Abstract
Depression is a severe psychiatric disorder that affects over 100 million people worldwide. 5-HT1A receptor agonists have been implicated in the treatment of a variety of central nervous system diseases, especially depression. In this study, based on FW01, a selective potent 5-HT1AR agonist discovered via dynamic pharmacophore-based virtual screening, a series of indolealkylpiperazine derivatives with a benzamide moiety were designed and synthesized by the modification of the amide tail group as well as indole head group of FW01. Among all tested compounds, 13m displayed potent agonistic activity towards 5-HT1AR with an EC50 value of 1.01 nM. Molecular docking studies were performed to disclose the mechanism of its potent agonistic activity and high selectivity. Finally, the activation model of 5-HT1AR induced by 13m was proposed.
1. Introduction
Depression is a severe psychiatric disorder that affects over 100 million people worldwide [1]. The main symptoms include low mood, lack of energy, sadness, insomnia, etc. [2]. Patients also suffer a high risk of suicide [3,4]. Though the etiology of depression is not clear, it is certain that genetic and psychological factors as well as environmental factors are all involved in the pathogenesis [5,6]. Selective serotonin reuptake inhibitors (SSRIs) and serotonin norepinephrine reuptake inhibitors (SNRIs) are common antidepressants clinically [7,8,9]. Though SSRIs are first-line drugs for the treatment of depression, they still have some drawbacks including delayed onset and limited efficacy [10,11]. Thus, drug discovery for new antidepressants with rapid onset and good efficacy is still urgent [12,13].
Serotonin (5-hydroxytryptamine, 5-HT), as an important neurotransmitter, is involved in multiple physiological and behavioral processes [14,15]. 5-HT mediates its physiological effects through at least 14 different receptor subtypes [16]. Among them, 5-HT1A receptor (5-HT1AR) plays a significant role in mood regulation and has been a hot target for various central nervous system disorders, especially depression [17,18,19,20]. Medicines like gepirone, tandospirone and vilazodone acting as 5-HT1AR partial agonists are representative antidepressants clinically [21]. Meanwhile, the combination of 5-HT1AR agonists and SSRIs not only accelerate the onset of SSRIs but also improve the therapeutic effect [10]. Therefore, based on our previous research, we aimed at discovering selective 5-HT1AR agonists with novel scaffold with the eventual aim to treat depression.
2. Results and Discussion
2.1. Compound Design
In our previous study, a series of lead compounds as 5-HT1AR agonists with novel scaffolds were obtained through the combination of pharmacophore-based virtual screening and biological assay [22]. Among them, FW01 (Figure 1) possessed high activity for 5-HT1AR (Ki = 51.9 ± 16.4 nM), medium activity for 5-HT2A receptor (5-HT2AR) (Ki = 206.71 ± 7.46 nM) and low activity for dopamine D2 receptor (D2R) (Ki = 2161.35 ± 25.55 nM). FW01 was bound to 5-HT1AR via multiple interaction forces [22].
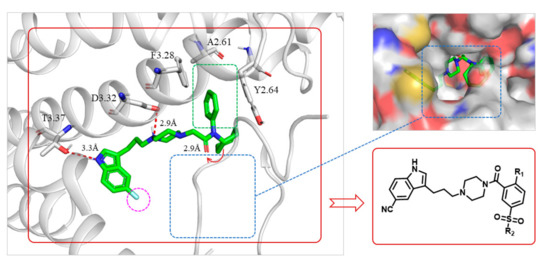
Figure 1.
Predicted binding mode of FW01 (green sticks) with 5-HT1AR (cartoon) and the design of indolealkylpiperazine derivatives. Ligands are shown as green sticks. The binding residues of 5-HT1AR are shown in gray. Hydrogen bond interactions are represented as dotted red lines. 5-HT1AR was obtained by homology modeling from the X-ray structure of 5-HT1BR (PDB code: 6G79).
The docking predicted binding mode of FW01 with 5-HT1AR (Figure 1) showed that the indole head group was inserted into the pocket formed by transmembrane helix 3 (TM3), TM5, TM6 and TM7 of 5-HT1AR. The NH group of the indole head formed hydrogen bonding with T3.37. The protonated nitrogen of piperazine participated in the salt bridge with D3.32. As for the amide tail group, the phenyl ring was inserted into the pocket formed by residues F3.28, Y2.64, and A6.21. However, there was an empty pocket formed by TM6 and TM7 which was not reached by FW01. Due to the presence of the upper pocket and the lower pocket formed by TM6 and TM7, the amide tail moiety of FW01 was replaced with different 2,5-substituted benzamide moieties. As for the indole moiety, the development of marketed antidepressant drug vilazodone showed that a 5-CN indole moiety could enhance the 5-HT1AR affinity, which was also proved in our previous study [23,24]. Therefore, the 5-F indole moiety of FW01 was replaced by 5-CN indole moiety to raise the agonistic activity of designed compounds towards 5-HT1AR.
