Epitope-Based Immunoinformatics Approach on Nucleocapsid Protein of Severe Acute Respiratory Syndrome-Coronavirus-2

 ,
,  ,
, 
 , and
, and 
Abstract
1. Introduction
2. Results
2.1. Sequence Retrieval and Analysis
2.2. Antigenic Protein Prediction
2.3. Toxicity Prediction
2.4. Protein Structure Prediction and Validation
2.5. CD8+ T-Cell Epitope Identification
2.6. Population Coverage
2.7. Allergenicity Assessment
2.8. Molecular Docking Analysis for HLA and Epitope Interaction
2.9. B-Cell Epitope Prediction
3. Discussion
4. Materials and Methods
4.1. Protein Sequence Retrieval
4.2. Sequence Analysis
4.3. Protein Antigenicity and Toxicity Prediction
4.4. Protein Secondary and Tertiary Structure Prediction
4.5. T-Cell Epitope Prediction
CD8+ T-Cell Epitope Prediction
4.6. Epitope Conservancy and Immunogenicity Prediction
4.7. Prediction of Population Coverage and Allergenicity Assessment
4.8. HLA and Epitope Interaction Analysis Using Molecular Docking Studies
4.8.1. Epitope Model Generation
4.8.2. Retrieval of the HLA Allele Molecule
4.8.3. Molecular Docking Analysis
4.9. B-Cell Epitope Identification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus--infected pneumonia in Wuhan, China. Jama 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N. Eng. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Gralinski, L.E.; Menachery, V.D. Return of the Coronavirus: 2019-nCoV. Viruses 2020, 12, 135. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Coronavirus Disease 2019 (COVID-19): Situation Report; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Yuan, S.; Kok, K.-H.; To, K.K.-W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.-Y.; Poon, R.W.-S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Hu, Y.; Song, Z.-G.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. Complete genome characterisation of a novel coronavirus associated with severe human respiratory disease in Wuhan, China. bioRxiv 2020. [Google Scholar] [CrossRef]
- Narayanan, K.; Huang, C.; Makino, S. SARS coronavirus accessory proteins. Virus Res. 2008, 133, 113–121. [Google Scholar] [CrossRef]
- Dermime, S.; Gilham, D.E.; Shaw, D.M.; Davidson, E.J.; Meziane, E.-K.; Armstrong, A.; Hawkins, R.E.; Stern, P.L. Vaccine and antibody-directed T cell tumour immunotherapy. Biochim. Biophys. Acta BBA-Rev. Cancer 2004, 1704, 11–35. [Google Scholar] [CrossRef]
- Meloen, R.H.; Langeveld, J.P.M.; Schaaper, W.M.M.; Slootstra, J.W. Synthetic peptide vaccines: Unexpected fulfillment of discarded hope? Biologicals 2001, 29, 233–236. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fett, C.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J. Virol. 2014, 88, 11034–11044. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R.; Black, S.; Bloom, D.E. Vaccines and global health: In search of a sustainable model for vaccine development and delivery. Sci. Transl. Med. 2019, 11, eaaw2888. [Google Scholar] [CrossRef] [PubMed]
- Olsson, S.-E.; Villa, L.L.; Costa, R.L.R.; Petta, C.A.; Andrade, R.P.; Malm, C.; Iversen, O.-E.; Høye, J.; Steinwall, M.; Riis-Johannessen, G.; et al. Induction of immune memory following administration of a prophylactic quadrivalent human papillomavirus (HPV) types 6/11/16/18 L1 virus-like particle (VLP) vaccine. Vaccine 2007, 25, 4931–4939. [Google Scholar] [CrossRef]
