Development of Diphenethylamines as Selective Kappa Opioid Receptor Ligands and Their Pharmacological Activities



Abstract
1. Introduction
2. Design and Synthesis of Diphenethylamines
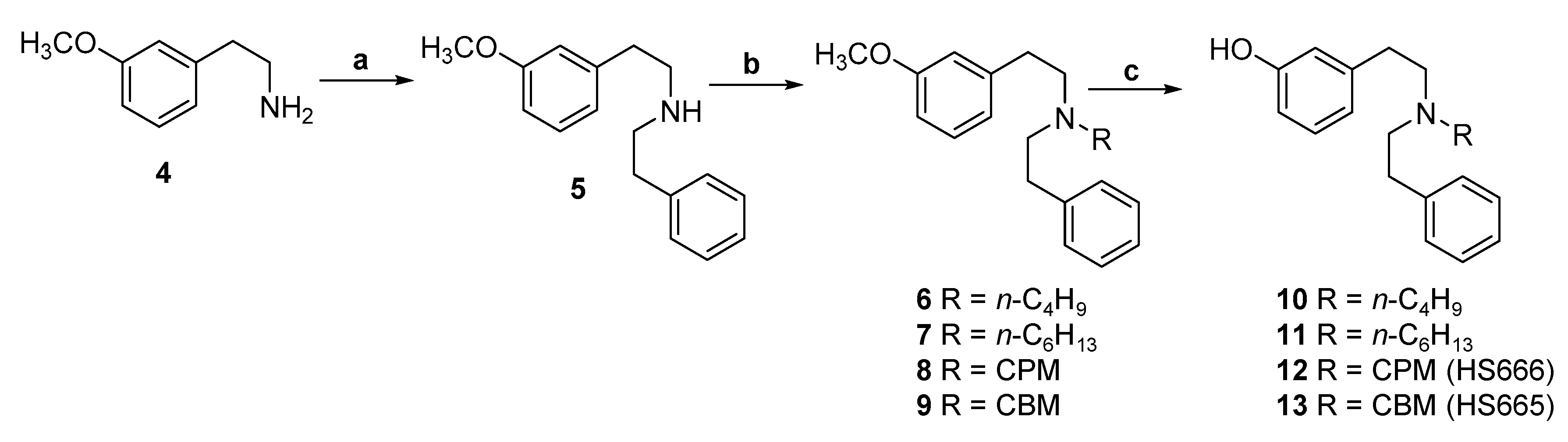
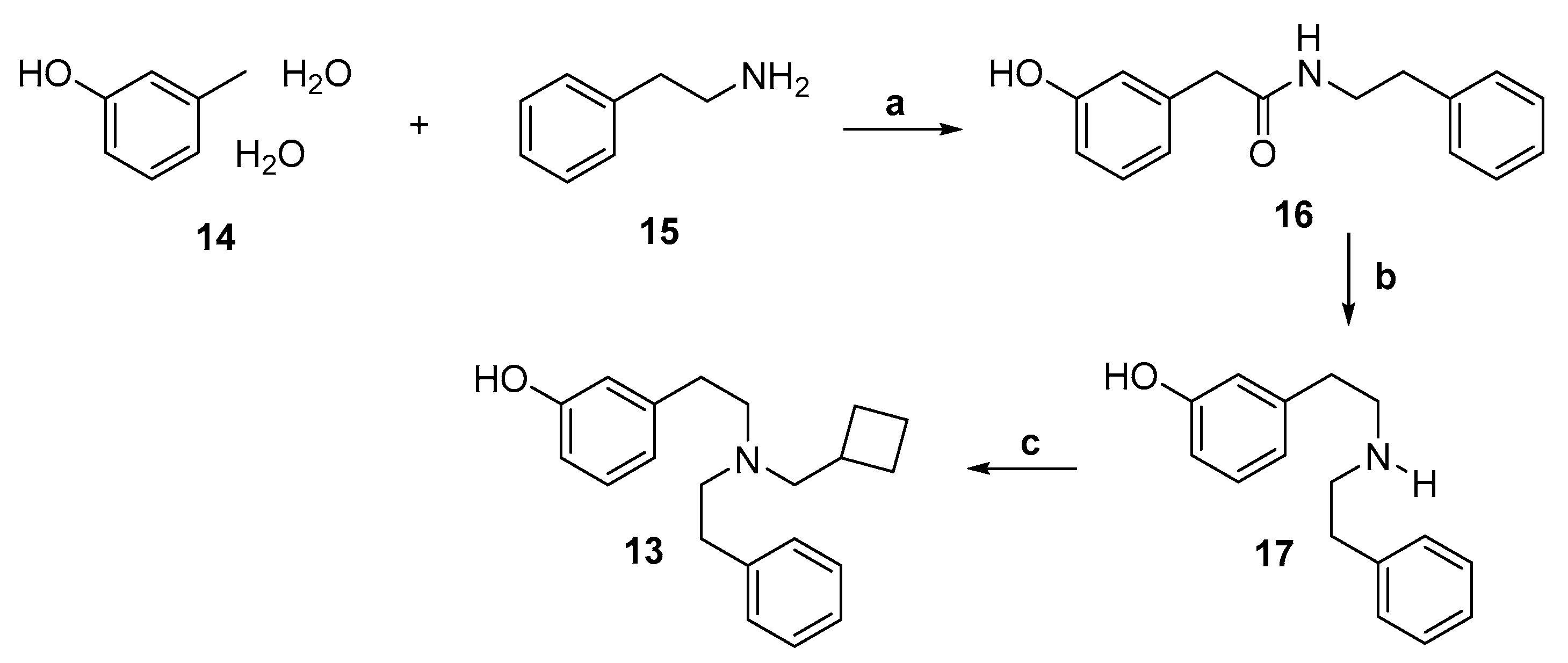
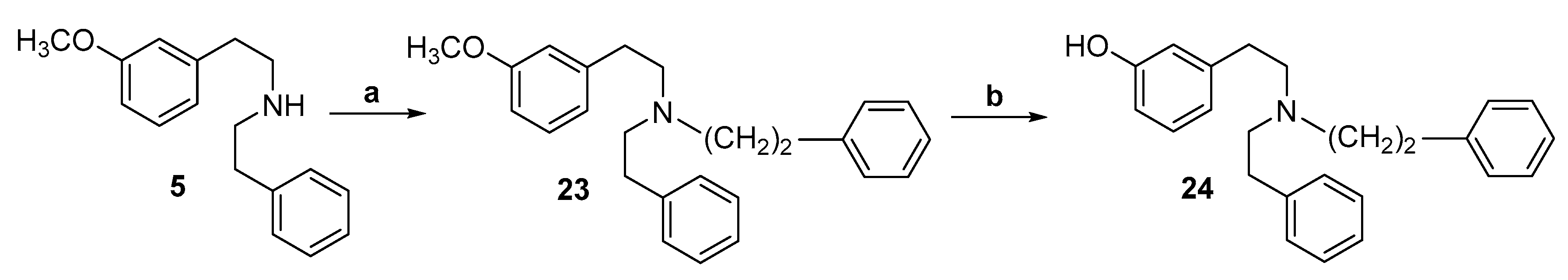
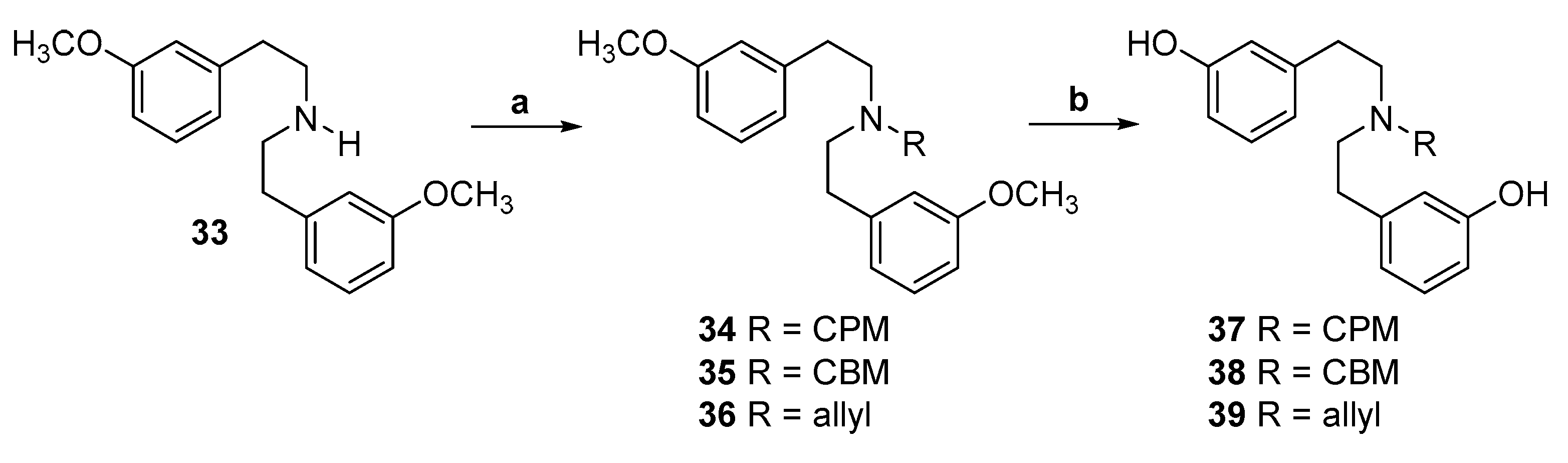
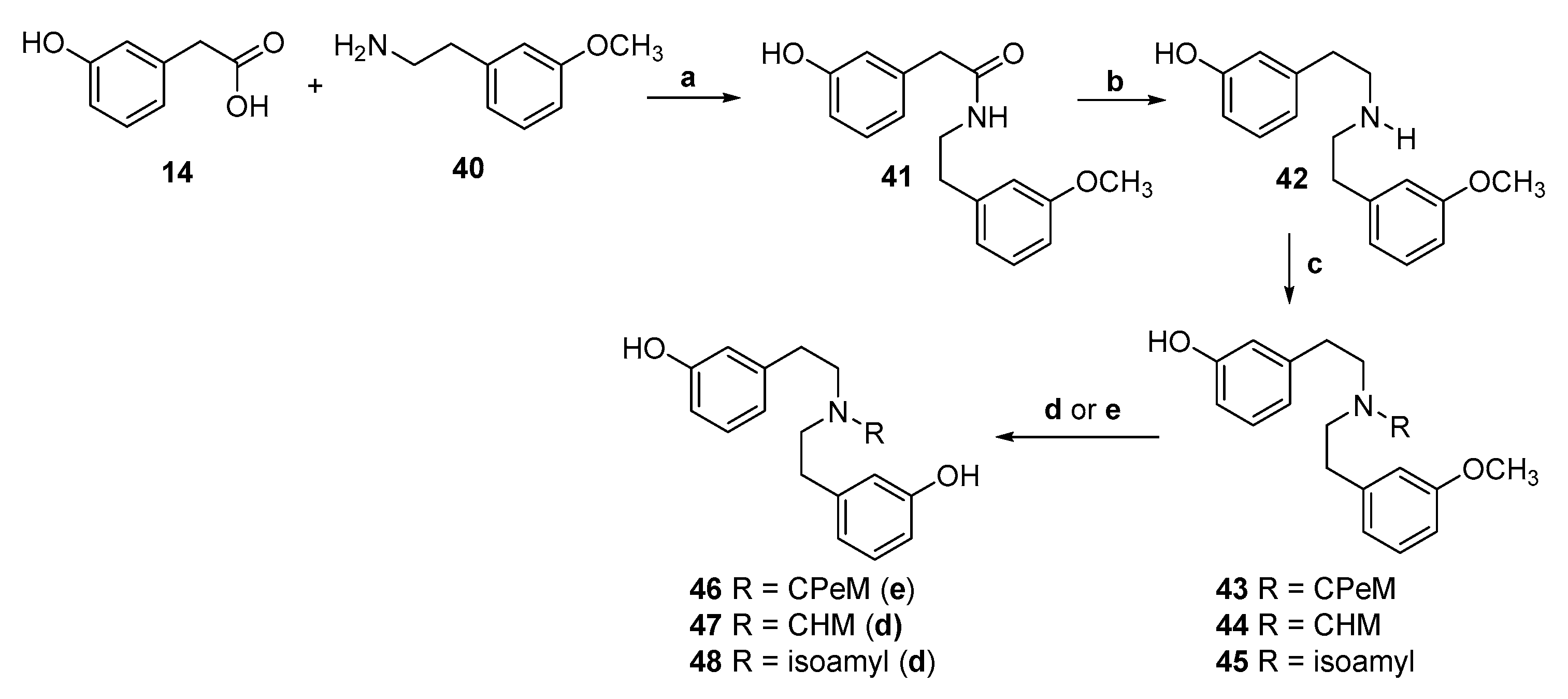
2.1. Synthesis of 3-Monohydroxy-Substituted Diphenethylamines
2.2. Synthesis of 4-Monohydroxy-Substituted Diphenethylamines
2.3. Synthesis of 3,3′-Dihydroxy-Substituted Diphenethylamines
2.4. Synthesis of 3,4′-Dihydroxy-Substituted Diphenethylamines
2.5. Synthesis of 2-Fluoro-Substituted Diphenethylamines
2.6. Synthesis of an Aromatic Unsubstituted Diphenethylamine
2.7. Synthesis of [3H]HS665
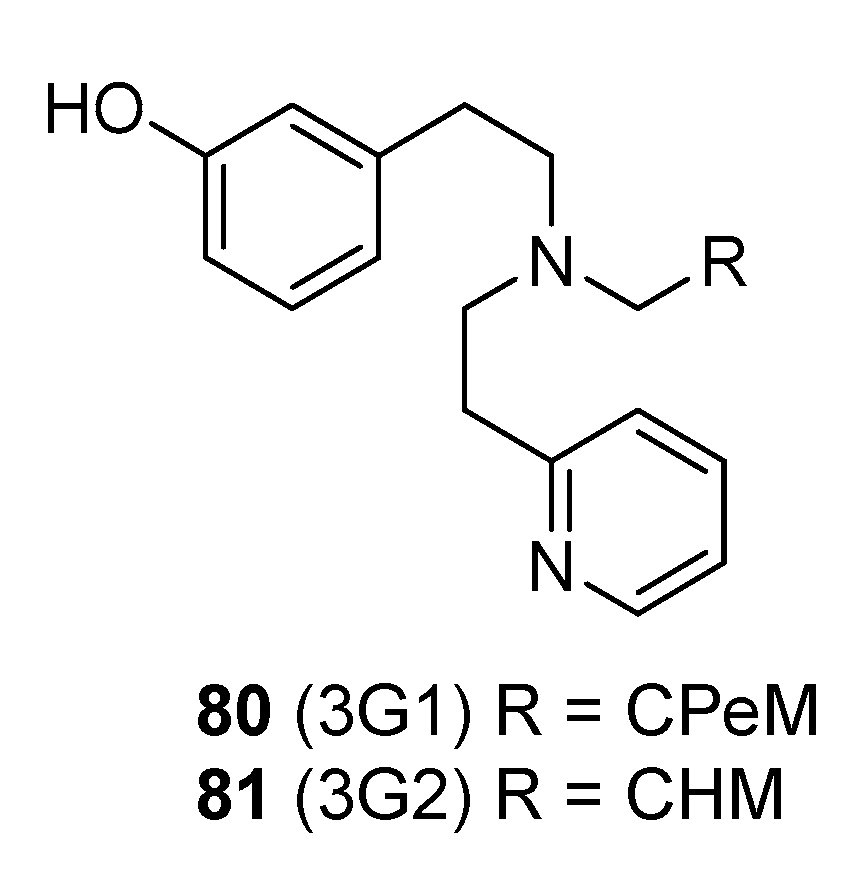
2.8. Synthesis of Structurally Related Diphenethylamines
3. Pharmacological Activities of Diphenethylamines at the KOR
3.1. Agonists and Partial Agonists
3.2. Biased Agonists
3.3. Antinociceptive and Behavioral Effects of Agonists from the Class of Diphenethylamines
3.4. Antagonists
4. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Evans, C.J. Secrets of opium poppy revealed. Neuropharmacology 2004, 47, 293–299. [Google Scholar] [CrossRef]
- Darcq, E.; Kieffer, B.L. Opioid receptors: Drivers to addiction. Nature 2018, 19, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Pert, C.B.; Snyder, S.H. Properties of opiate-receptor binding in rat brain. Science 1973, 70, 2243–2247. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.J.; Hiller, J.M.; Edelman, I. Stereospecific binding of the potent narcotic analgesic [3H]etorphine to rat-brain homogenate. Proc. Natl. Acad. Sci. USA 1973, 70, 1947–1949. [Google Scholar] [CrossRef]
- Terenius, L. Stereospecific interaction between narcotic analgesics and a synaptic plasma membrane fraction of rat cerebral cortex. Acta Pharmacol. Toxicol. 1973, 32, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar] [PubMed]
- Stein, C. Opioid receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Corder, G.; Castro, D.C.; Bruchas, M.R.; Scherrer, G. Endogenous and exogenous opioids in pain. Annu. Rev. Neurosci. 2018, 41, 453–473. [Google Scholar] [CrossRef]
- Waldoher, M.; Bartlett, S.E.; Whistler, J.L. Opioid receptors. Annu. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef] [PubMed]
- Casy, A.F.; Parfitt, R.T. Opioid Analgesics: Chemistry and Receptors; Plenum Press: New York, NY, USA, 1986. [Google Scholar]
- Fürst, S.; Hosztafi, S. The chemical and pharmacological importance of morphine analogues. Acta Physiol. Hung. 2008, 95, 3–44. [Google Scholar] [CrossRef]
- Spetea, M.; Asim, M.F.; Wolber, G.; Schmidhammer, H. The μ opioid receptor and ligands acting at the μ opioid receptor, as therapeutics and potential therapeutics. Curr. Pharm. Des. 2013, 19, 7415–7434. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W. Mu opioid pharmacology: 40 years to the promised land. Adv. Pharmacol. 2018, 82, 261–291. [Google Scholar]
- Volkow, N.D.; Jones, E.B.; Einstein, E.B.; Wargo, E.M. Prevention and treatment of opioid misuse and addiction: A review. JAMA Psychiatry 2019, 76, 208–216. [Google Scholar] [CrossRef]
- Stevens, G.W. Receptor-centric solutions for the opioid epidemic: Making the opioid user impervious to overdose death. J. Neurosci. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lemos, C.J.; Chavkin, C. Kappa opioid receptor function. In The Opiate Receptors, 2nd ed.; Pasternak, C.W., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 265–305. [Google Scholar]