Finally, a series of indolealkylpiperazine derivatives with a benzamide moiety were designed and synthesized. Their cellular function activities on dopamine and serotonin receptors were evaluated by Ultra Lance cAMP assay and Fluorometric Imaging Plate Reader (FLIPR) assay.
2.2. Chemical Synthesis
Synthesis of intermediate 5 was shown in Scheme 1. Commercially available 5-cyanoindole was first acylated with 3-chloropropionyl chloride via Friedel-Crafts acylation to obtain 2, which was reduced by NaBH4/CF3COOH system to give compound 3. Compound 4, which was obtained via the reaction of 3 and N-Boc-piperazine, was deprotected to give 5.
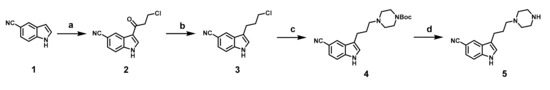
Scheme 1.
Synthesis of intermediate 5. Reagents and conditions: (a) 3-chloropropionyl chloride, AlCl3, anhydrous DCM, r.t., overnight; (b) NaBH4, CF3COOH, anhydrous THF, 0 °C, 1 h; (c) Boc-piperazine, CH3CN, K2CO3, 75 °C, 36 h; (d) CF3COOH, DCM, r.t., 4 h.
Synthesis of the 5-sulfonyl benzoic acid was outlined in Scheme 2. Compound 7a was obtained by the sulfonation reaction of 6a and chlorosulfonic acid. 7a reacted with sodium sulfite and then alkylated by iodomethane to give 8a. 8a was deprotected by lithium hydroxide to give 10a. Synthesis of 10b–10d was accomplished by reaction of 8a and corresponding reagents and then deprotection by lithium hydroxide. For 8b–8d, the starting material was methyl salicylate and the synthetic route was similar to 8a. Compounds 11a–11m were obtained by Mitsunobu reaction of 8b–8d and corresponding alcohol or alkylation of 8b–8d and 3-iodo-oxetane, which were then deprotected by lithium hydroxide to give compounds 12a–12m.
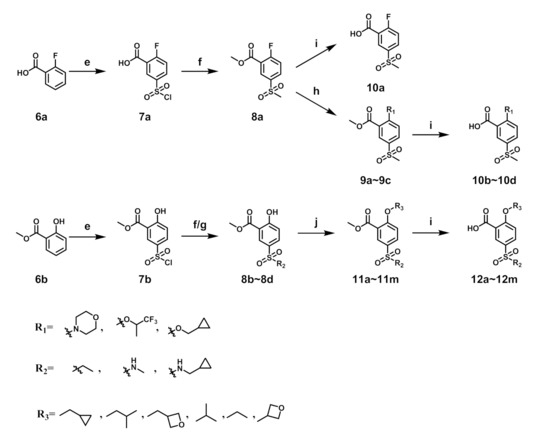
Scheme 2.
Synthesis of benzoic acid intermediates. Reagents and conditions: (e) HClSO3, 40 °C, 30 min; (f) i. Na2SO3, H2SO4, H2O, r.t., 2 h; ii. iodide alkanes, K2CO3, DMF, r.t., 3 h; (g) amine, DCM, r.t., 2 h; (h) corresponding reagent, K2CO3, DMF, 150 °C, 18 h; (i) LiOH·H2O, THF:H2O = 4:1, r.t., 5 h; (j) alcohol, PPh3, DBAD, THF, r.t., 6 h or 3-iodo-oxetane, Cs2CO3, DMF, 80 °C, 10 h.
As shown in Scheme 3, compounds 13a–13q were obtained by condensation of 5 and benzoic acid intermediate. Structure of all compounds were confirmed by ESI-MS, HRMS, 1H NMR and 13C NMR.
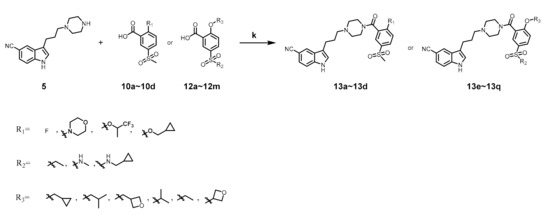
Scheme 3.