- Suarez, D.L.; Schultz-Cherry, S. Immunology of avian influenza virus: A review. Dev. Comp. Immunol. 2000, 24, 269–283. [Google Scholar] [CrossRef]
- Briney, B.; Sok, D.; Jardine, J.G.; Kulp, D.W.; Skog, P.; Menis, S.; Jacak, R.; Kalyuzhniy, O.; De Val, N.; Sesterhenn, F.; et al. Tailored immunogens direct affinity maturation toward HIV neutralizing antibodies. Cell 2016, 166, 1459–1470. [Google Scholar] [CrossRef]
- Pedersen, S.R.; Christensen, J.P.; Buus, S.; Rasmussen, M.; Korsholm, K.S.; Nielsen, M.; Claesson, M.H. Immunogenicity of HLA class I and II double restricted influenza A-derived peptides. PLoS ONE 2016, 11, e0145629. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.I.N.; Xu, S.; Yang, R.F.; Li, Y.X.; Ji, Y.Y.; He, Y.Y.; De Shi, M.; Wei, L.U.; Shi, T.L.; Jin, W.; et al. Identification of an epitope of SARS-coronavirus nucleocapsid protein. Cell Res. 2003, 13, 141–145. [Google Scholar]
- Rakib, A.; Sami, S.A.; Mimi, N.J.; Chowdhury, M.M.; Eva, T.A.; Nainu, F.; Paul, A.; Shahriar, A.; Tareq, A.M.; Emon, N.U.; et al. Immunoinformatics-guided design of an epitope-based vaccine against severe acute respiratory syndrome coronavirus 2 spike glycoprotein. Comput. Biol. Med. 2020, 124, 103967. [Google Scholar] [CrossRef] [PubMed]
- Kharisma, V.D.; Ansori, A.N.M. Construction of epitope-based peptide vaccine against SARS-CoV-2: Immunoinformatics study. J. Pure Appl. Microbiol. 2020, 14, 999–1005. [Google Scholar] [CrossRef]
- Peng, H.; Yang, L.T.; Wang, L.Y.; Li, J.; Huang, J.; Lu, Z.Q.; Koup, R.A.; Bailer, R.T.; Wu, C. Long-lived memory T lymphocyte responses against SARS coronavirus nucleocapsid protein in SARS-recovered patients. Virology 2006, 351, 466–475. [Google Scholar] [CrossRef]
- Seah, J.N.; Yu, L.; Kwang, J. Localization of linear B-cell epitopes on infectious bronchitis virus nucleocapsid protein. Vet. Microbiol. 2000, 75, 11–16. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Parashar, N.C.; Poddar, J.; Chakrabarti, S.; Parashar, G. Repurposing of SARS-CoV nucleocapsid protein specific nuclease resistant RNA aptamer for therapeutics against SARS-CoV-2. Infect. Genet. Evol. 2020, 85, 104497. [Google Scholar] [CrossRef] [PubMed]
- Dalsass, M.; Brozzi, A.; Medini, D.; Rappuoli, R. Comparison of open-source reverse vaccinology programs for bacterial vaccine antigen discovery. Front. Immunol. 2019, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Geourjon, C.; Deleage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP-a server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Liu, Y.; Gayle, A.A.; Wilder-Smith, A.; Rocklöv, J. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J. Travel Med. 2020, 27, taaa021. [Google Scholar] [CrossRef]
- Wang, T.; Du, Z.; Zhu, F.; Cao, Z.; An, Y.; Gao, Y.; Jiang, B. Comorbidities and multi-organ injuries in the treatment of COVID-19. Lancet 2020, 395, e52. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A trial of lopinavir--ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Chen, L.; Xiong, J.; Bao, L.; Shi, Y. Convalescent plasma as a potential therapy for COVID-19. Lancet Infect. Dis. 2020, 20, 398–400. [Google Scholar] [CrossRef]