- Imam, M.Z.; Kuo, A.; Ghassabian, S.; Smith, M.T. Progress in understanding mechanisms of opioid-induced gastrointestinal adverse effects and respiratory depression. Neuropharmacology 2018, 131, 238–255. [Google Scholar] [CrossRef]
- Goldstein, A.; Tachibana, S.; Lowney, L.I.; Hunkapiller, M.; Hood, L. Dynorphin-(1-13), an extraordinarily potent opioid peptide. Proc. Natl. Acad. Sci. USA 1979, 76, 6666–6670. [Google Scholar] [CrossRef]
- Stein, C.; Millan, M.J.; Shippenberg, T.S.; Peter, K.; Herz, A. Peripheral opioid receptors mediating antinociception in inflammation. Evidence for involvement of mu, delta and kappa receptors. J. Pharmacol. Exp. Ther. 1989, 248, 1269–1275. [Google Scholar]
- Ji, R.R.; Zhang, Q.; Law, P.Y.; Low, H.H.; Elde, R.; Hökfelt, T. Expression of µ-, δ-, and κ-opioid receptor-like immunoreactivities in rat dorsal root ganglia after carrageenan-induced inflammation. J. Neurosci. 1995, 15, 8156–8166. [Google Scholar] [CrossRef]
- Mansour, A.; Fox, C.A.; Akil, H.; Watson, S.J. Opioid-receptor mRNA expression in the rat CNS: Anatomical and functional implications. Trends Neurosci. 1995, 18, 22–29. [Google Scholar] [CrossRef]
- McCarthy, L.; Wetzel, M.; Sliker, J.K.; Eisenstein, T.K.; Rogers, T.J. Opioids, opioid receptors, and the immune response. Drug Alcohol Depend. 2001, 62, 111–123. [Google Scholar] [CrossRef]
- Holzer, P. Opioid receptors in the gastrointestinal tract. Regul. Pept. 2009, 155, 11–17. [Google Scholar] [CrossRef]
- Peng, J.; Sarkar, S.; Chang, S. Opioid receptor expression in human brain and peripheral tissues using absolute quantitative real-time RT-PCR. Drug Alcohol Depend. 2012, 124, 223–228. [Google Scholar] [CrossRef]
- Cowan, A.; Kehner, G.B.; Inan, S. Targeting itch with ligands selective for κ opioid receptors. Handb. Exp. Pharmacol. 2015, 226, 291–314. [Google Scholar]
- Snyder, L.M.; Chiang, M.C.; Loeza-Alcocer, E.; Omori, Y.; Hachisuka, J.; Sheahan, T.D.; Gale, J.R.; Adelman, P.C.; Sypek, E.I.; Fulton, S.A.; et al. Kappa opioid receptor distribution and function in primary afferents. Neuron 2018, 99, 1274–1288. [Google Scholar] [CrossRef]
- Van’t Veer, A.; Carlezon, W.A. Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology 2013, 229, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Cahill, C.M.; Taylor, A.M.W.; Cook, C.; Ong, E.; Morón, J.A.; Evans, C.J. Does the kappa opioid receptor system contribute to pain aversion? Front. Pharmacol. 2014, 253, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lalanne, L.; Ayranci, G.; Kieffer, B.L.; Lutz, P.E. The kappa opioid receptor: From addiction to depression, and back. Front. Psychiatry 2014, 170, 1–17. [Google Scholar] [CrossRef]
- Crowley, N.A.; Kash, T.L. Kappa opioid receptor signaling in the brain: Circuitry and implications for treatment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 62, 51–60. [Google Scholar] [CrossRef]
- Burtscher, J.; Schwarzer, C. The opioid system in temporal lobe epilepsy: Functional role and therapeutic potential. Front. Mol. Neurosci. 2017, 10, 245. [Google Scholar] [CrossRef]
- Kivell, B.; Prisinzano, T.E. Kappa opioids and the modulation of pain. Psychopharmacology 2010, 210, 109–119. [Google Scholar] [CrossRef]
- Albert-Vartanian, A.; Boyd, M.R.; Hall, A.L.; Morgado, S.J.; Nguyen, E.; Nguyen, V.P.; Patel, S.P.; Russo, L.J.; Shao, A.J.; Raffa, R.B. Will peripherally restricted κ opioid receptor agonists (pKORAs) relieve pain with less opioid adverse effects and abuse potential? J. Clin. Pharm. Ther. 2016, 41, 371–382. [Google Scholar] [CrossRef]
