Identification of Iminium Intermediates Generation in the Metabolism of Tepotinib Using LC-MS/MS: In Silico and Practical Approaches to Bioactivation Pathway Elucidation


Abstract

1. Introduction
2. Results and Discussion
2.1. Results of In Silico TEP Metabolites Prediction
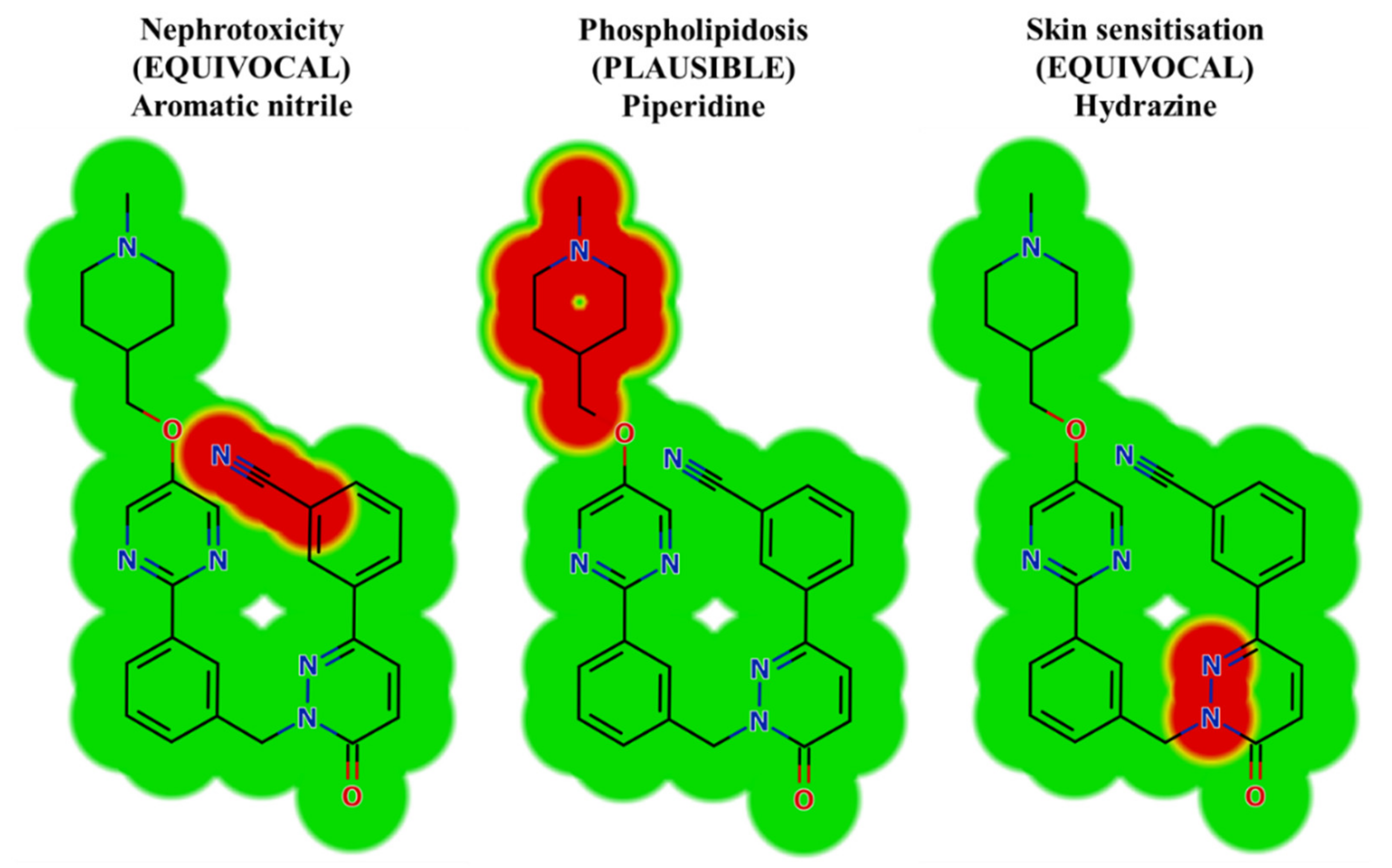
2.2. Results of In Silico TEP Structural Alerts Sites and Toxicity Prediction
2.3. Identification of In Vitro Phase I TEP Metabolites
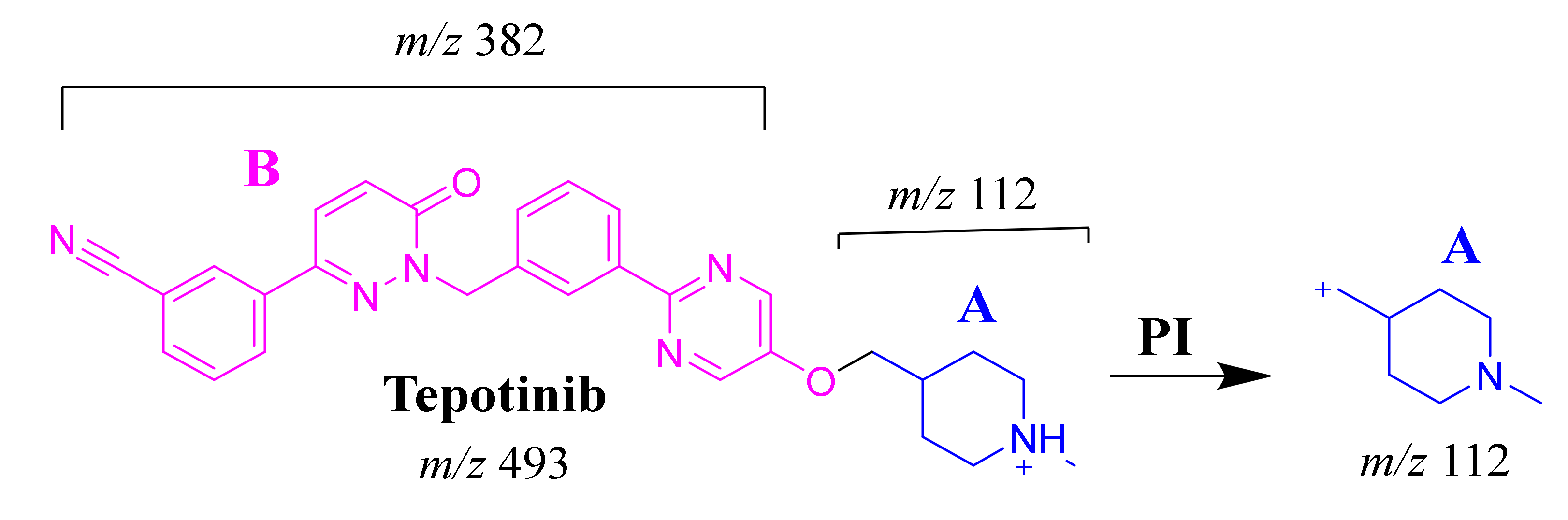
2.3.1. TEP Fragmentation Pattern
2.3.2. M1 Fragment Ions
2.3.3. M2 Fragment Ions
2.3.4. M3 Fragment Ions
2.3.5. M4 Fragment Ions
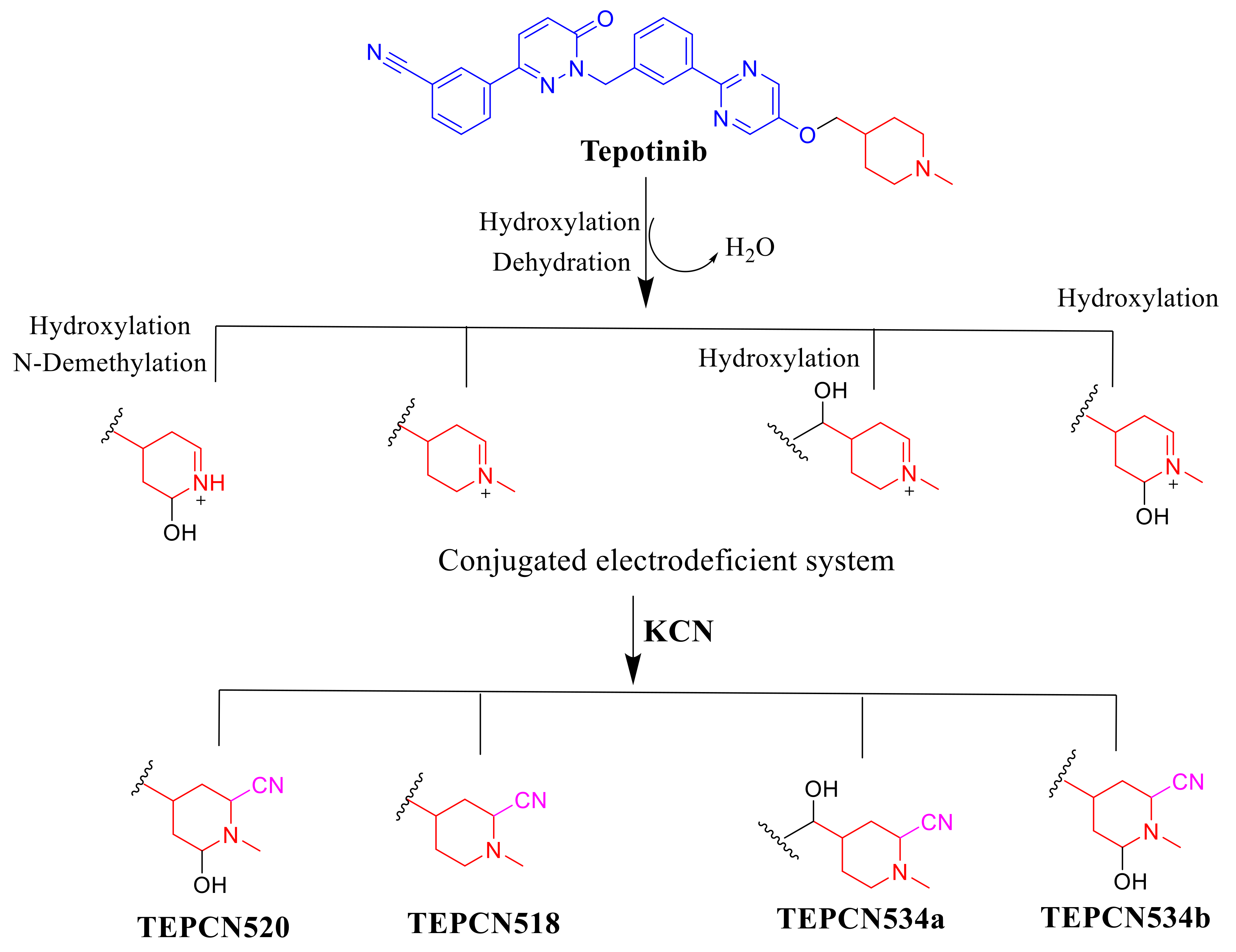
2.4. Reactive Metabolites
2.4.1. TEPCN518 Fragment Ions
2.4.2. TEPCN520 Fragment Ions
2.4.3. TEP534a and TEP534b Fragment Ions
2.5. Proposed TEP Bioactivation Mechanism
3. Chemicals and Methods
3.1. Chemicals
3.2. Chromatography Conditions
3.3. In Silico Prediction of TEP Metabolism Using WhichP450TM Module of StarDrop Software
3.4. In Silico Prediction of the Toxicity of TEP Metabolites and Reactivity Using DEREK Software and Xenosite Reactivity Model
3.5. HLM Incubation
3.6. Identification of TEP-Reactive Metabolites
4. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bylicki, O.; Paleiron, N.; Assié, J.-B.; Chouaïd, C. Targeting the met-signaling pathway in non-small-cell lung cancer: Evidence to date. Onco Targets Ther. 2020, 13, 5691–5706. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Veillon, R.; Cortot, A.B.; Felip, E.; Sakai, H.; Mazieres, J.; Griesinger, F.; Horn, L.; Senellart, H.; Van Meerbeeck, J.P. Phase II study of tepotinib in nsclc patients with met EX14 mutations. J. Clin. Oncol. 2019, 37, 9005. [Google Scholar] [CrossRef]
- Ruiz-Cordero, R.; Devine, W.P. Targeted therapy and checkpoint immunotherapy in lung cancer. Surg. Pathol. Clin. 2020, 13, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Tepotinib: First approval. Drugs 2020, 80, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Knowles, S.R.; Uetrecht, J.; Shear, N.H. Idiosyncratic drug reactions: The reactive metabolite syndromes. Lancet 2000, 356, 1587–1591. [Google Scholar] [CrossRef]