- Chhabra, H.S.; Bagaraia, V.; Keny, S.; Kalidindi, K.K.V.; Mallepally, A.; Dhillon, M.S.; Malhotra, R.; Rajasekharan, S. COVID-19: Current Knowledge and Best Practices for Orthopaedic Surgeons. Indian J. Orthop. 2020, 54, 917–918. [Google Scholar] [CrossRef] [PubMed]
- Oany, A.R.; Emran, A.-A.; Jyoti, T.P. Design of an epitope-based peptide vaccine against spike protein of human coronavirus: An in silico approach. Drug Des. Devel. Ther. 2014, 8, 1139. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Hossain, M.; Alam, J. A computational assay to design an epitope-based Peptide vaccine against Saint Louis encephalitis virus. Bioinform. Biol. Insights 2013, 7, BBI-S13402. [Google Scholar] [CrossRef]
- Chakraborty, S.; Chakravorty, R.; Ahmed, M.; Rahman, A.; Waise, T.M.; Hassan, F.; Rahman, M.; Shamsuzzaman, S. A computational approach for identification of epitopes in dengue virus envelope protein: A step towards designing a universal dengue vaccine targeting endemic regions. Silico Biol. 2010, 10, 235–246. [Google Scholar] [CrossRef]
- Islam, R.; Sakib, M.S.; Zaman, A. A computational assay to design an epitope-based peptide vaccine against chikungunya virus. Future Virol. 2012, 7, 1029–1042. [Google Scholar] [CrossRef]
- Huang, Y.; Khorchid, A.; Wang, J.; Parniak, M.A.; Darlix, J.-L.; Wainberg, M.A.; Kleiman, L. Effect of mutations in the nucleocapsid protein (NCp7) upon Pr160 (gag-pol) and tRNA (Lys) incorporation into human immunodeficiency virus type 1. J. Virol. 1997, 71, 4378–4384. [Google Scholar] [CrossRef]
- Thomas, J.A.; Gorelick, R.J. Nucleocapsid protein function in early infection processes. Virus Res. 2008, 134, 39–63. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, J.H.; Hung, C.-F.; Peng, S.; Roden, R.; Wang, M.-C.; Viscidi, R.; Tsai, Y.-C.; He, L.; Chen, P.-J.; et al. Generation and characterization of DNA vaccines targeting the nucleocapsid protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004, 78, 4638–4645. [Google Scholar] [CrossRef] [PubMed]
- Sabara, M.; Frenchick, P.J.; Mullin-Ready, K.F. Rotavirus nucleocapsid protein VP6 in vaccine compositions. Biotechnol. Adv. 1995, 13, 803–804. [Google Scholar]
- Arthur, L.O.; Bess, J.W., Jr.; Chertova, E.N.; Rossio, J.L.; Esser, M.T.; Benveniste, R.E.; Henderson, L.E.; Lifson, J.D. Chemical inactivation of retroviral infectivity by targeting nucleocapsid protein zinc fingers: A candidate SIV vaccine. AIDS Res. Hum. Retrovir. 1998, 14, S311–S319. [Google Scholar] [PubMed]
- Zhao, P.; Cao, J.; Zhao, L.-J.; Qin, Z.-L.; Ke, J.-S.; Pan, W.; Ren, H.; Yu, J.-G.; Qi, Z.-T. Immune responses against SARS-coronavirus nucleocapsid protein induced by DNA vaccine. Virology 2005, 331, 128–135. [Google Scholar] [CrossRef]
- Chiou, S.-S.; Fan, Y.-C.; Crill, W.D.; Chang, R.-Y.; Chang, G.-J.J. Mutation analysis of the cross-reactive epitopes of Japanese encephalitis virus envelope glycoprotein. J. Gen. Virol. 2012, 93, 1185–1192. [Google Scholar] [CrossRef]