- Beck, T.C.; Dix, T.A. Targeting peripheral κ-opioid receptors for the non-addictive treatment of pain. Future Drug Discov. 2019, 1, FDD17. [Google Scholar] [CrossRef] [PubMed]
- Shigeki, I. Nalfurafine hydrochloride to treat pruritus: A review. Clin. Cosmet. Investig. Dermatol. 2015, 8, 249–255. [Google Scholar]
- Zangrandi, L.; Burtscher, J.; MacKay, J.P.; Colmers, W.F.; Schwarzer, C. The G-protein biased partial κ opioid receptor agonist 6′GNTI blocks hippocampal paroxysmal discharges without inducing aversion. Br. J. Pharmacol. 2016, 173, 1756–1767. [Google Scholar] [CrossRef]
- Spetea, M.; Asim, M.F.; Noha, S.; Wolber, G.; Schmidhammer, H. Current ĸ-opioid receptor ligands and discovery of a new molecular scaffold as a ĸ-opioid receptor antagonist using pharmacophore-based virtual screening. Curr. Pharm. Des. 2013, 19, 7362–7372. [Google Scholar] [CrossRef]
- Carroll, F.I.; Carlezon, W.A., Jr. Development of κ opioid receptor antagonists. J. Med. Chem. 2013, 56, 2178–2195. [Google Scholar] [CrossRef]
- Rorick-Kehn, L.M.; Witkin, J.M.; Statnick, M.A.; Eberle, E.L.; McKinzie, J.H.; Kahl, S.D.; Forster, B.M.; Wong, C.J.; Li, X.; Crile, R.S.; et al. LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology 2014, 77, 131–144. [Google Scholar] [CrossRef]
- Urbano, M.; Guerrero, M.; Rosen, H.; Roberts, E. Antagonists of the kappa opioid receptor. Bioorg. Med. Chem. Lett. 2014, 24, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Simonson, B.; Morani, A.S.; Ewald, A.W.; Walker, L.; Kumar, N.; Simpson, D.; Miller, J.H.; Prisinzano, T.E.; Kivell, B.M. Pharmacology and anti-addiction effects of the novel κ opioid receptor agonist Mesyl Sal B, a potent and long-acting analogue of salvinorin A. Br. J. Pharmacol. 2015, 172, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Carlezon, W.A., Jr.; Krystal, A.D. Kappa-opioid antagonists for psychiatric disorders: From bench to clinical trials. Depress. Anxiety 2016, 33, 895–906. [Google Scholar] [CrossRef]
- Browne, C.A.; Lucki, I. Targeting opioid dysregulation in depression for the development of novel therapeutics. Pharmacol. Ther. 2019, 201, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.L.; Browne, C.A.; Lucki, I. Kappa opioid receptor antagonists as potential therapeutics for stress-related disorders. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 615–636. [Google Scholar] [CrossRef] [PubMed]
- Pande, A.C.; Pyke, R.E.; Greiner, M.; Wideman, G.L.; Benjamin, R.; Pierce, M.W. Analgesic efficacy of enadoline versus placebo or morphine in postsurgical pain. Clin. Neuropharmacol. 1996, 19, 451–456. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Brantl, V.; Herz, A.; Emrich, H.M. Psychotomimesis mediated by kappa opiate receptors. Science 1986, 233, 774–776. [Google Scholar] [CrossRef]
- Ranganathan, M.; Schnakenberg, A.; Skosnik, P.D.; Cohen, B.M.; Pittman, B.; Sewell, R.A.; D’Souza, D.C. Dose-related behavioral, subjective, endocrine, and psychophysiological effects of the κ opioid agonist Salvinorin A in humans. Biol. Psychiatry 2012, 72, 871–879. [Google Scholar] [CrossRef]
- Land, B.B.; Bruchas, M.R.; Lemos, J.C.; Xu, M.; Melief, E.J.; Chavkin, C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J. Neurosci. 2008, 28, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Tejeda, H.A.; Counotte, D.S.; Oh, E.; Ramamoorthy, S.; Schultz-Kuszak, K.N.; Bäckman, C.M.; Chefer, V.; O’Donnell, P.; Shippenberg, T.S. Prefrontal cortical kappa-opioid receptor modulation of local neurotransmission and conditioned place aversion. Neuropsychopharmacology 2013, 38, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, Z.; Brust, T.F.; Bohn, L.M. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett. 2016, 26, 241–250. [Google Scholar] [CrossRef]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef]