- Ju, C.; Uetrecht, J. Mechanism of idiosyncratic drug reactions: Reactive metabolites formation, protein binding and the regulation of the immune system. Curr. Drug Metab. 2002, 3, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S. Detection and characterization of olmutinib reactive metabolites by lc–ms/ms: Elucidation of bioactivation pathways. J. Sep. Sci. 2020, 43, 708–718. [Google Scholar] [CrossRef]
- Evans, D.C.; Watt, A.P.; Nicoll-Griffith, D.A.; Baillie, T.A. Drug-protein adducts: An industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem. Res. Toxicol. 2004, 17, 3–16. [Google Scholar] [CrossRef]
- Kalgutkar, A.S.; Dalvie, D.K.; O’Donnell, J.P.; Taylor, T.J.; Sahakian, D.C. On the diversity of oxidative bioactivation reactions on nitrogen-containing xenobiotics. Curr. Drug Metab. 2002, 3, 379–424. [Google Scholar] [CrossRef]
- Boelsterli, U.A. Xenobiotic acyl glucuronides and acyl coa thioesters as protein-reactive metabolites with the potential to cause idiosyncratic drug reactions. Curr. Drug Metab. 2002, 3, 439–450. [Google Scholar] [CrossRef]
- AlRabiah, H.; Kadi, A.A.; Attwa, M.W.; Abdelhameed, A.S.; Mostafa, G.A. Reactive intermediates in copanlisib metabolism identified by LC-MS/MS: Phase I metabolic profiling. RSC Adv. 2019, 9, 6409–6418. [Google Scholar] [CrossRef]
- Ma, S.; Zhu, M. Recent advances in applications of liquid chromatography-tandem mass spectrometry to the analysis of reactive drug metabolites. Chem. Biol. Interact. 2009, 179, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Stepan, A.F.; Walker, D.P.; Bauman, J.; Price, D.A.; Baillie, T.A.; Kalgutkar, A.S.; Aleo, M.D. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: A perspective based on the critical examination of trends in the top 200 drugs marketed in the united states. Chem. Res. Toxicol. 2011, 24, 1345–1410. [Google Scholar] [CrossRef]