Synthesis of compounds 13a–13q. Reagents and conditions: (k) EDC·HCl, HOBT, DIEA, DCM, r.t., 8 h.
2.3. In Vitro Biological Activity Evaluation
After these new derivatives were synthesized, they were all subjected to in vitro evaluation of functional activities towards 5-HT1A, 5-HT2A and D2 receptors. As shown in Table 1, indolealkylpiperazine derivatives displayed varied selectivity for 5-HT1AR over D2R and 5-HT2AR. When R2 was methyl group, R1 was subjected to substituents of different size to examine the SAR. Compound 13a with an F atom displayed reduced agonistic activity for 5-HT1AR. The introduction of morpholine group (13b) resulted in drastically decreased activity towards three receptors, indicating that large size group of R1 position was not compatible with these receptors. Replacing morpholine group with smaller size 1,1,1-trifluoro-2-propyl or cyclopropylmethoxy group afforded 13c and 13d, respectively. By comparing 13b, they showed increased activity for 5-HT1AR and 5-HT2AR. We postulated that the cyclopropylmethoxy group extended into the hydrophobic pocket where the phenyl ring of FW01 located, thus stabilizing the binding conformation of 13d in the active pockets of 5-HT1AR and 5-HT2AR. Then, the ethyl, methylamino and cyclopropylmethylamino groups were employed to replace methyl of 13d and compounds 13e–13g were designed. When adopting the sulfonamide substituents (13f and 13g), the activity for 5-HT1AR showed a remarkable elevation in comparison to compound 13a–13e, presumably due to the less flexible nitrogen chain posing a limited conformation to form a more stable hydrophobic interaction.

Table 1.
Functional activities of compounds 13a–13g for dopamine receptor (D2R) and serotonin receptors (5-HT1AR and 5-HT2AR).
Encouraged by the results above, we next extended our efforts to investigate the effect of substituents of position 2 of the phenyl ring on compounds 13f and 13g. Overall, compounds 13f–13q showed selective agonistic activity for 5-HT1AR. Among these compounds, 13g and 13m–13q bearing cyclopropylmethylamino moiety displayed more potent activity for 5-HT1AR than their corresponding compounds 13f and 13h–13l bearing methylamino moiety, suggesting that cyclopropylmethylamino moiety was more favorable than methylamino moiety at position 2 as shown in Table 2. Compounds 13h and 13m bearing the isobutyl moiety were found to demonstrate the most potent agonistic activity for 5-HT1AR among methylamine and cyclopropylmethylamino moiety possessing compounds, respectively. Compound 13j with isopropyl group was found to completely lose activity for 5-HT1AR, which was due to the isopropyl moiety of 13j forming steric hindrance with 5-HT1AR. As for 13o which showed potent activity for 5-HT1AR, the presence of the N-(cyclopropylmethyl)sulfamoyl moiety limited the conformation of the compound. By contrast, the introduction of a polar atom to the R3 group resulted in a decreased activity for 5-HT1AR by an order of magnitude, indicating that a polar group was unfavorable to the binding affinity towards 5-HT1AR (13i, 13l, 13n, 13q).
Table 2.
Functional activities of compounds 13f–13q for dopamine receptor (D2R) and serotonin receptors (5-HT1AR and 5-HT2AR).
2.4. Binding Mode
We next studied the binding mode of 13m with agonistic conformation of 5-HT1AR. We used the GOLD 5.1 program to dock 13m into the binding site of the 5-HT1AR which was obtained by homology modeling from the X-ray structure of 5-HT1B receptor (PDB code: 6G79) [25]. The results of molecular modeling were shown in Figure 2. The protonated nitrogen atom of 13m formed a strong salt bridge with D3.32 in 5HT1AR. The NH group of the indole ring formed hydrogen bonding with T3.37. The N-(cyclopropylmethyl)sulfamoyl moiety interacted with A2.61, Y2.64 and F3.28 through hydrophobic packing, which was in accordance with the binding mode of FW01 with 5-HT1AR. The isobutoxy group was inserted into another hydrophobic pocket formed by F6.51, V6.54 and I7.38.