- Mores, K.L.; Cassell, R.J.; van Rijn, R.M. Arrestin recruitment and signaling by G protein-coupled receptor heteromers. Neuropharmacology 2019, 152, 15–21. [Google Scholar] [CrossRef]
- Seyedabadi, M.; Ghahremani, M.H.; Albert, P.R. Biased signaling of G protein coupled receptors (GPCRs): Molecular determinants of GPCR/transducer selectivity and therapeutic potential. Pharmacol. Ther. 2019, 200, 148–178. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Schindler, A.G.; Shankar, H.; Messinger, D.I.; Miyatake, M.; Land, B.B.; Lemos, J.C.; Hagan, C.E.; Neumaier, J.F.; Quintana, A.; et al. Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron 2011, 71, 498–511. [Google Scholar] [CrossRef]
- Ehrich, J.M.; Messinger, D.I.; Knakal, C.R.; Kuhar, J.R.; Schattauer, S.S.; Bruchas, M.R.; Zweifel, L.S.; Kieffer, B.L.; Phillips, P.E.; Chavkin, C. Kappa opioid receptor-induced aversion requires p38 MAPK activation in VTA dopamine neurons. J. Neurosci. 2015, 35, 1291–12931. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Roth, B.L. New technologies for elucidating opioid receptor function. Trends Pharmacol. Sci. 2016, 37, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Aubé, J. Seeking (and finding) biased ligands of the kappa opioid receptor. ACS Med. Chem. Lett. 2017, 8, 694–700. [Google Scholar] [CrossRef]
- Mores, K.L.; Cummins, B.R.; Cassell, R.J.; van Rijn, R.M. A review of the therapeutic potential of recently developed G protein-biased kappa agonists. Front. Pharmacol. 2019, 10, 407. [Google Scholar] [CrossRef]
- Turnaturi, R.; Chiechio, S.; Salerno, L.; Rescifina, A.; Pittalà, V.; Cantarella, G.; Tomarchio, E.; Parenti, C.; Pasquinucci, L. Progress in the development of more effective and safer analgesics for pain management. Eur. J. Med. Chem. 2019, 183, 111701. [Google Scholar] [CrossRef]
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased opioid ligands. Molecules 2020, 25, 4257. [Google Scholar] [CrossRef]
- Aldrich, J.V.; McLaughlin, J.P. Opioid peptides: Potential for drug development. Drug Discov. Today Technol. 2012, 9, e23–e31. [Google Scholar] [CrossRef]
- Chavkin, C.; Martinez, D. Kappa antagonist JDTic in phase 1 clinical trial. Neuropsychopharmacology 2015, 40, 2057–2058. [Google Scholar] [CrossRef]
- Helal, M.A.; Habib, E.S.; Chittiboyina, A.G. Selective kappa opioid antagonists for treatment of addiction, are we there yet? Eur. J. Med. Chem. 2017, 141, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Roach, J.J.; Shenvi, R.A. A review of salvinorin analogs and their kappa-opioid receptor activity. Bioorg. Med. Chem. Lett. 2018, 28, 1436–1445. [Google Scholar] [CrossRef]
- Turnaturi, R.; Marrazzo, A.; Parenti, C.; Pasquinucci, L. Benzomorphan scaffold for opioid analgesics and pharmacological tools development: A comprehensive review. Eur. J. Med. Chem. 2018, 148, 410–422. [Google Scholar] [CrossRef]
- Coffeen, U.; Pellicer, F. Salvia divinorum: From recreational hallucinogenic use to analgesic and anti-inflammatory action. J. Pain Res. 2019, 12, 1069–1076. [Google Scholar] [CrossRef]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Won Han, G.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the nanobody-stabilized active state of the κ opioid receptor. Cell 2018, 172, 55–67. [Google Scholar] [CrossRef]
- Ferré, G.; Czaplicki, G.; Demange, P.; Milon, A. Structure and dynamics of dynorphin peptide and its receptor. Vitam. Horm. 2019, 111, 17–47. [Google Scholar]
