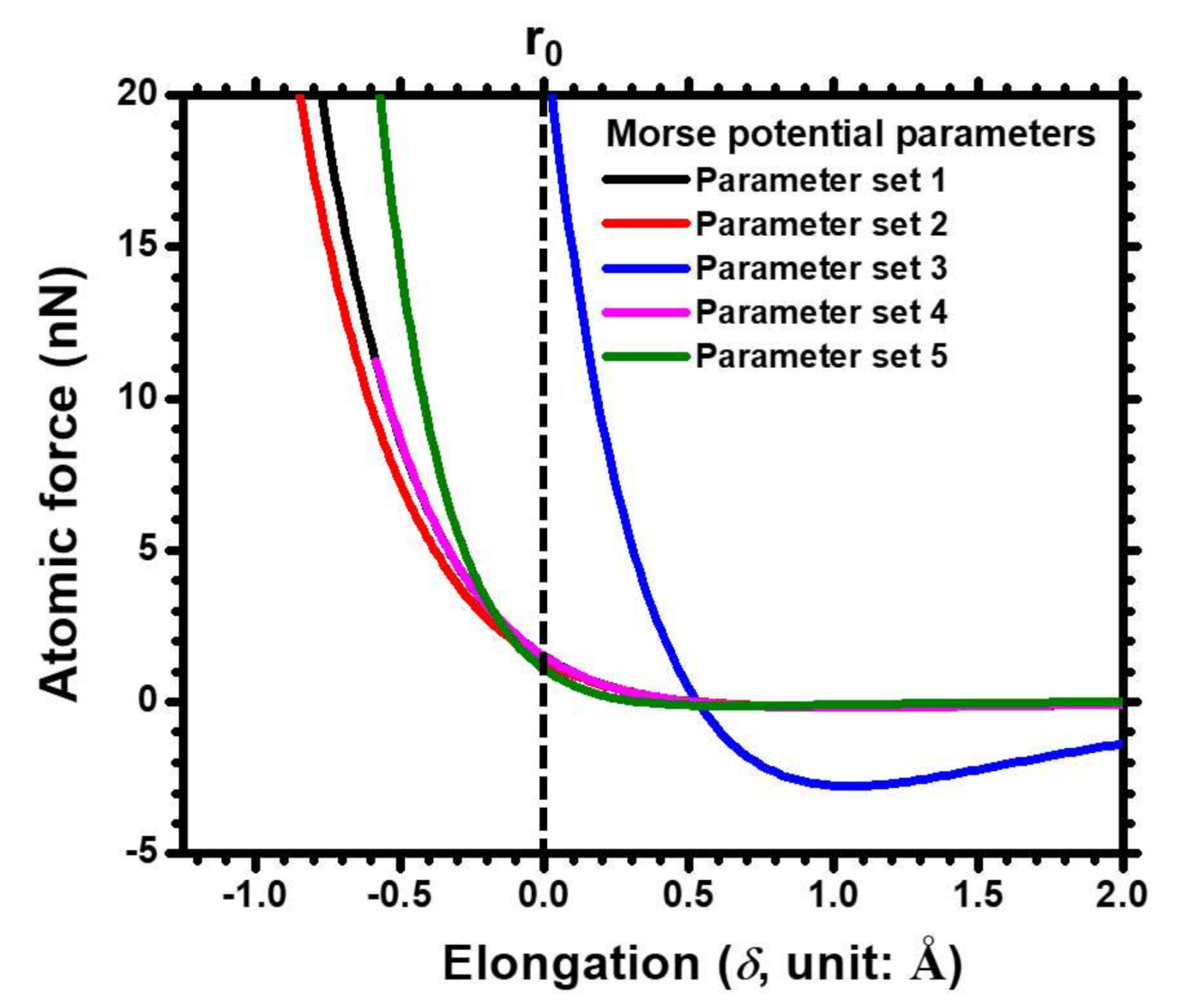
2.2. Comparisons of Selected Morse Parameters
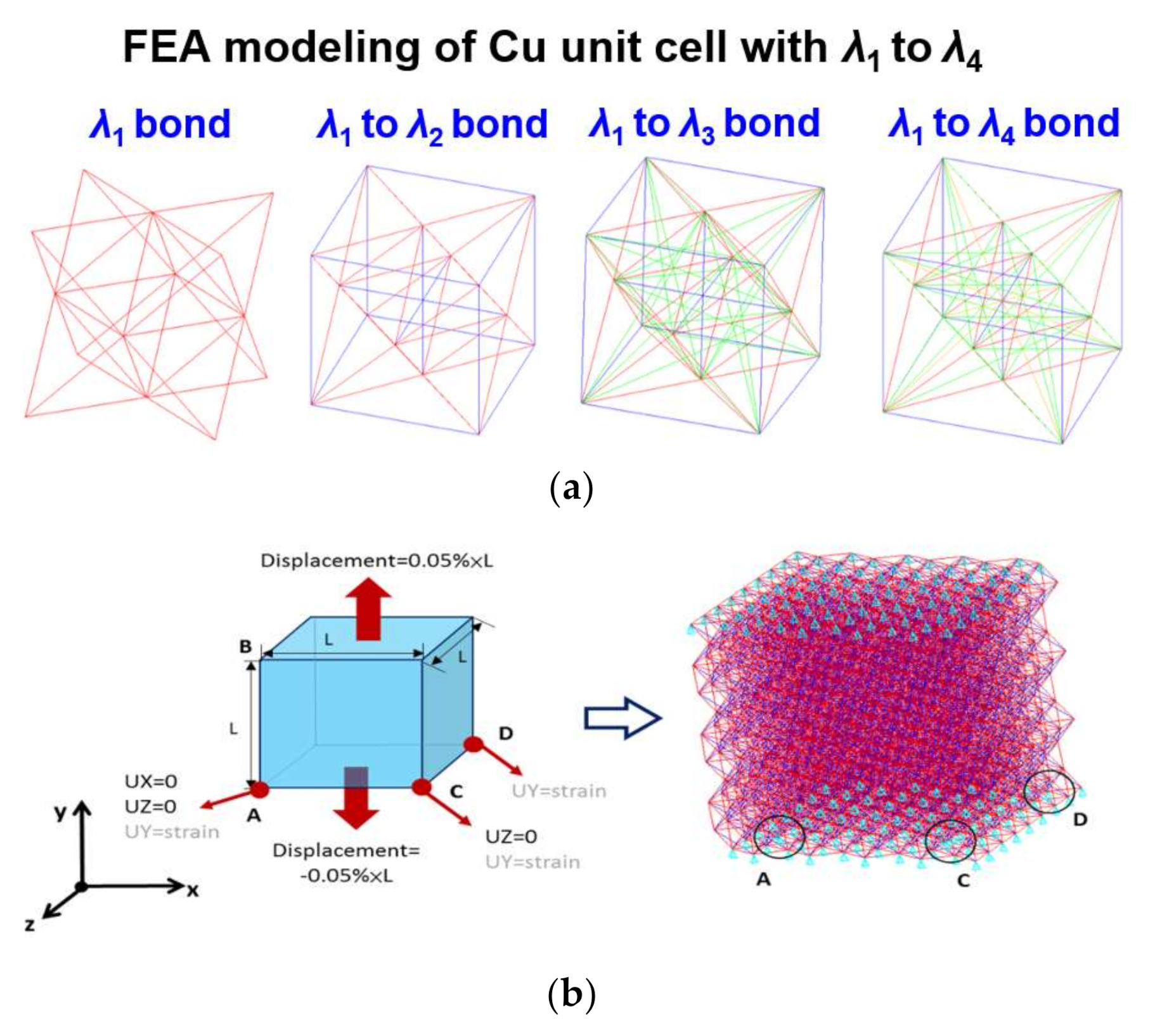
3. Atomistic-Continuum Method in FEA Modeling
4. Results and Discussion
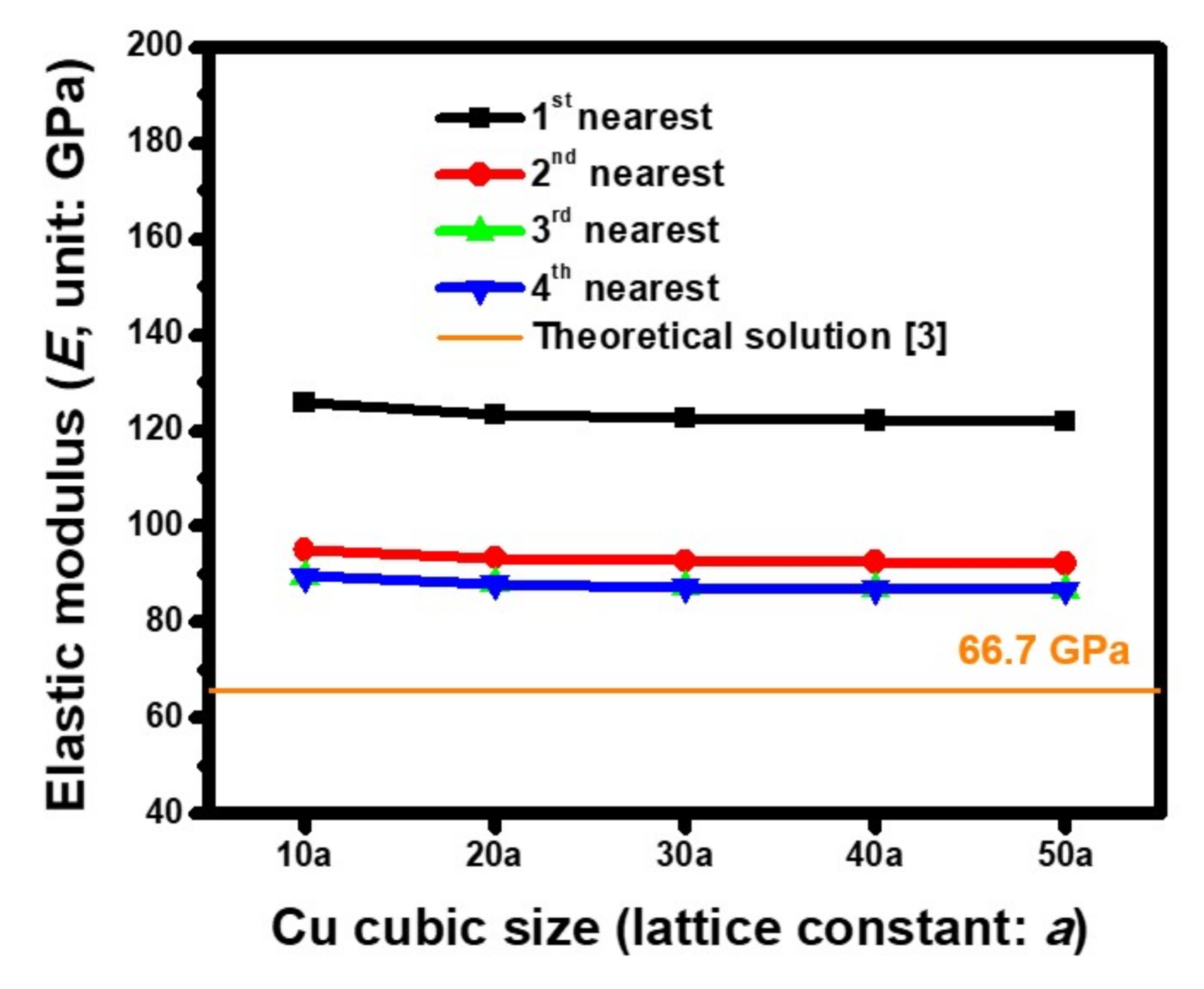
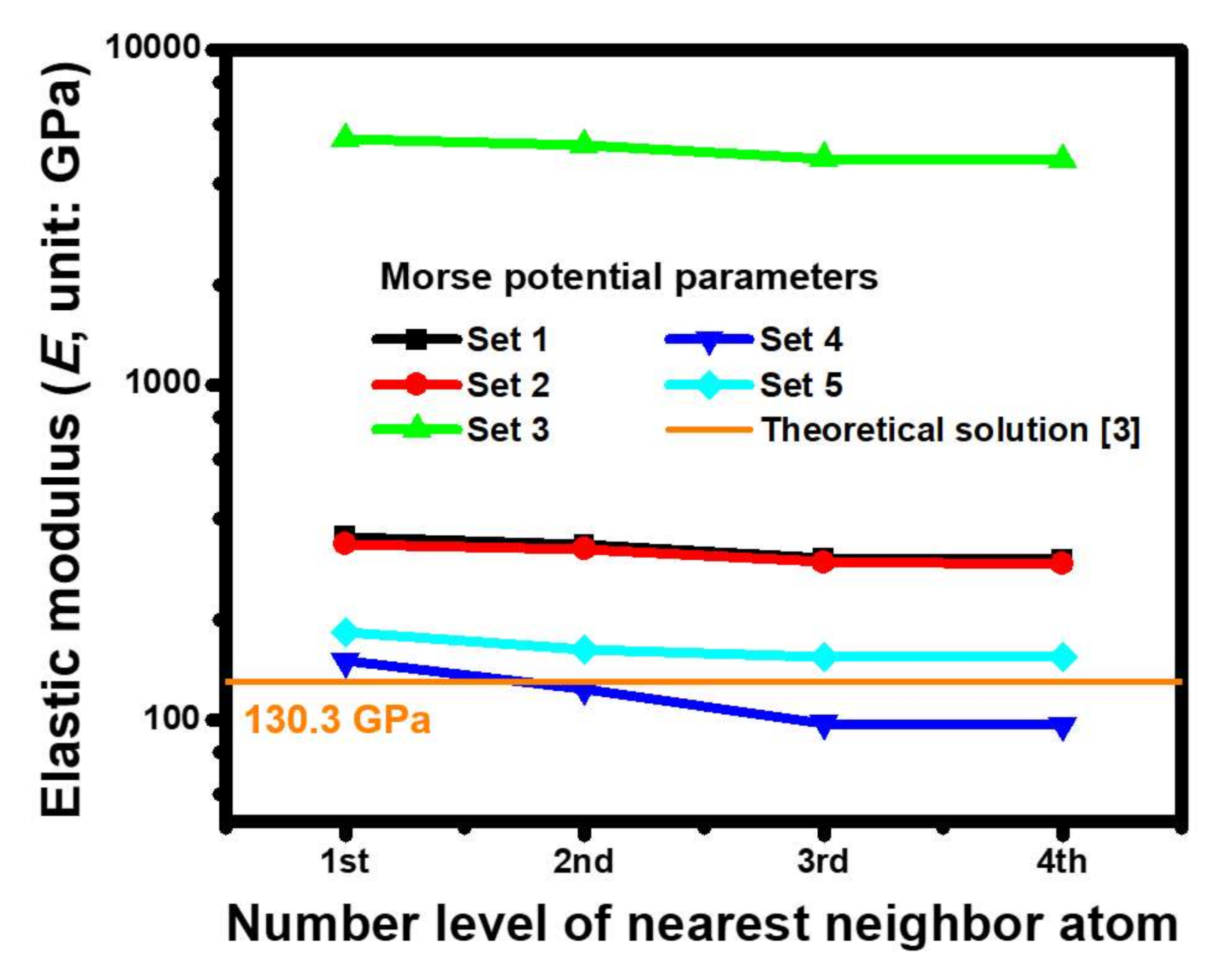
4.1. Estimated Accuracies of the Morse Parameters
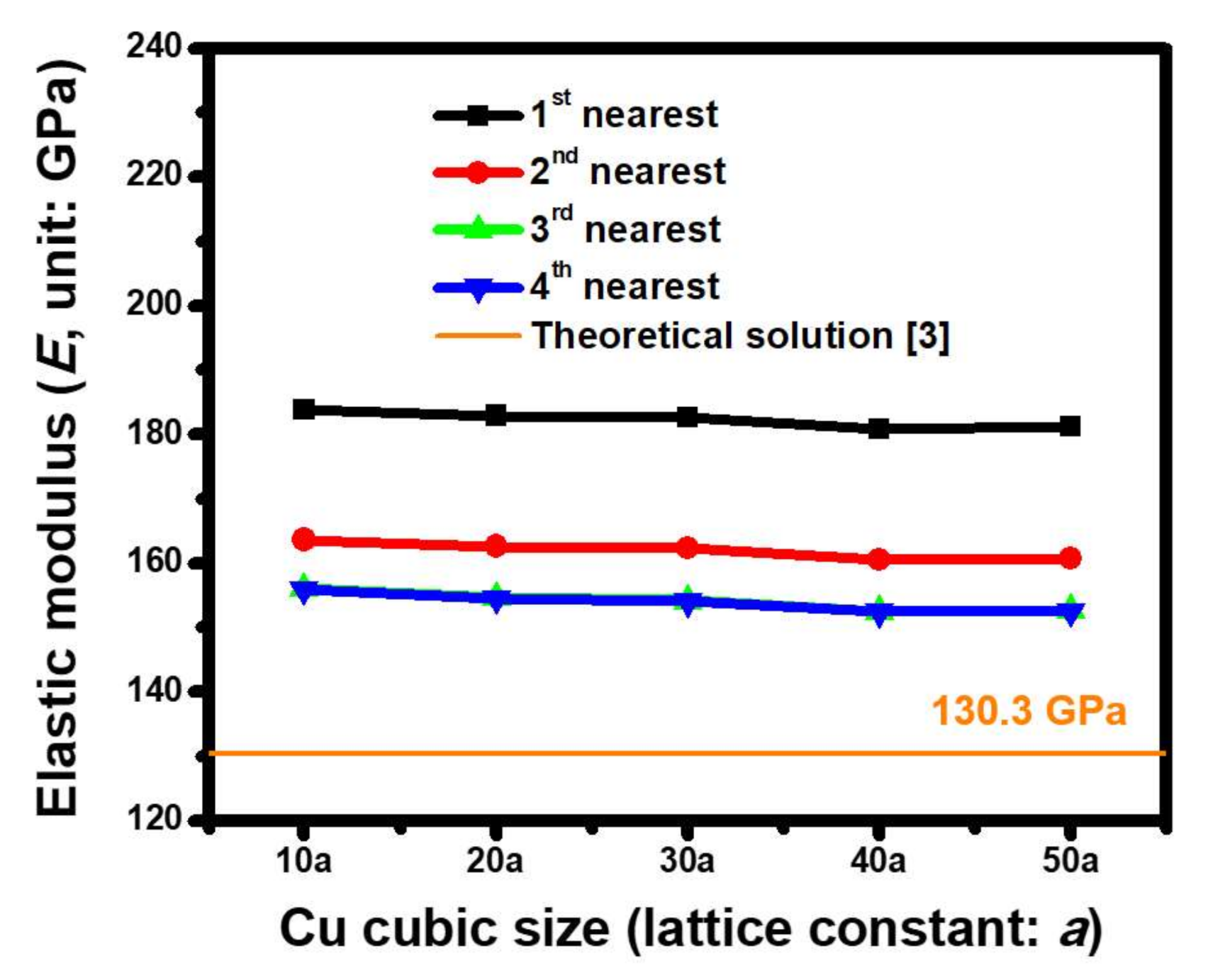
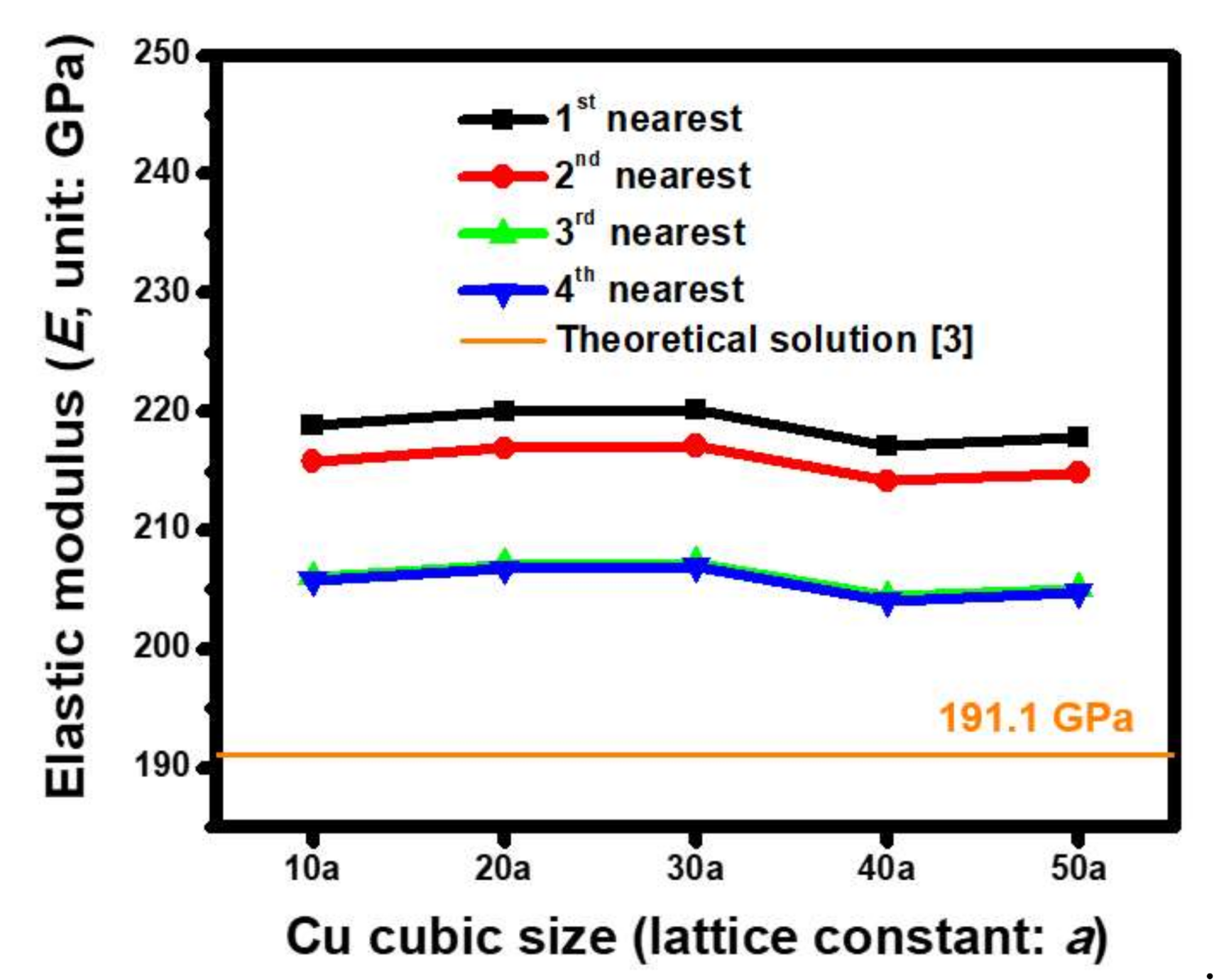
4.2. Oriented Dependence of Atomic-Scale Elastic Modulus (E) Estimated based on ACM Simulations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kwon, Y.; Lee, Y.; Kim, S.; Lee, K.; Choa, Y. Full densification of inkjet-printed copper conductive tracks on a flexible substrate utilizing a hydrogen plasma sintering. Appl. Surf. Sci. 2017, 396, 1239–1244. [Google Scholar] [CrossRef]
- Liao, Y.; Kao, Z. Direct writing patterns for electroless plated copper thin film on plastic substrates. ACS Appl. Mater. Interfaces 2012, 4, 5109–5113. [Google Scholar] [CrossRef] [PubMed]