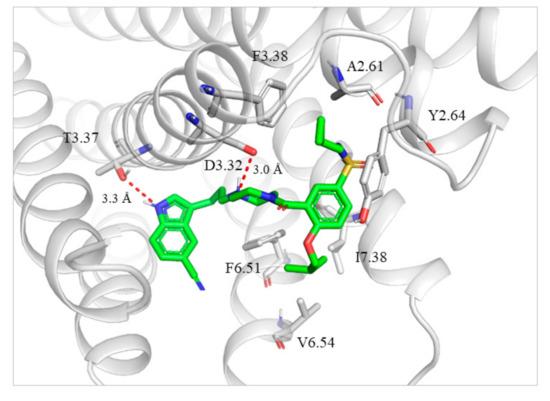
Figure 2.
Binding mode of 13m with 5-HT1AR. The ligand is shown as green sticks. 5-HT1AR and the surrounding residues are in gray. Hydrogen bond interactions are represented as dotted red lines.
To explain the high selectivity of 13m toward 5-HT1AR, the sequences of 5-HT1AR, 5-HT2AR and D2R at the binding site were aligned. As shown in Figure 3A, the residues of the active site in three receptors differed greatly and thus the conformations of the three receptors at the binding site were different. The 3D structures of 13m-bounded 5-HT1AR and 5-HT2AR were superimposed to explain the selectivity of 13m for 5-HT1AR and 5-HT2AR. The PDB code of 5-HT2AR was 6A93 [26]. As shown in Figure 3B, compared with 5-HT1AR, the amino acid residues in the hydrophobic pocket of 5-HT2AR stretched out further. Thus the N-(cyclopropylmethyl)sulfamoyl of 13m collapsed with W3.28 in the active pocket of 5-HT2AR, which is obvious to be observed in Figure 3C. It accounted for the reduced affinity of 13m for 5-HT2AR. The same phenomenon was found in the case of D2R when superposing 5-HT1AR and D2R (Figure 3D). The PDB code of D2R was 6CM4 [27]. The amino acid residues in the active pocket of D2R stretched out further than those in the active pocket of 5HT1AR. Thus the indole head and the N-(cyclopropylmethyl)sulfamoyl of 13m collapsed with S5.42 and L2.64 in the active pocket of D2R. This explains why 13m showed high selectivity toward 5-HT1AR over 5-HT2AR and D2R.
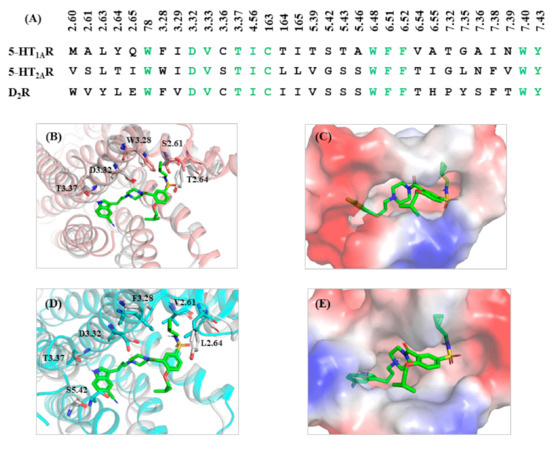
Figure 3.
(A) Sequences of 5-HT1AR, 5-HT2AR, and D2R at the binding site. (B) The superimposition of 5-HT2AR with 13m-bound 5-HT1AR. (C) Electrostatic potential energy diagram of 13m and 5-HT2AR at the binding site. (D) The superimposition of D2R with 13m-bound 5-HT1AR. (E) Electrostatic potential energy diagram of 13m and D2R at the binding site. The ligand is shown as green sticks. 5-HT1AR and the surrounding residues are in gray. 5-HT2AR and the surrounding residues are in pink. D2R and the surrounding residues are in blue.
2.5. 13m Induced Activation Mechanism of 5-HT1AR
Molecular dynamic simulations were carried out to explore the activation process of 5-HT1AR by 13m. Two systems, 13m bound to the active-state 5-HT1AR and the inactive-state 5-HT1AR without ligand (apo), were performed for 200 ns simulation, respectively. The inactive-state 5-HT1AR was modelled from the X-ray structure of 5-HT1BR (PDB code: 5V54). The Gromos conformational cluster analysis was used to obtain the representative structures of two systems in the course of simulation.