- Filizola, M. Insights from molecular dynamics simulations to exploit new trends for the development of improved opioid drugs. Neurosci. Lett. 2019, 700, 50–55. [Google Scholar] [CrossRef]
- Manglik, A. Molecular basis of opioid action: From structures to new leads. Biol. Psychiatry 2020, 87, 6–14. [Google Scholar] [CrossRef]
- Nedelec, L.; Dumont, C.; Oberlander, C.; Frechet, D.; Laurent, J.; Boissier, J.R. Synthèse et étude de l’activité dopaminergique de dérivés de la di(phénéthyl)amine. Eur. J. Med. Chem. Chim. Ther. 1978, 13, 553–563. [Google Scholar]
- Euvrard, C.; Ferland, L.; Di Paolo, T.; Beaulieu, M.; Labrie, F.; Oberlander, C.; Raynaud, J.P.; Boissier, J.R. Activity of two new dopaminergic agonists at the striatal and anterio pituitary levels. Neuropharmacology 1980, 19, 379–386. [Google Scholar] [CrossRef]
- Fortin, M.; Degryse, M.; Petit, F.; Hunt, P.F. The dopamine D2 agonist RU 241213 and RU 24926 are also kappa-opioid receptor antagonists. Neuropharmacology 1991, 30, 409–412. [Google Scholar] [CrossRef]
- Cosquer, P.; Delevallee, F.; Droux, S.; Fortin, M.; Petit, F. Amine Compounds. US Patent 5,141,962, 25 August 1992. [Google Scholar]
- Spetea, M.; Berzetei-Gurske, I.P.; Guerrieri, E.; Schmidhammer, H. Discovery and pharmacological evaluation of a diphenethylamine derivative (HS665), a highly potent and selective κ opioid receptor agonist. J. Med. Chem. 2012, 55, 10302–10306. [Google Scholar] [CrossRef]
- Guerrieri, E.; Bermudez, M.; Wolber, G.; Berzetei-Gurske, I.P.; Schmidhammer, H.; Spetea, M. Structural determinants of diphenethylamines for interaction with the κ opioid receptor: Synthesis, pharmacology and molecular modeling studies. Bioorg. Med. Chem. Lett. 2016, 26, 4769–4774. [Google Scholar] [CrossRef]
- Erli, F.; Guerrieri, E.; Ben Haddou, T.; Lantero, A.; Mairegger, M.; Schmidhammer, H.; Spetea, M. Highly potent and selective new diphenethylamines interacting with the κ-opioid receptor: Synthesis, pharmacology, and structure-activity relationships. J. Med. Chem. 2017, 60, 7579–7590. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.D.; Reed, B.; Guariglia, C.; Dunn, A.M.; Hillman, J.M.; Kreek, M.J. Structurally related kappa opioid receptor agonists with substantial differential signaling bias: Neuroendocrine and behavioral effects in C57BL6 mice. Int. J. Neuropsychopharmacol. 2018, 21, 847–857. [Google Scholar] [CrossRef]
- Dunn, A.D.; Reed, B.; Erazo, J.; Ben-Ezra, A.; Kreek, M.J. Signaling properties of structurally diverse kappa opioid receptor ligands: Toward in vitro models of in vivo responses. ACS Chem. Neurosci. 2019, 10, 3590–3600. [Google Scholar] [CrossRef]
- Schmidhammer, H.; Spetea, M.; Guerrieri, E. Diphenethylamine Derivatives which are Inter Alia Useful as Analgesics and Methods for their Production. US Patent 10,377,698, 13 August 2019. [Google Scholar]
- Rice, K.C. A rapid, high-yield conversion of codeine to morphine. J. Med. Chem. 1977, 20, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.M.; Shu, Y.-Z.; Zhuo, X.; Meanwell, N.A. Metabolic and pharmaceutical aspects of fluorinated compounds. J. Med. Chem. 2020, 63, 6315–6386. [Google Scholar] [CrossRef]
- Tóth, G.; Mallareddy, J.R.; Tóth, F.; Lipkowski, A.W.; Tourwe, D. Radiotracers, tritium labeling of neuropeptides. ARKIVOC 2012, 163–174. [Google Scholar] [CrossRef]
- Tóth, G.; Mallareddy, J.R. Tritiated opioid receptor ligands as radiotracers. Curr. Pharm. Des. 2013, 19, 7461–7472. [Google Scholar] [CrossRef]