- Yu, Z.; Wu, S.; Zhao, W.; Ding, L.; Shiuan, D.; Chen, F.; Li, J.; Liu, J. Identification and the molecular mechanism of a novel myosin-derived ACE inhibitory peptide. Food Funct. 2018, 9, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S. Designing of cytotoxic and helper T cell epitope map provides insights into the highly contagious nature of the pandemic novel coronavirus SARS-CoV2. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Liniger, M.; Zuniga, A.; Tamin, A.; Azzouz-Morin, T.N.; Knuchel, M.; Marty, R.R.; Wiegand, M.; Weibel, S.; Kelvin, D.; Rota, P.A.; et al. Induction of neutralising antibodies and cellular immune responses against SARS coronavirus by recombinant measles viruses. Vaccine 2008, 26, 2164–2174. [Google Scholar] [CrossRef]
- Mishra, S. T cell epitope-based vaccine design for pandemic novel coronavirus 2019-nCoV. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Noorimotlagh, Z.; Karami, C.; Mirzaee, S.A.; Kaffashian, M.; Mami, S.; Azizi, M. Immune and bioinformatics identification of T cell and B cell epitopes in the protein structure of SARS-CoV-2: A systematic review. Int. Immunopharmacol. 2020, 86, 106738. [Google Scholar] [CrossRef]
- Caubet, J.-C.; Ponvert, C. Vaccine allergy. Immunol. Allergy Clin. 2014, 34, 597–613. [Google Scholar] [CrossRef]
- Kallinich, T.; Beier, K.C.; Wahn, U.; Stock, P.; Hamelmann, E. T-cell co-stimulatory molecules: Their role in allergic immune reactions. Eur. Respir. J. 2007, 29, 1246–1255. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Ahmed, S.; Rakib, A.; Islam, M.A.; Khanam, B.H.; Faiz, F.B.; Paul, A.; Chy, M.N.U.; Bhuiya, N.M.M.A.; Uddin, M.M.N.; Ullah, S.M.A.; et al. In vivo and in vitro pharmacological activities of Tacca integrifolia rhizome and investigation of possible lead compounds against breast cancer through in silico approaches. Clin. Phytosci. 2019, 5, 36. [Google Scholar] [CrossRef]
- Zhang, X.W. A combination of epitope prediction and molecular docking allows for good identification of MHC class I restricted T-cell epitopes. Comput. Biol. Chem. 2013, 45, 30–35. [Google Scholar] [CrossRef]
- Knudsen, N.P.H.; Nørskov-Lauritsen, S.; Dolganov, G.M.; Schoolnik, G.K.; Lindenstrøm, T.; Andersen, P.; Agger, E.M.; Aagaard, C. Tuberculosis vaccine with high predicted population coverage and compatibility with modern diagnostics. Proc. Natl. Acad. Sci. USA 2014, 111, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Lazoura, E.; Lodding, J.; Farrugia, W.; Ramsland, P.A.; Stevens, J.; Wilson, I.A.; Pietersz, G.A.; Apostolopoulos, V. Enhanced major histocompatibility complex class I binding and immune responses through anchor modification of the non-canonical tumour-associated mucin 1-8 peptide. Immunology 2006, 119, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Pietersz, G.A.; Pouniotis, D.S.; Apostolopoulos, V. Design of peptide-based vaccines for cancer. Curr. Med. Chem. 2006, 13, 1591–1607. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, Y.; Chen, Y.-H. Epitope-vaccine strategy against HIV-1: Today and tomorrow. Immunobiology 2003, 208, 423–428. [Google Scholar] [CrossRef]