- Hertzberg, R.W.; Vinci, R.P.; Hertzberg, J.L. Deformation and Fracture Mechanics of Engineering Materials; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Armstrong, D.; Wilkinson, A.; Roberts, S. Measuring anisotropy in Young’s modulus of copper using microcantilever testing. J. Mater. Res. 2009, 24, 3268–3276. [Google Scholar] [CrossRef]
- Wang, W.; Lu, K. Nanoindentation study on elastic and plastic anisotropies of Cu single crystals. Philos. Mag. 2006, 86, 5309–5320. [Google Scholar] [CrossRef]
- Dub, S.; Lim, Y.; Chaudhri, M. Nanohardness of high purity Cu (111) single crystals: The effect of indenter load and prior plastic sample strain. J. Appl. Phys. 2010, 107, 043510. [Google Scholar] [CrossRef]
- Ahadi, A.; Melin, S. Size dependence of the Poisson’s ratio in single-crystal fcc copper nanobeams. Comput. Mater. Sci. 2016, 111, 322–327. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, G.; Luo, G. Mechanical properties of copper nanocube under three-axial tensile loadings. Chin. Phys. B 2015, 24, 066203. [Google Scholar] [CrossRef]
- Wu, H. Molecular dynamics study of the mechanics of metal nanowires at finite temperature. Eur. J. Mech. A-Solids 2006, 25, 370–377. [Google Scholar] [CrossRef]
- Li, C.; Chou, T. A structural mechanics approach for the analysis of carbon nanotubes. Int. J. Solids Struct. 2003, 40, 2487–2499. [Google Scholar] [CrossRef]
- Tserpes, K.; Papanikos, P. Finite element modeling of single-walled carbon nanotubes. Compos. Pt. B-Eng. 2005, 36, 468–477. [Google Scholar] [CrossRef]
- Giannopoulos, G.; Kakavas, P.; Anifantis, N. Evaluation of the effective mechanical properties of single walled carbon nanotubes using a spring based finite element approach. Comput. Mater. Sci. 2008, 41, 561–569. [Google Scholar] [CrossRef]
- Fan, F.; Liu, Y.; Hwu, C. Finite element simulation for estimating the mechanical properties of multi-walled carbon nanotubes. Appl. Phys. A-Mater. Sci. Process 2009, 95, 819–831. [Google Scholar] [CrossRef]
- Lu, X.; Hu, Z. Mechanical property evaluation of single-walled carbon nanotubes by finite element modeling. Compos. Part B Eng. 2012, 43, 1902–1913. [Google Scholar] [CrossRef]