- Guerrieri, E.; Mallareddy, J.R.; Tóth, G.; Schmidhammer, H.; Spetea, M. Synthesis and pharmacological evaluation of [3H]HS665, a novel, highly selective radioligand for the kappa opioid receptor. ACS Chem. Neurosci. 2015, 6, 456–463. [Google Scholar] [CrossRef]
- Dumitrascuta, M.; Ben Haddou, T.; Guerrieri, E.; Noha, S.M.; Schläfer, L.; Schmidhammer, H.; Spetea, M. Synthesis, pharmacology, and molecular docking studies on 6-desoxo-N-methylmorphinans as potent μ-opioid receptor agonists. J. Med. Chem. 2017, 60, 9407–9412. [Google Scholar] [CrossRef] [PubMed]
- Erdei, A.I.; Borbély, A.; Magyar, A.; Taricska, N.; Perczel, A.; Zsíros, O.; Garab, G.; Szűcs, E.; Ötvös, F.; Zádor, F.; et al. Biochemical and pharmacological characterization of three opioid-nociceptin hybrid peptide ligands reveals substantially differing modes of their actions. Peptides 2018, 99, 205–216. [Google Scholar] [CrossRef]
- Martin, C.; Dumitrascuta, M.; Mannes, M.; Lantero, A.; Bucher, D.; Walker, K.; Van Wanseele, Y.; Oyen, E.; Hernot, S.; Van Eeckhaut, A.; et al. Biodegradable amphipathic peptide hydrogels as extended-release system for opioid peptides. Med. Chem. 2018, 61, 9784–9789. [Google Scholar] [CrossRef]
- Dumitrascuta, M.; Bermudez, M.; Ben Haddou, T.; Guerrieri, E.; Schläfer, L.; Ritsch, A.; Hosztafi, S.; Lantero, A.; Kreutz, C.; Massotte, D.; et al. N-Phenethyl substitution in 14-methoxy-N-methylmorphinan-6-ones turns selective µ opioid receptor ligands into dual µ/δ opioid receptor agonists. Sci. Rep. 2020, 10, 5653. [Google Scholar] [CrossRef]
- Szűcs, E.; Marton, J.; Szabó, Z.; Hosztafi, S.; Kékesi, G.; Tuboly, G.; Bánki, L.; Horváth, G.; Szabó, P.T.; Tömböly, C.; et al. Synthesis, biochemical, pharmacological characterization and in silico profile modelling of highly potent opioid orvinol and thevinol derivatives. Eur. J. Med. Chem. 2020, 191, 112–145. [Google Scholar] [CrossRef]
- Szűcs, E.; Stefanucci, A.; Dimmito, M.P.; Zádor, F.; Pieretti, S.; Zengin, G.; Vécsei, L.; Benyhe, S.; Nalli, M.; Mollica, A. Discovery of kynurenines containing oligopeptides as potent opioid receptor agonists. Biomolecules 2020, 10, 284. [Google Scholar] [CrossRef]
- Spetea, M.; Eans, S.O.; Ganno, M.L.; Lantero, A.; Mairegger, M.; Toll, L.; Schmidhammer, H.; McLaughlin, J.P. Selective κ receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice. Br. J. Pharmacol. 2017, 174, 2444–2456. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Robinson, J.E.; Zhu, H.; DiBerto, J.F.; Polepally, P.R.; Zjawiony, J.K.; Nichols, D.E.; Malanga, C.J.; Roth, B.L. The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J. Pharmacol. Exp. Ther. 2015, 352, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Sharma, K.; Zangrandi, L.; Chen, C.; Humphrey, S.J.; Chiu, Y.-T.; Spetea, M.; Liu-Chen, L.Y.; Schwarzer, C.; Mann, M. In vivo brain GPCR signaling elucidated by phosphoproteomics. Science 2018, 360, eaao4927. [Google Scholar] [CrossRef]
- Liu, J.J.; Chiu, Y.T.; DiMattio, K.M.; Chen, C.; Huang, P.; Gentile, T.A.; Muschamp, J.W.; Cowan, A.; Mann, M.; Liu-Chen, L.Y. Phosphoproteomic approach for agonist-specific signaling in mouse brains: mTOR pathway is involved in κ opioid aversion. Neuropsychopharmacology 2019, 44, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Von, P.F.V.; Lewis, R.A. U-50,488, a selective kappa opioid agonist: Comparison to other reputed kappa agonists. Prog. Neuropsychopharmacol. Biol. Psychiatry 1982, 6, 467–470. [Google Scholar]
- Sharma, S.K.; Jones, R.M.; Metzger, T.G.; Ferguson, D.M.; Portoghese, P.S. Transformation of a kappa-opioid receptor antagonist to a kappa agonist by transfer of a guanidinium group from the 5′- to 6′-position of naltrindole. J. Med. Chem. 2001, 44, 2073–2079. [Google Scholar] [CrossRef]