In 13m-5-HT1AR system, a conserved hydrogen bond was formed between the NH group of the indole ring in 13m and the oxygen atom of hydroxyl in T3.37 during the simulation (Figure 4A,D). At the same time, the protonated nitrogen atom in 13m maintained a stable hydrogen bond with the carbonyl oxygen of D3.32 and a relatively weak interaction with the hydroxyl oxygen of Y7.43 (Figure 4B). Therefore, the interaction between D3.32 and Y7.43 (also called 3-7 lock) stayed unbroken in 13m-5-HT1AR system but experienced big fluctuation in apo system (Figure 4C). This phenomenon was in agreement with the previous study of our group [28]. The oxygen atom on the sulfonyl group of 13m formed a hydrogen bond with Q2.65 initially, but it fluctuates greatly due to the swing of the molecular tail after 80 ns (Figure 4E).
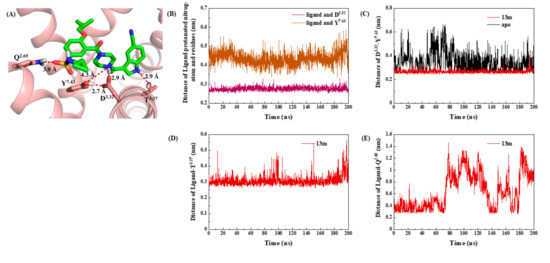
Figure 4.
(A) The interaction between 13m and D3.32, Y7.43, Q2.65 and T3.37. The ligand is shown as green sticks. 5-HT1AR and the surrounding residues are in pink. Hydrogen bond interactions are represented as dotted red lines. (B) Distance of the ligand with D3.32 and Y7.43. (C) Distance between D3.32 and Y7.43. (D) Distance between ligand and T3.37. (E) The interaction between ligand and Q2.65 in 13m-5-HT1AR system.
The strong interaction between 13m and 5-HT1AR induced the movement of TM3, TM5 and TM6. The triplets P5.50, I3.40 and F6.44 were located under the binding pockets of small molecules, their arrangement was important for stabilizing the antagonistic conformation of 5-HT1AR. The interaction of 13m with D3.32 and T3.37 adjusted the side chain of I3.40 to flip up, subsequently leading to the relocation of P5.50 and F6.44 (Figure 5A). Their rearrangement was closely related to the movement of the helices. At the same time, W6.48 moved outward due to the steric restraints between 13m and residue W6.48. As a rotamer toggle switch, W6.48 further pushed the intracellular end of the TM6 to move inward and the external end of the TM6 to move outward (Figure 5B). Finally, TM3, TM5 and TM6 underwent large movement to create space for the binding of Gα/s protein (Figure 5C). It was worth noting that the TM6 moved outward 11.5 Å.
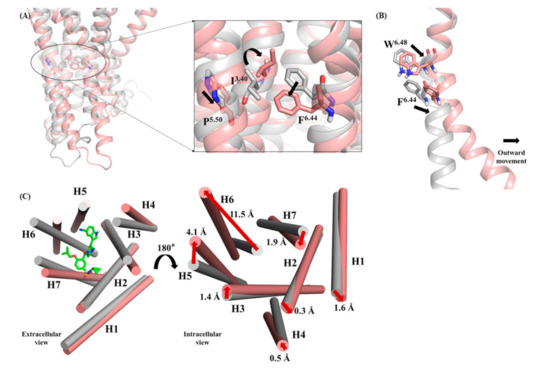
Figure 5.
(A) Superimposed structures of apo and 13m-5-HT1AR systems and the rearrangements of I3.40, P5.50 and F6.44 when 13m was bound to 5-HT1AR. (B) the movement of rotamer toggle switch W6.48 in 13m-5-HT1AR system contributed to the movement of the TM6. (C) Superimposed structures of apo and 13m-5-HT1AR systems and the rearrangements of I3.40, P5.50, and F6.44 when 13m was bound to 5-HT1AR. The apo system and surrounding residues are represented as grey. The 13m-5-HT1AR system and surrounding residues are represented as pink.
On the basis of simulation trajectory and analysis results, a possible activation process of 5-HT1AR could be proposed (Figure 6). Typically, unbound 5-HT1AR maintained in an inactive state or a basic active state. Owing to the intramolecular interaction and inherent flexibility, the molecular thermodynamic motion would lead to the dynamic equilibrium of several energy minima conformations of 5-HT1AR. When 13m got close to the binding pocket of 5-HT1AR, the electrostatic effect between them promoted the ligand recognition and binding process. Then, 13m stably stayed in the pocket enclosed by TM3, TM5, TM6 and TM7 through several anchoring interactions with the residues D3.32, Q2.65 and T3.37. These residues played the role of molecular triggers, propagating the conformational changes to the inner-middle part of the seven helices. The conformational changes induced the rearrangement of P5.50, I3.40 and F6.44 as well as the adjustment of W6.48, ultimately leading to the outward movement of TM3 and TM6, along with TM5 getting closer to TM6.