- Sakhaee-Pour, A.; Ahmadian, M.; Vafai, A. Vibrational analysis of single-walled carbon nanotubes using beam element. Thin Walled Struct. 2009, 47, 646–652. [Google Scholar] [CrossRef]
- Chandra, S.; Samal, M.K.; Chavan, V.M.; Raghunathan, S. Hierarchical multiscale modeling of plasticity in copper: From single crystals to polycrystalline aggregates. Int. J. Plast. 2018, 101, 188–212. [Google Scholar] [CrossRef]
- Chandra, S.; Samal, M.K.; Chavan, V.M.; Raghunathan, S. Void growth in single crystal Copper-an atomistic modeling and statistical analysis study. Philos. Mag. 2018, 98, 577–604. [Google Scholar] [CrossRef]
- Lee, G.H.; Chung, Y.J.; Na, S.M.; Beom, H.G. Atomistic investigation of the T-stress effect on fracture toughness of copper and aluminum single crystals. J. Mech. Sci. Technol. 2018, 32, 3765–3774. [Google Scholar] [CrossRef]
- Dai, Y.; Lin, J.; Huang, J. Fractal contact behavior of single crystal copper substrate and rigid plane. J. Dispers. Sci. Technol. 2019, 40, 1504–1512. [Google Scholar] [CrossRef]