- Vivona, S.; Gardy, J.L.; Ramachandran, S.; Brinkman, F.S.L.; Raghava, G.P.S.; Flower, D.R.; Filippini, F. Computer-aided biotechnology: From immuno-informatics to reverse vaccinology. Trends Biotechnol. 2008, 26, 190–200. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Bulik, S.; Tampe, R.; Van Endert, P.M.; Holzhütter, H.-G. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J. Immunol. 2003, 171, 1741–1749. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Sette, A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform. 2005, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Koohy, H. In silico identification of vaccine targets for 2019-nCoV. F1000Research 2020, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xu, D.; Li, X.; Li, H.; Shan, M.; Tang, J.; Wang, M.; Wang, F.-S.; Zhu, X.; Tao, H.; et al. Screening and identification of severe acute respiratory syndrome-associated coronavirus-specific CTL epitopes. J. Immunol. 2006, 177, 2138–2145. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, K.; Toyoshima, Y.; Nemoto, K.; Nakamura, Y. Bioinformatic prediction of potential T cell epitopes for SARS-Cov-2. J. Hum. Genet. 2020, 65, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Khalili, S.; Jahangiri, A.; Borna, H.; Ahmadi Zanoos, K.; Amani, J. Computational vaccinology and epitope vaccine design by immunoinformatics. Acta Microbiol. Immunol. Hung. 2014, 61, 285–307. [Google Scholar] [CrossRef]
- Vihinen, M.; Torkkila, E.; Riikonen, P. Accuracy of protein flexibility predictions. Proteins Struct. Funct. Bioinform. 1994, 19, 141–149. [Google Scholar] [CrossRef]
- Hopp, T.P.; Woods, K.R. Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 3824–3828. [Google Scholar] [CrossRef]
- Nardin, E.H.; Calvo-Calle, J.M.; Oliveira, G.A.; Nussenzweig, R.S.; Schneider, M.; Tiercy, J.-M.; Loutan, L.; Hochstrasser, D.; Rose, K. A totally synthetic polyoxime malaria vaccine containing Plasmodium falciparum B cell and universal T cell epitopes elicits immune responses in volunteers of diverse HLA types. J. Immunol. 2001, 166, 481–489. [Google Scholar] [CrossRef]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef]
- Grifoni, A.; Sidney, J.; Zhang, Y.; Scheuermann, R.H.; Peters, B.; Sette, A. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe 2020, 27, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Amrun, S.N.; Lee, C.Y.-P.; Lee, B.; Fong, S.-W.; Young, B.E.; Chee, R.S.-L.; Yeo, N.K.-W.; Torres-Ruesta, A.; Carissimo, G.; Poh, C.M.; et al. Linear B-cell epitopes in the spike and nucleocapsid proteins as markers of SARS-CoV-2 exposure and disease severity. EBioMedicine 2020, 58, 102911. [Google Scholar] [CrossRef]