- Yan, F.; Bikbulatov, R.V.; Mocanu, V.; Dicheva, N.; Parker, C.E.; Wetsel, W.C.; Mosier, P.D.; Westkaemper, R.B.; Allen, J.A.; Zjawiony, J.K.; et al. Structure-based design, synthesis, and biochemical and pharmacological characterization of novel salvinorin A analogues as active state probes of the kappa-opioid receptor. Biochemistry 2009, 48, 6898–6908. [Google Scholar] [CrossRef]
- White, K.L.; Scopton, A.P.; Rives, M.L.; Bikbulatov, R.V.; Polepally, P.R.; Brown, P.J.; Kenakin, T.; Javitch, J.A.; Zjawiony, J.K.; Roth, B.L. Identification of novel functionally selective κ-opioid receptor scaffolds. Mol. Pharmacol. 2014, 85, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Rives, M.L.; Rossillo, M.; Liu-Chen, L.Y.; Javitch, J.A. 6′-Guanidinonaltrindole (6′-GNTI) is a G protein-biased κ-opioid receptor agonist that inhibits arrestin recruitment. J. Biol. Chem. 2012, 287, 27050–27054. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.L.; Streicher, J.M.; Groer, C.E.; Munro, T.A.; Zhou, L.; Bohn, L.M. Functional selectivity of 6′-guanidinonaltrindole (6′-GNTI) at κ-opioid receptors in striatal neurons. J. Biol. Chem. 2013, 288, 22387–22398. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Ligand | R1, R2, R3, R4, R5 | KOR Binding a | KOR Activity b | ||
|---|---|---|---|---|---|
| Ki (nM) | Ki ratio KOR/MOR/DOR | EC50 (nM) | %Stim. | ||
| 3-Monohydroxy-substituted | |||||
| 1 (RU 24213) | n-C3H7, H, OH, H, H | 8.13 | 1/73/457 | 49.1 | 21.1 |
| 3 | n-C5H11, H, OH, H, H | 12.6 | 1/26/104 | 86.4 | 36.2 |
| 10 | n-C4H9, H, OH, H, H | 10.9 | 1/38/223 | 46.2 | 45.5 |
| 11 | n-C6H13, H, OH, H, H | 141 | 1/5.6/25 | 647 | 24.0 |
| 12 (HS666) | CPM, H, OH, H, H | 5.90 | 1/140/>1700 | 35.0 | 53.4 |
| 13 (HS665) | CBM, H, OH, H, H | 0.49 | 1/1106/>20000 | 3.62 | 90.0 |
| 18 | CPeM, H, OH, H, H | 0.017 | 1/16118/133471 | 3.87 | 82.8 |
| 19 | CHM, H, OH, H, H | 0.061 | 1/8803/35066 | 0.23 | 61.9 |
| 20 | Bn, H, OH, H, H | 0.71 | 1/652/2623 | 4.65 | 79.5 |
| 21 | CB, H, OH, H, H | 10.3 | 1/65/344 | 46.1 | 50.7 |
| 22 | isoamyl, H, OH, H, H | 2.69 | 1/96/1020 | 22.1 | 74.7 |
| 4-Monohydroxy-substituted | |||||
| 28 | CBM, H, OH, H, H | 36.3 | 1/20/59 | 109 | 43.8 |
| 3,3′-Dihydroxy-substituted | |||||
| 37 | CPM, H, OH, OH, H | 4.62 | 1/137/617 | 20.6 | 51.3 |
| 38 | CBM, H, OH, OH, H | 0.38 | 1/605/8789 | 4.44 | 71.1 |
| 39 | allyl, H, OH, OH, H | 19.1 | 1/19/39 | 154 | 37.5 |
| 46 | CPeM, H, OH, OH, H | 0.31 | 1/1884/8952 | 13.7 | 80.4 |
| 47 | CHM, H, OH, OH, H | 0.14 | 1/1193/10229 | 17.6 | 91.1 |
| 48 | isoamyl, H, OH, OH, H | 2.10 | 1/100/699 | 16.6 | 65.6 |
| 3,4′-Dihydroxy-substituted | |||||
| 55 | allyl, H, OH, H, OH | 43.5 | 1/4/25 | 248 | 29.8 |
| 57 | CBM, H, OH, H, OH | 3.43 | 1/5/125 | 22.2 | 76.4 |
| 58 | CHM, H, OH, H, OH | 1.85 | 1/126/885 | 22.2 | 84.3 |
| 2-Fluoro-substituted | |||||
| 64 | CBM, F, OH, H, H | 0.072 | 1/5529/>138000 | 6.90 | 66.1 |
| 65 | CHM, F, OH, H, H | 0.040 | 1/21275/>250000 | 2.77 | 88.9 |
| 69 | CBM, F, OH, OH, H | 0.12 | 1/4642/>83000 | 1.49 | 57.5 |
| 74 | CBM, F, OH, H, OH | 3.37 | 1/155/389 | 36.7 | 69.1 |
| Unsubstituted | |||||
| 78 | CBM, H, H, H, H | 79.1 | 1/13/28 | 359 | 91.9 |
| Ligand | G Protein Activation a | β-Arrestin2 Recruitment b | References | ||
|---|---|---|---|---|---|
| EC50 (nM) | Emax (%) c | EC50 (nM) | Emax (%) c | ||
| 10 | 14 (GTPγS, U2OS-hKOR) | 94 | –d (PathHunter, U2OS-hKOR-β-arrestin2) | –d | [79] |
| 12 (HS666) | 35.7 (GTPγS, CHO-hKOR) | 50 | 449 (PathHunter, U2OS-hKOR-β-arrestin2) | 24 | [93] |