- Cui, C.B.; Lee, G.H.; Beom, H.G. Mixed-mode fracture toughness evaluation of a copper single crystal using atomistic simulations. Comput. Mater. Sci. 2017, 136, 216–222. [Google Scholar] [CrossRef]
- Lin, Y.; Shiu, Y. Effect of crystallographic orientation on single crystal copper nanogrooving behaviors by MD method. Int. J. Adv. Manuf. Technol. 2017, 89, 3207–3215. [Google Scholar] [CrossRef]
- Pei, Q.; Lu, C.; Lee, H. Large scale molecular dynamics study of nanometric machining of copper. Comput. Mater. Sci. 2007, 41, 177–185. [Google Scholar] [CrossRef]
- Fang, T.; Weng, C. Three-dimensional molecular dynamics analysis of processing using a pin tool on the atomic scale. Nanotechnology 2000, 11, 148–153. [Google Scholar] [CrossRef]
- Hsu, Q.; Wu, C.; Fang, T. Deformation mechanism and punch taper effects on nanoimprint process by molecular dynamics. Jpn. J. Appl. Phys. 2004, 43, 7665–7669. [Google Scholar] [CrossRef]
- Fang, T.; Jian, S.; Chuu, D. Molecular dynamics analysis of effects of velocity and loading on the nanoindentation. Jpn. J. Appl. Phys. 2002, 41, L1328–L1331. [Google Scholar] [CrossRef]
- Girifalco, L.; Weizer, V. Application of the Morse Potential function to cubic metals. Phys. Rev. 1959, 114, 687–690. [Google Scholar] [CrossRef]
- Cotterill, R.; Doyama, M. Energy and atomic configuration of complete and dissociated dislocations. I. Edge Dislocation in an fcc Metal. Phys. Rev. 1966, 145, 465–478. [Google Scholar] [CrossRef]
- Lincoln, R.; Koliwad, K.; Ghate, P. Morse-potential evaluation of second and third-order elastic constants of some cubic metals. Phys. Rev. 1967, 157, 463–466. [Google Scholar] [CrossRef]
- Maekawa, K.; Itoh, A. Friction and tool wear in nano-scale machining- a molecular dynamics approach. Wear 1995, 188, 115–122. [Google Scholar] [CrossRef]
- Wette, F.D.; Cotterill, R.; Doyama, M. Lattice dynamics of copper with a Morse potential. Phys. Lett. 1966, 23, 309–311. [Google Scholar] [CrossRef]