- Vapalahti, O.; Kallio-Kokko, H.; Närvänen, A.; Julkunen, I.; Lundkvist, Å.; Plyusnin, A.; Lehvaslaiho, H.; Brummer-Korvenkontio, M.; Vaheri, A.; Lankinen, H. Human B-cell epitopes of Puumala virus nucleocapsid protein, the major antigen in early serological response. J. Med. Virol. 1995, 46, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Lundkvist, Å.K.E.; Kallio-Kokko, H.; Sjölander, K.B.; Lankinen, H.; Niklasson, B.O.; Vaheri, A.; Vapalahti, O. Characterization of Puumala virus nucleocapsid protein: Identification of B-cell epitopes and domains involved in protective immunity. Virology 1996, 216, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-S.; Nah, J.-J.; Ko, Y.-J.; Kang, S.-Y.; Yoon, K.-J.; Jo, N.-I. Antigenic and immunogenic investigation of B-cell epitopes in the nucleocapsid protein of peste des petits ruminants virus. Clin. Diagn. Lab. Immunol. 2005, 12, 114–121. [Google Scholar] [CrossRef]
- Khan, M.K.; Zaman, S.; Chakraborty, S.; Chakravorty, R.; Alam, M.M.; Bhuiyan, T.R.; Rahman, M.J.; Fernández, C.; Qadri, F.; Seraj, Z.I. In silico predicted mycobacterial epitope elicits in vitro T-cell responses. Mol. Immunol. 2014, 61, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Srivastava, V.; Kumar, A. Epitope based peptide prediction from proteome of enterotoxigenic E. coli. Int. J. Pept. Res. Ther. 2018, 24, 323–336. [Google Scholar] [CrossRef]
- Lundegaard, C.; Lamberth, K.; Harndahl, M.; Buus, S.; Lund, O.; Nielsen, M. NetMHC-3.0: Accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008, 36, W509–W512. [Google Scholar] [CrossRef]
- Flower, D.R. Immunoinformatics and the in Silico Prediction of Immunogenicity. In Immunoinformatics; Springer: Berlin/Heidelberg, Germany, 2007; pp. 1–15. [Google Scholar]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. Peptide toxicity prediction. In Computational Peptidology; Springer: New York, NY, USA, 2015; pp. 143–157. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Buus, S.; Lauemøller, S.L.; Worning, P.; Kesmir, C.; Frimurer, T.; Corbet, S.; Fomsgaard, A.; Hilden, J.; Holm, A.; Brunak, S. Sensitive quantitative predictions of peptide-MHC binding by a ’Query by Committee’artificial neural network approach. Tissue Antigens 2003, 62, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Giguère, S.; Drouin, A.; Lacoste, A.; Marchand, M.; Corbeil, J.; Laviolette, F. MHC-NP: Predicting peptides naturally processed by the MHC. J. Immunol. Methods 2013, 400, 30–36. [Google Scholar] [CrossRef]
- Bui, H.-H.; Sidney, J.; Li, W.; Fusseder, N.; Sette, A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinform. 2007, 8, 361. [Google Scholar] [CrossRef]
- Moutaftsi, M.; Peters, B.; Pasquetto, V.; Tscharke, D.C.; Sidney, J.; Bui, H.-H.; Grey, H.; Sette, A. A consensus epitope prediction approach identifies the breadth of murine T CD8+-cell responses to vaccinia virus. Nat. Biotechnol. 2006, 24, 817–819. [Google Scholar] [CrossRef]
- Maupetit, J.; Derreumaux, P.; Tuffery, P. PEP-FOLD: An online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009, 37, W498–W503. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Dallakyan, S. PyRx-python prescription v. 0.8. Scripps Res. Inst. 2008, 2010. [Google Scholar] [CrossRef]
- Dunbrack, R.L. Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol. 2002, 12, 431–440. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Rakib, A.; Paul, A.; Chy, M.N.U.; Sami, S.A.; Baral, S.K.; Majumder, M.; Tareq, A.M.; Amin, M.N.; Shahriar, A.; Uddin, M.Z.; et al. Biochemical and Computational Approach of Selected Phytocompounds from Tinospora crispa in the Management of COVID-19. Molecules 2020, 25, 3936. [Google Scholar] [CrossRef]