| 13 (HS665) | 4.987 (GTPγS, CHO-hKOR) | 88 | 463 (PathHunter, U2OS-hKOR-β-arrestin2) | 55 | [93] |
| 18 | 0.64 (GTPγS, U2OS-hKOR) | 100 | 720 (PathHunter, U2OS-hKOR-β-arrestin2) | 55 | [79] |
| 80 | 8.2 (GTPγS, U2OS-hKOR) | 86.7 | 3956 (PathHunter, U2OShKOR-β-arrestin2) | 61.2 | [80] |
| 81 | 11.8 (GTPγS, U2OS-hKOR) | 81.6 | 2082 (PathHunter, U2OShKOR-β-arrestin2) | 57.7 | [80] |

| Ligand | R1, R2, R3, R4, R5 | Antinociception a ED50 (mg/kg, s.c.) a |
|---|---|---|
| U50,488 | - | 1.54 |
| 12 (HS666) | CPM, H, OH, H, H | 3.23 |
| 13 (HS665) | CBM, H, OH, H, H | 1.91 |
| 18 | CPeM, H, OH, H, H | 0.49 |
| 19 | CHM, H, OH, H, H | 1.01 |
| 20 | Bn, H, OH, H, H | 1.21 |
| 22 | isoamyl, H, OH, H, H | 2.78 |
| 37 | CPM, H, OH, OH, H | 4.73 |
| 38 | CBM, H, OH, OH, H | 1.71 |
| 46 | CPeM, H, OH, OH, H | 1.19 |
| 47 | CHM, H, OH, OH, H | 0.95 |
| 48 | isoamyl, H, OH, OH, H | 2.63 |
| 57 | CBM, H, OH, H, OH | 1.73 |
| 58 | CHM, H, OH, H, OH | 1.90 |
| 64 | CBM, F, OH, H, H | 2.64 |
| 65 | CHM, F, OH, H, H | 1.33 |
| 69 | CBM, F, OH, OH, H | 2.25 |
| 74 | CBM, F, OH, H, OH | 2.14 |
| Ligand | Antinociception (Test, ED50, Route, Strain) | Locomotor Activity (rotarod test) (Dose, Route, Strain) | Aversion (CPA) (Dose, Route, Strain) | References |
|---|---|---|---|---|
| 10 | n.d. | 30 mg/kg, i.p., C57/BL6 | n.d. | [79,80] |
| 12 (HS666) | tail-withdrawal, 6.02 nmol, i.c.v., C57/BL6J writhing, 3.23 mg/kg, s.c., CD1 | 30 nmol, i.c.v., C57/BL6J 10 and 20 mg/kg, s.c., CD1 | CPA, 150 nmol, i.c.v., C57/BL6J n.d. | [93] [78] |
| 13 (HS665) | tail-withdrawal, 3.74 nmol, i.c.v., C57/BL6J writhing, 1.91 mg/kg, s.c., CD1 | 10 nmol, i.c.v., C57/BL6J 5 and 10 mg/kg, s.c., CD1 30 mg/kg, i.p., C57/BL6 | CPA, 30 nmol, i.c.v. C57/BL6J n.d. | [93] [76,78] [79,80] |
| 18 | writhing, 0.49 mg/kg, s.c., CD1 | 2.5 mg/kg, s.c., CD1 30 mg/kg, i.p., C57/BL6 | n.d. | [78] [79,80] |
| 19 | writhing, 1.01 mg/kg, s.c., CD1 | 5 mg/kg, s.c., CD1 | n.d. | [78] |
| 64 | writhing, 2.64 mg/kg, s.c., CD1 | 15 mg/kg, s.c., CD1 | n.d. | [78] |
| 65 | writhing, 1.33 mg/kg, s.c., CD1 | 7.5 mg/kg, s.c., CD1 | n.d. | [78] |
| 80 | n.d. | 30 mg/kg, i.p., C57/BL6 | n.d. | [80] |
| 81 | n.d. | 30 mg/kg, i.p., C57/BL6 | n.d. | [80] |

| Ligand | R1, R2, R3, R4, R5 | KOR Binding a | KOR Activity b | |
|---|---|---|---|---|
| Ki (nM) | Ki Ratio KOR/MOR/DOR | Ke (nM) | ||
| 24 | (CH2)2Ph, H, OH, H, H | 211 | 1/>47/>47 | 1311 |
| 32 | CPM, H, H, H, OH | 218 | 1/8/10 | 32.1 |
| 56 | CPM, H, OH, H, OH | 3.56 | 1/129/>2800 | 24.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidhammer, H.; Erli, F.; Guerrieri, E.; Spetea, M. Development of Diphenethylamines as Selective Kappa Opioid Receptor Ligands and Their Pharmacological Activities. Molecules 2020, 25, 5092. https://doi.org/10.3390/molecules25215092
Schmidhammer H, Erli F, Guerrieri E, Spetea M. Development of Diphenethylamines as Selective Kappa Opioid Receptor Ligands and Their Pharmacological Activities. Molecules. 2020; 25(21):5092. https://doi.org/10.3390/molecules25215092
Chicago/Turabian StyleSchmidhammer, Helmut, Filippo Erli, Elena Guerrieri, and Mariana Spetea. 2020. "Development of Diphenethylamines as Selective Kappa Opioid Receptor Ligands and Their Pharmacological Activities" Molecules 25, no. 21: 5092. https://doi.org/10.3390/molecules25215092
APA StyleSchmidhammer, H., Erli, F., Guerrieri, E., & Spetea, M. (2020). Development of Diphenethylamines as Selective Kappa Opioid Receptor Ligands and Their Pharmacological Activities. Molecules, 25(21), 5092. https://doi.org/10.3390/molecules25215092