Sample Availability: The material properties regarding the samples of the single-crystal copper are surveyed and available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter Set No. | D (eV) | α (Å−1) | r0 (Å) | Ref. |
|---|---|---|---|---|
| 1 | 0.343 | 1.356 | 2.866 | [26] |
| 2 | 0.324 | 1.294 | 2.913 | [27] |
| 3 | 5.259 | 1.312 | 2.899 | [28] |
| 4 | 0.343 | 1.356 | 2.626 | [29] |
| 5 | 0.162 | 2.093 | 2.616 | [30] |
| Parameter Sets No. | k1 (nN/Å) | k2 (nN/Å) | k3 (nN/Å) | k4 (nN/Å) |
|---|---|---|---|---|
| 1 | 6.286 | −0.202 | −0.185 | −0.020 |
| 2 | 5.998 | −0.135 | −0.176 | −0.022 |
| 3 | 97.090 | −2.480 | −2.853 | −0.343 |
| 4 | 2.676 | −0.253 | −0.145 | −0.014 |
| 5 | 3.262 | −0.211 | −0.049 | −0.001 |
| Cu(100) | Cu(110) | Cu(111) | Ref. |
|---|---|---|---|
| 66.7 | 130.3 | 191.1 | Theoretical solution [3] |
| 51~55 | 121~134 | 161~181 | [4] |
| 115~142 | 147~167 | 140~169 | [5] |
| 52 | None | 170 | [6] |
| 86.8 | 152.6 | 205.2 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-C.; He, J.-Y. Interactive Field Effect of Atomic Bonding Forces on the Equivalent Elastic Modulus Estimation of Micro-Level Single-Crystal Copper by Utilizing Atomistic-Continuum Finite Element Simulation. Molecules 2020, 25, 5107. https://doi.org/10.3390/molecules25215107
Lee C-C, He J-Y. Interactive Field Effect of Atomic Bonding Forces on the Equivalent Elastic Modulus Estimation of Micro-Level Single-Crystal Copper by Utilizing Atomistic-Continuum Finite Element Simulation. Molecules. 2020; 25(21):5107. https://doi.org/10.3390/molecules25215107
Chicago/Turabian StyleLee, Chang-Chun, and Jing-Yan He. 2020. "Interactive Field Effect of Atomic Bonding Forces on the Equivalent Elastic Modulus Estimation of Micro-Level Single-Crystal Copper by Utilizing Atomistic-Continuum Finite Element Simulation" Molecules 25, no. 21: 5107. https://doi.org/10.3390/molecules25215107
APA StyleLee, C.-C., & He, J.-Y. (2020). Interactive Field Effect of Atomic Bonding Forces on the Equivalent Elastic Modulus Estimation of Micro-Level Single-Crystal Copper by Utilizing Atomistic-Continuum Finite Element Simulation. Molecules, 25(21), 5107. https://doi.org/10.3390/molecules25215107