- Jahan, I.; Tona, M.R.; Sharmin, S.; Sayeed, M.A.; Tania, F.Z.; Paul, A.; Chy, M.N.U.; Rakib, A.; Emran, T.B.; Simal-Gandara, J. GC-MS Phytochemical Profiling, Pharmacological Properties, and In Silico Studies of Chukrasia velutina Leaves: A Novel Source for Bioactive Agents. Molecules 2020, 25, 3536. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. Empirical predictions of protein conformation. Annu. Rev. Biochem. 1978, 47, 251–276. [Google Scholar]
- Emini, E.A.; Hughes, J.V.; Perlow, D.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef]
- Kolaskar, A.S.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef]
- Karplus, P.A.; Schulz, G.E. Prediction of chain flexibility in proteins. Naturwissenschaften 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Larsen, J.E.P.; Lund, O.; Nielsen, M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006, 2, 2. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rini, J.M.; Schulze-Gahmen, U.; Wilson, I.A. Structural evidence for induced fit as a mechanism for antibody-antigen recognition. Science 1992, 255, 959–965. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
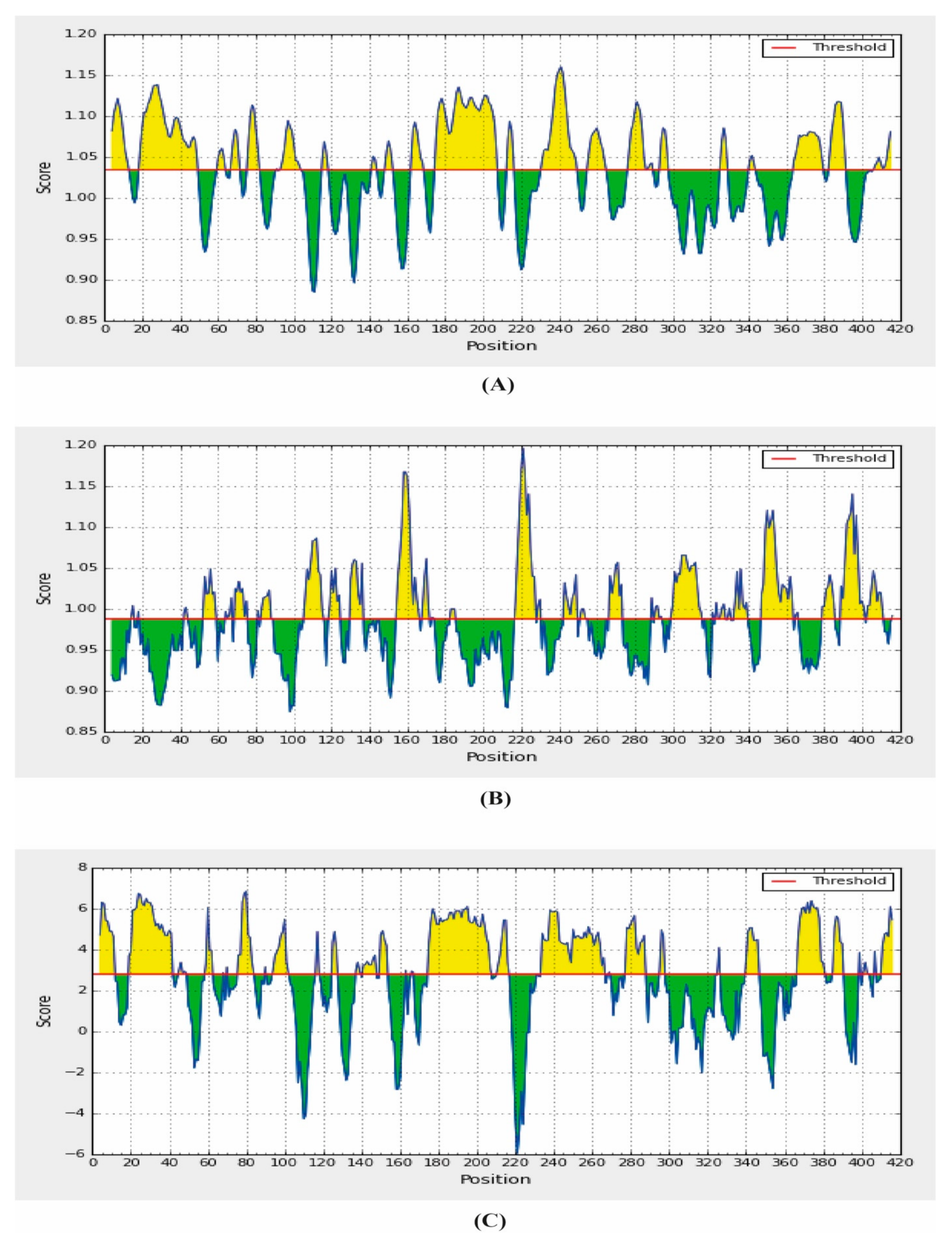{kind=link}
| Epitopes | Toxicity Prediction | SVM Score | Hydrophobicity | Hydrophilicity | Molecular Weight |
|---|---|---|---|---|---|
| LSPRWYFYY | Non-Toxin | −1.08 | −0.06 | −1.26 | 1294.59 |
| GTTLPKGFY | Non-Toxin | −1.13 | −0.01 | −0.49 | 983.26 |
| DLSPRWYFY | Non-Toxin | −1.18 | −0.14 | −0.67 | 1246.5 |
| SSPDDQIGY | Non-Toxin | −0.33 | −0.2 | 0.3 | 981.1 |
| LLNKHIDAY | Non-Toxin | −0.81 | −0.09 | −0.28 | 1086.39 |
| GTDYKHWPQ | Non-Toxin | −0.29 | −0.29 | −0.04 | 1131.34 |
| SPDDQIGYY | Non-Toxin | −0.54 | −0.17 | 0.01 | 1057.2 |
| NTASWFTAL | Non-Toxin | −1 | 0.08 | -1 | 1010.23 |
| Epitopes | NetCTL Combined Score | Epitope Conservancy Hit (MAX. Identity %) | MHC-I Interaction with an Affinity of IC50 < 200 and the Total Score (Proteasome Score, TAP Score, MHC-I Score, Processing Score) | pMHC-I Immunogenicity Score |
|---|---|---|---|---|
| LSPRWYFYY | 2.3408 | 100 | HLA-A*29:02 (1.32), HLA-A*30:02 (0.8), HLA-A*01:01 (0.66), HLA-C*16:01 (0.26) | 0.35734 |
| NTASWFTAL | 0.9521 | 100 | HLA-A*68:02 (1.11), HLA-C*16:01 (0.18), HLA-C*03:03 (0.12), HLA-C*03:04 (0.12), HLA-C*12:03 (0.10), HLA-A*02:06 (0.04), HLA-C*03:02 (−0.07), HLA-A*26:01 (−0.13), HLA-C*14:02 (−0.34) | 0.22775 |
| DLSPRWYFY | 1.4994 | 100 | HLA-A*29:02 (0.99) | 0.25933 |
| SPDDQIGYY | 1.1404 | 100 | HLA-B*35:01 (0.52) | 0.06844 |
| SSPDDQIGY | 0.6895 | 100 | 0.0634 |
| Population | Coverage (%) a | Average Hit b | PC90 c |
|---|---|---|---|
| Central Africa | 35.31 | 0.40 | 0.15 |
| East Africa | 39.25 | 0.45 | 0.16 |
| East Asia | 57.56 | 0.71 | 0.24 |
| Europe | 42.95 | 0.50 | 0.18 |
| North Africa | 42.15 | 0.49 | 0.17 |
| North America | 45.32 | 0.53 | 0.18 |
| Northeast Asia | 48.11 | 0.55 | 0.19 |
| Oceania | 31.43 | 0.34 | 0.15 |
| South Africa | 33.91 | 0.38 | 0.15 |
| South America | 38.66 | 0.44 | 0.16 |
| South Asia | 36.53 | 0.41 | 0.16 |