- Masic, L.P. Role of cyclic tertiary amine bioactivation to reactive iminium species: Structure toxicity relationship. Curr. Drug Metab. 2011, 12, 35–50. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Q.; Li, Y.; Doss, G.A.; Dean, B.J.; Ngui, J.S.; Silva Elipe, M.; Kim, S.; Wu, J.Y.; Dininno, F.; et al. In vitro bioactivation of dihydrobenzoxathiin selective estrogen receptor modulators by cytochrome p450 3a4 in human liver microsomes: Formation of reactive iminium and quinone type metabolites. Chem. Res. Toxicol. 2005, 18, 675–685. [Google Scholar] [CrossRef]
- Park, B.K.; Boobis, A.; Clarke, S.; Goldring, C.E.; Jones, D.; Kenna, J.G.; Lambert, C.; Laverty, H.G.; Naisbitt, D.J.; Nelson, S. Managing the challenge of chemically reactive metabolites in drug development. Nat. Rev. Drug Discov. 2011, 10, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S. Characterization of reactive intermediates formation in dacomitinib metabolism and bioactivation pathways elucidation by LC-MS/MS: In vitro phase I metabolic investigation. RSC Adv. 2018, 8, 38733–38744. [Google Scholar] [CrossRef]
- Ma, S.; Subramanian, R. Detecting and characterizing reactive metabolites by liquid chromatography/tandem mass spectrometry. J. Mass Spectrum. 2006, 41, 1121–1139. [Google Scholar] [CrossRef]
- Tolonen, A.; Turpeinen, M.; Pelkonen, O. Liquid chromatography–mass spectrometry in in vitro drug metabolite screening. Drug Discov. Today 2009, 14, 120–133. [Google Scholar] [CrossRef]
- Li, F.; MacKenzie, K.R.; Nyshadham, P.; Kerlec, K.A.; Matzuk, M.M. Identifying metabolic pathways of c-met tyrosine kinase inhibitor tepotinib in human and mouse liver microsomes. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A. Sapitinib: Reactive intermediates and bioactivation pathways characterized by lc-ms/ms. RSC Adv. 2019, 9, 32995–33006. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A.; AlRabiah, H.; Darwish, H.W. Reactive intermediates in naquotinib metabolism identified by liquid chromatography-tandem mass spectrometry: Phase I metabolic profiling. RSC Adv. 2019, 9, 10211–10225. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A.; Darwish, H.W.; Amer, S.M.; Al-shakliah, N.S. Identification and characterization of in vivo, in vitro and reactive metabolites of vandetanib using LC–ESI–MS/MS. Chem. Cent. J. 2018, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- T’jollyn, H.; Boussery, K.; Mortishire-Smith, R.; Coe, K.; De Boeck, B.; Van Bocxlaer, J.; Mannens, G. Evaluation of three state-of-the-art metabolite prediction software packages (meteor, metasite, and stardrop) through independent and synergistic use. Drug Metab. Dispos. 2011, 39, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Marchant, C.A.; Briggs, K.A.; Long, A. In silico tools for sharing data and knowledge on toxicity and metabolism: Derek for windows, meteor, and vitic. Toxicol. Mech. Methods 2008, 18, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Matlock, M.K.; Hughes, T.B.; Swamidass, S.J. Xenosite server: A web-available site of metabolism prediction tool. Bioinformatics 2015, 31, 1136–1137. [Google Scholar] [CrossRef] [PubMed]
- Zaretzki, J.; Matlock, M.; Swamidass, S.J. Xenosite: Accurately predicting cyp-mediated sites of metabolism with neural networks. J. Chem. Inf. Model. 2013, 53, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- Abdelhameed, A.S.; Attwa, M.W.; Kadi, A.A. Liquid chromatography-tandem mass spectrometry metabolic profiling of nazartinib reveals the formation of unexpected reactive metabolites. R. Soc. Open Sci. 2019, 6, 190852. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S. Phase I metabolic profiling and unexpected reactive metabolites in human liver microsome incubations of X-376 using LC-MS/MS: Bioactivation pathway elucidation and in silico toxicity studies of its metabolites. RSC Adv. 2020, 10, 5412–5427. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S.; Alhazmi, H.A. Metabolic stability assessment of new parp inhibitor talazoparib using validated lc-ms/ms methodology: In silico metabolic vulnerability and toxicity studies. Drug Des. Dev. Ther. 2020, 14, 783–793. [Google Scholar] [CrossRef]
- Lüllmann, H.; Lüllmann-Rauch, R.; Wassermann, O.; de la Iglesia, F.A. Drug-induced phospholipidoses. CRC Crit. Rev. Toxicol. 1975, 4, 185–218. [Google Scholar] [CrossRef] [PubMed]