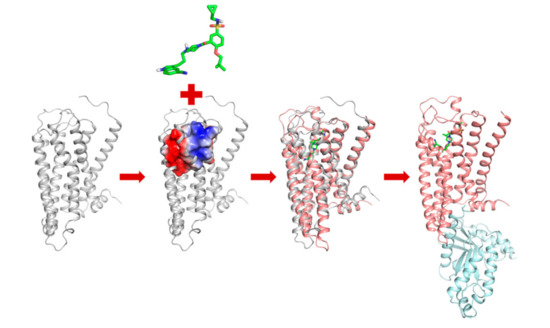
Figure 6.
Proposed activation process of 5-HT1AR. The apo system and surrounding residues are represented as grey. The 13m-5-HT1AR system and surrounding residues are represented as pink. The Gα/s protein is represented as blue.
The synergic helix movement of TM3, TM5, and TM6 created space for the binding of Gα/s protein. Then, 5-HT1AR was fully activated, subsequently leading to the downstream signal transduction.
3. Experiment Protocols
3.1. Chemistry and Biological Evaluation
The compound synthesis methods, NMR data and biological evaluation methods were provided in the Supplementary Materials.
3.2. Molecular Modeling
3.2.1. Homology Modeling
The amino acid sequence of 5-HT1AR were downloaded from the UniProtKB database (Entry code: P08908), and sequence similarity search was performed using NCBI BLAST server (Bethesda, MD, USA) [29]. The structure (PDB code: 6G79 and 5V54) of 5-HT1B receptor were selected as the templates to construct the agonistic and antagonistic conformation of 5-HT1A receptor. Sequence alignment of 5-HT1B receptor and 5-HT1AR was carried out using Discovery Studio 2016 (hereafter abbreviated to DS) (Waltham, MA, USA). Homology modeling was performed with DS. Ten models were generated after loop refinement and the one with the lowest Discrete Optimized Protein Energy (DOPE) score was submitted to energy minimization (100 steps steepest descent with backbone constrained). The PROCHECK program (Cambridge, UK) was used to evaluate the stereochemical quality of 5-HT1AR [30].
3.2.2. Molecular Docking Operations
Molecular docking was carried out using GOLD 5.0.1 (Cambridge, UK). The binding site was defined to include all residues within a 15.0 Å radius of the conserved D3.32 Cγ carbon atom of 5-HT1AR. Ten conformations were produced for each ligand, and Gold-Score was used as scoring function. Other parameters were set as standard default. High-scoring complexes were inspected visually to identify the most reasonable solution.
3.3. Molecular Dynamic Simulations
The molecular dynamic (MD) simulations were performed using the GROMACS 5.1.2 package (Uppsala, Sweden) [31]. Two systems, 13m bound to active-state 5-HT1AR and inactive-state 5-HT1AR without ligand (apo), were embedded in the hydrated, equilibrated palmitoyloleoylphosphatidylcholine (POPC) bilayer via the CHARMM-GUI server. The TIP3P water model was used for the MD simulation. Sodium and chloride ions were added to neutralize two systems to an ionic concentration of 0.15 mol/L. Both systems were minimized and gradually equilibrated in an NPT ensemble at 310 K and 1 bar. Periodic boundary conditions were applied to these simulation systems. Bonds connected to hydrogen atoms were restrained with the LINCS algorithm to allow an integration time step of 2 fs. Electrostatic interactions were calculated using the particle mesh Ewald (PME) method. Finally, the MD simulations for both systems were performed for 200 ns. All analysis of MD trajectories was performed using tools implemented in the GROMACS 5.1.2 package (Uppsala, Sweden). All the structural graphics were processed with PyMOL software (San Carlos, CA, USA) [32].
4. Conclusions
In summary, a series of indolealkylpiperazine derivatives with a benzamide moiety were designed and synthesized. Their activities on dopamine and serotonin receptors were evaluated and these compounds were found to be selective 5-HT1AR agonists. Among all tested compounds, 13m displayed the most potent agonistic activity over 5-HT1AR with an EC50 value of 1.01 nM. The investigation of the binding mode uncovered the mechanism of 13m’s potent agonistic activity and high selectivity. In addition, molecular dynamic simulations were carried out to propose 13m induced activation model of 5-HT1AR. The electrostatic interaction between the negatively charged active site of the 5-HT1AR and the positively protonated charged 13m is the initial driving force for the ligand recognition. 13m formed anchoring interactions with residues D3.32, Q2.65, T3.37 and induced conformation changes of 5-HT1AR. This leads to the rearrangement of P5.50, I3.40 and F6.44 as well as the adjustment of W6.48, causing the outward movement of TM3 and TM6, along with TM5 getting closer to TM6. Then, 5-HT1AR was ready to bind Gα/s protein, leading itself to a fully activated state. This model would further promote structure-based agonist design of 5-HT1AR for the treatment of depression.