| Southeast Asia | 49.45 | 0.57 | 0.20 |
| Southwest Asia | 28.53 | 0.32 | 0.14 |
| West Africa | 56.22 | 0.67 | 0.23 |
| West Indies | 12.89 | 0.13 | 0.11 |
| Epitopes | Docking Score (kcal/mol) |
|---|---|
| NTASWFTAL | −9.4 |
| Positive Control | −8.2 |
| Start | End | Peptide | Length |
|---|---|---|---|
| 361 | 390 | KTFPPTEPKKDKKKKADETQALPQRQKKQQ | 30 |
| 338 | 347 | KLDDKDPNFK | 10 |
| 323 | 331 | EVTPSGTWL | 9 |
| 273 | 287 | AFGRRGPEQTQGNFG | 15 |
| 232 | 269 | SKMSGKGQQQQGQTVTKKSAAEASKKPRQKRTATKAYN | 38 |
| 164 | 216 | GTTLPKGFYAEGSRGGSQASSRSSSRSRNSSRNSTPGSSRGTSPARMAGNGGD | 53 |
| 137 | 154 | GALNTPKDHIGTRNPANN | 18 |
| 115 | 127 | TGPEAGLPYGANK | 13 |
| 93 | 104 | RIRGGDGKMKDL | 12 |
| 58 | 85 | QHGKEDLKFPRGQGVPINTNSSPDDQIG | 28 |
| 1 | 51 | MSDNGPQNQRNAPRITFGGPSDSTGSNQNGERSGARSKQRRPQGLPNNTAS | 51 |
| Start | End | Peptide | Length |
|---|---|---|---|
| 52 | 59 | WFTALTQH | 8 |
| 69 | 75 | GQGVPIN | 7 |
| 83 | 89 | QIGYYRR | 7 |
| 106 | 115 | PRWYFYYLGT | 10 |
| 130 | 136 | IIWVATE | 7 |
| 154 | 166 | NAAIVLQLPQGTT | 13 |
| 217 | 227 | AALALLLLDRL | 11 |
| 243 | 249 | GQTVTKK | 7 |
| 267 | 273 | AYNVTQA | 7 |
| 299 | 315 | KHWPQIAQFAPSASAFF | 17 |
| 333 | 339 | YTGAIKL | 7 |
| 347 | 363 | KDQVILLNKHIDAYKTF | 17 |
| 379 | 385 | TQALPQR | 7 |
| 389 | 401 | QQTVTLLPAADLD | 13 |
| 403 | 411 | FSKQLQQSM | 9 |
| Start | End | Peptide | Length |
|---|---|---|---|
| 4 | 11 | NGPQNQRN | 8 |
| 36 | 42 | RSKQRRP | 7 |
| 185 | 197 | RSSSRSRNSSRNS | 13 |
| 254 | 264 | ASKKPRQKRTA | 11 |
| 277 | 282 | RGPEQT | 6 |
| 295 | 300 | GTDYKH | 6 |
| 340 | 346 | DDKDPNF | 7 |
| 365 | 377 | PTEPKKDKKKKAD | 13 |
| 384 | 390 | QRQKKQQ | 7 |
Sample Availability: Samples of the compounds are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakib, A.; Sami, S.A.; Islam, M.A.; Ahmed, S.; Faiz, F.B.; Khanam, B.H.; Marma, K.K.S.; Rahman, M.; Uddin, M.M.N.; Nainu, F.; et al. Epitope-Based Immunoinformatics Approach on Nucleocapsid Protein of Severe Acute Respiratory Syndrome-Coronavirus-2. Molecules 2020, 25, 5088. https://doi.org/10.3390/molecules25215088
Rakib A, Sami SA, Islam MA, Ahmed S, Faiz FB, Khanam BH, Marma KKS, Rahman M, Uddin MMN, Nainu F, et al. Epitope-Based Immunoinformatics Approach on Nucleocapsid Protein of Severe Acute Respiratory Syndrome-Coronavirus-2. Molecules. 2020; 25(21):5088. https://doi.org/10.3390/molecules25215088
Chicago/Turabian StyleRakib, Ahmed, Saad Ahmed Sami, Md. Ashiqul Islam, Shahriar Ahmed, Farhana Binta Faiz, Bibi Humayra Khanam, Kay Kay Shain Marma, Maksuda Rahman, Mir Muhammad Nasir Uddin, Firzan Nainu, and et al. 2020. "Epitope-Based Immunoinformatics Approach on Nucleocapsid Protein of Severe Acute Respiratory Syndrome-Coronavirus-2" Molecules 25, no. 21: 5088. https://doi.org/10.3390/molecules25215088
APA StyleRakib, A., Sami, S. A., Islam, M. A., Ahmed, S., Faiz, F. B., Khanam, B. H., Marma, K. K. S., Rahman, M., Uddin, M. M. N., Nainu, F., Emran, T. B., & Simal-Gandara, J. (2020). Epitope-Based Immunoinformatics Approach on Nucleocapsid Protein of Severe Acute Respiratory Syndrome-Coronavirus-2. Molecules, 25(21), 5088. https://doi.org/10.3390/molecules25215088