- Attwa, M.W.; Kadi, A.A.; Alrabiah, H.; Darwish, H.W. Lc–ms/ms reveals the formation of iminium and quinone methide reactive intermediates in entrectinib metabolism: In vivo and in vitro metabolic investigation. J. Pharm. Biomed. Anal. 2018, 160, 19–30. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S. Reactive intermediates and bioactivation pathways characterization of avitinib by lc–ms/ms: In vitro metabolic investigation. J. Pharm. Biomed. Anal. 2019, 164, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Abdelhameed, A.S.; Kadi, A.A.; Attwa, M.W.; AlRabiah, H. Validated LC-MS/MS assay for quantification of the newly approved tyrosine kinase inhibitor, dacomitinib, and application to investigating its metabolic stability. PLoS ONE 2019, 14, e4598. [Google Scholar] [CrossRef] [PubMed]
- Alrabiah, H.; Kadi, A.A.; Attwa, M.W.; Abdelhameed, A.S. A simple liquid chromatography-tandem mass spectrometry method to accurately determine the novel third-generation egfr-tki naquotinib with its applicability to metabolic stability assessment. RSC Adv. 2019, 9, 4862–4869. [Google Scholar] [CrossRef]
- Abdelhameed, A.S.; Attwa, M.W.; Al-Shaklia, N.S.; Kadi, A.A. A highly sensitive LC-MS/MS method to determine novel bruton’s tyrosine kinase inhibitor spebrutinib: Application to metabolic stability evaluation. Royal Soc. Open Sci. 2019, 6, 190434. [Google Scholar] [CrossRef] [PubMed]
- Attwa, M.W.; Kadi, A.A.; Darwish, H.W.; Abdelhameed, A.S. Investigation of the metabolic stability of olmutinib by validated LC-MS/MS: Quantification in human plasma. RSC Adv. 2018, 8, 40387–40394. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TEP and Its Phase I Metabolites | Phospholipidosis | Skin Sensitization | Nephrotoxicity | Chromosome Damage, Teratogenicity, Carcinogencity, Mutagenicity and Genotoxicity |
|---|---|---|---|---|
| Piperidine | Hydrazine | Halogenated nitrile | ||
| TEP | Plausible | Equivocal | Equivocal | NA |
| M1 | NA * | Plausible | Equivocal | NA |
| M2 | NA | Equivocal | Equivocal | NA |
| M3 | NA | Equivocal | Equivocal | NA |
| M4 | NA | Equivocal | Equivocal | NA |
| MS Scan | Fragment Ions | Elution Time (min) | Metabolic and Bioactivation Pathways | |
|---|---|---|---|---|
| TEP | 493 | 112 | 25.1 | Main drug |