Supplementary Materials
The following are available online, the compound synthesis methods, NMR data, biological evaluation methods.
Author Contributions
C.Z., X.L. and W.P. contributed equally in this work. Compound design, C.Z. and W.F.; compound synthesis, C.Z., X.L. and W.P.; writing—original draft preparation, C.Z. and X.L.; writing—review and editing, W.F.; funding acquisition, W.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by grants from National Natural Science Foundation of China (NO. 81773635, NO. 82073765) and Shanghai Science and Technology Development Funds (14431900500, 20S11902400).
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Oliveira, M.R.; Chenet, A.L.; Duarte, A.R.; Scaini, G.; Quevedo, J. Molecular Mechanisms Underlying the Anti-depressant Effects of Resveratrol: A Review. Mol. Neurobiol. 2017, 55, 4543–4559. [Google Scholar] [CrossRef] [PubMed]
- Cui, R. Editorial (Thematic Selection: A Systematic Review of Depression). Curr. Neuropharmacol. 2015, 13, 480. [Google Scholar] [CrossRef] [PubMed]
- Hawton, K.; I Comabella, C.C.; Haw, C.; Saunders, K.E.A. Risk factors for suicide in individuals with depression: A systematic review. J. Affect. Disord. 2013, 147, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Rihmer, Z. Antidepressants, depression and suicide. Neuropsychopharmacol. Hung. 2013, 15, 157–164. [Google Scholar]
- Lacerda-Pinheiro, S.F.; Junior, R.F.F.P.; De Lima, M.A.P.; Da Silva, C.G.L.; Dos Santos, M.D.S.V.; Júnior, A.G.T.; De Oliveira, P.N.L.; Ribeiro, K.D.B.; Neto, M.L.R.; Bianco, B. Are there depression and anxiety genetic markers and mutations? A systematic review. J. Affect. Disord. 2014, 168, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Vrijsen, J.N.; Van Oostrom, I.; Franke, B.; Becker, E.S.; Speckens, A.; Arias-Vásquez, A. Association between genes, stressful childhood events and processing bias in depression vulnerable individuals. Genes Brain Behav. 2014, 13, 508–516. [Google Scholar] [CrossRef]
- Latendresse, G.; Elmore, C.; Deneris, A. Selective Serotonin Reuptake Inhibitors as First-Line Antidepressant Therapy for Perinatal Depression. J. Midwifery Women’s Health 2017, 62, 317–328. [Google Scholar] [CrossRef]
- Mago, R.; Mahajan, R.E.; Thase, M. Levomilnacipran: A newly approved drug for treatment of major depressive disorder. Expert Rev. Clin. Pharmacol. 2014, 7, 137–145. [Google Scholar] [CrossRef]
- Kornstein, S.G.; McIntyre, R.S.; Thase, M.E.; Boucher, M. Desvenlafaxine for the treatment of major depressive disorder. Expert Opin. Pharmacother. 2014, 15, 1449–1463. [Google Scholar] [CrossRef]
- Yoshinaga, H.; Nishida, T.; Sasaki, I.; Kato, T.; Oki, H.; Yabuuchi, K.; Toyoda, T. Discovery of DSP-1053, a novel benzylpiperidine derivative with potent serotonin transporter inhibitory activity and partial 5-HT 1A receptor agonistic activity. Bioorganic Med. Chem. 2018, 26, 1614–1627. [Google Scholar] [CrossRef]
- Seabrook, E.; Kern, M.L.; Rickard, N.S.; Cha, M.; Rice, S.; Gritton, J. Social Networking Sites, Depression, and Anxiety: A Systematic Review. JMIR Ment. Health 2016, 3, e50. [Google Scholar] [CrossRef] [PubMed]
- Cowen, P.J. Backing into the future: Pharmacological approaches to the management of resistant depression. Psychol. Med. 2017, 47, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Penn, E.; Tracy, D.K. The drugs don’t work? Antidepressants and the current and future pharmacological management of depression. Ther. Adv. Psychopharmacol. 2012, 2, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Marston, O.J.; Garfield, A.S.; Heisler, L.K. Role of central serotonin and melanocortin systems in the control of energy balance. Eur. J. Pharmacol. 2011, 660, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Gray, J.A.; Roth, B.L. The Expanded Biology of Serotonin. Annu. Rev. Med. 2009, 60, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.E.; Nichols, C.D. Serotonin Receptors. Chem. Rev. 2008, 108, 1614–1641. [Google Scholar] [CrossRef] [PubMed]
- Czopek, A.; Byrtus, H.; Kolaczkowski, M.; Pawłowski, M.; Dybała, M.; Nowak, G.; Tatarczynska, E.; Wesołowska, A.; Chojnacka-Wójcik, E. Synthesis and pharmacological evaluation of new 5-(cyclo)alkyl-5-phenyl- and 5-spiroimidazolidine-2,4-dione derivatives. Novel 5-HT1A receptor agonist with potential antidepressant and anxiolytic activity. Eur. J. Med. Chem. 2010, 45, 1295–1303. [Google Scholar] [CrossRef]
- Shimizu, S.; Tatara, A.; Imaki, J.; Ohno, Y. Role of cortical and striatal 5-HT1A receptors in alleviating antipsychotic-induced extrapyramidal disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 877–881. [Google Scholar] [CrossRef]
- Blier, P.; Ward, N.M. Is There A Role for 5-HT1A Agonists in the Treatment of Depression? Biol. Psychiatry 2003, 53, 193–203. [Google Scholar] [CrossRef]
- Bara-Jimenez, W.; Bibbiani, F.; Morris, M.J.; Dimitrova, T.; Sherzai, A.; Mouradian, M.M.; Chase, T.N. Effects of serotonin 5-HT1A agonist in advanced Parkinson’s disease. Mov. Disord. 2005, 20, 932–936. [Google Scholar] [CrossRef]
- Blier, P.; De Montigny, C. Current advances and trends in the treatment of depression. Trends Pharmacol. Sci. 1994, 15, 220–226. [Google Scholar] [CrossRef]
- Xu, L.; Zhou, S.; Yu, K.; Gao, B.; Jiang, H.; Zhen, X.; Fu, W. Molecular Modeling of the 3D Structure of 5-HT1AR: Discovery of Novel 5-HT1AR Agonists via Dynamic Pharmacophore-Based Virtual Screening. J. Chem. Inf. Model. 2013, 53, 3202–3211. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.; Böttcher, H.; Gericke, R.; Bartoszyk, G.D.; Anzali, S.; Seyfried, C.A.; Greiner, H.E.; Van Amsterdam, C. Synthesis and Structure−Activity Relationship in a Class of Indolebutylpiperazines as Dual 5-HT1A Receptor Agonists and Serotonin Reuptake Inhibitors. J. Med. Chem. 2004, 47, 4684–4692. [Google Scholar] [CrossRef] [PubMed]
- Lian, P.; Li, L.; Geng, C.; Zhen, X.; Fu, W. Higher-Affinity Agonists of 5-HT1AR Discovered through Tuning the Binding-Site Flexibility. J. Chem. Inf. Model. 2015, 55, 1616–1627. [Google Scholar] [CrossRef]
- García-Nafría, J.; Nehmé, R.; Edwards, P.C.; Tate, C.G. Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go. Nature 2018, 558, 620–623. [Google Scholar] [CrossRef]
- Kimura, K.T.; Asada, H.; Inoue, A.; Kadji, F.M.N.; Im, D.; Mori, C.; Arakawa, T.; Hirata, K.; Nomura, Y.; Nomura, N.; et al. Structures of the 5-HT2A receptor in complex with the antipsychotics risperidone and zotepine. Nat. Struct. Mol. Biol. 2019, 26, 121–128. [Google Scholar] [CrossRef]
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, L.; Li, W.; Zhu, C.; Peng, W.; Han, B.; Fu, W. Design, Synthesis, and Structure–Activity Relationship Studies of Novel Indolyalkylpiperazine Derivatives as Selective 5-HT1A Receptor Agonists. J. Chem. Inf. Model. 2020, 60, 235–248. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, w5–w9. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System, version 1.2r3pre; Schrödinger, LLC.: San Carlos, CA, USA, 2002; 3p. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |


































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).