| Phase-I Metabolites | ||||
| M1 | 493 | 382, 296, 185, 112, 72 | 26.6 | N-demethylation and α-oxidation at piperidine ring |
| M2 | 479 | 382, 185, 98 | 24.6 | N-demethylation at piperidine ring |
| M3 | 507 | 310, 126 | 28.3 | α-Oxidation at piperidine ring |
| M4 | 509 | 128 | 28.4 | α-Hydroxylation at piperidine ring |
| Reactive Metabolites | ||||
| TEPCN518 | 518 | 491, 459, 137, 110, 104 | 39.2 | Cyano attack at bioactivated α-carbon of piperidine ring |
| TEPCN520 | 520 | 493, 475, 278, 139 | 42.9 | N-demethylation, hydroxylation and cyano attack at bioactivated α-carbon of piperidine ring |
| TEPCN534a | 534 | 516, 475, 296, 153 | 28.9 | Hydroxylation and cyano addition at α-carbon of piperidine ring |
| TEPCN534b | 534 | 491, 382, 210, 153, 110 | Hydroxylation at methylene carbon attached to piperidine ring and cyano attack at bioactivated α-carbon of piperidine ring | |
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelhameed, A.S.; Attwa, M.W.; Kadi, A.A. Identification of Iminium Intermediates Generation in the Metabolism of Tepotinib Using LC-MS/MS: In Silico and Practical Approaches to Bioactivation Pathway Elucidation. Molecules 2020, 25, 5004. https://doi.org/10.3390/molecules25215004
Abdelhameed AS, Attwa MW, Kadi AA. Identification of Iminium Intermediates Generation in the Metabolism of Tepotinib Using LC-MS/MS: In Silico and Practical Approaches to Bioactivation Pathway Elucidation. Molecules. 2020; 25(21):5004. https://doi.org/10.3390/molecules25215004
Chicago/Turabian StyleAbdelhameed, Ali S., Mohamed W. Attwa, and Adnan A. Kadi. 2020. "Identification of Iminium Intermediates Generation in the Metabolism of Tepotinib Using LC-MS/MS: In Silico and Practical Approaches to Bioactivation Pathway Elucidation" Molecules 25, no. 21: 5004. https://doi.org/10.3390/molecules25215004
APA StyleAbdelhameed, A. S., Attwa, M. W., & Kadi, A. A. (2020). Identification of Iminium Intermediates Generation in the Metabolism of Tepotinib Using LC-MS/MS: In Silico and Practical Approaches to Bioactivation Pathway Elucidation. Molecules, 25(21), 5004. https://doi.org/10.3390/molecules25215004