Phthalocyanine-Cored Fluorophores with Fluorene-Containing Peripheral Two-Photon Antennae as Photosensitizers for Singlet Oxygen Generation




 and
and
Abstract
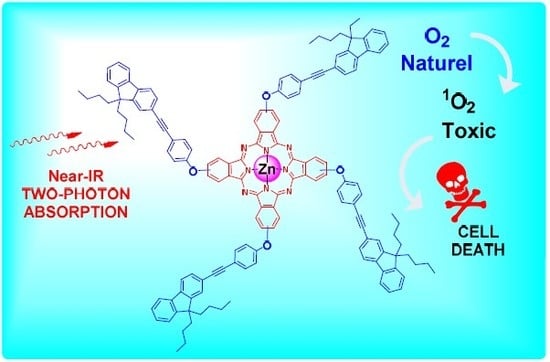
1. Introduction
2. Results
2.1. Synthesis of the Phthalonitrile Precursors
2.2. Synthesis of Phthalocyanine Macrocycles
2.3. Photophysical Properties
2.4. Absorption Properties
2.5. Emission Properties
2.6. Energy Transfer from the Fluorene Units to the Phthalocyanine Core
2.7. Oxygen Photosensitization
2.8. Two-Photon Absorption
2.9. DFT Studies
3. Discussion
3.1. Structural Effects on the Linear and Nonlinear Optical Properties
3.2. Comparison with Porphyrin Analogues
4. Materials and Methods
4.1. General
4.2. Synthesis of Precursors
4.3. Phthalocyanine Synthesis
4.4. Spectroscopic Measurements
4.5. Two-Photon Absorption Experiments
4.6. Measurement of Singlet Oxygen Quantum Yields (ΦΔ)
4.7. DFT Computations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Boudon, J.; Paris, J.; Bernhard, Y.; Popova, E.; Decreau, R.A.; Millot, N. Magneto-optical nanomaterials: A SPIO–phthalocyanine scaffold built step-by-step towards bimodal imaging. Chem. Commun. 2013, 49, 7394. [Google Scholar] [CrossRef] [PubMed]
- Kuzyniak, W.; Ermilov, E.A.; Atilla, D.; Gürek, A.G.; Nitzsche, B.; Derkow, K.; Hoffmann, B.; Steinemann, G.; Ahsen, V.; Höpfner, M. Tetra-triethyleneoxysulfonyl substituted zinc phthalocyanine for photodynamic cancer therapy. Photodiag. Photodyn. Therapy 2016, 13, 148. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.; Jenni, S.; Sour, A.; Heitz, V.; Bolze, F.; Pallier, A.; Bonnet, C.S.; Tóth, É.; Ventura, B. A Porphyrin Dimer-GdDOTA Conjugate as a Theranostic Agent for One- and Two-Photon Photodynamic Therapy and MRI. Bioconjugate Chem. 2018, 29, 3726. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Nguyen, C.; Daurat, M.; Dhieb, A.C.; Smirani, W.; Blanchard-Desce, M.; Gary-Bobo, M.; Mongin, O.; Paul-Roth, C.; Paul, F. Biocompatible Conjugated Fluorenylporphyrins for Two-photon Photodynamic Therapy and Fluorescence Imaging. Chem. Commun. 2019, 55, 12231. [Google Scholar] [CrossRef] [PubMed]
- Josefsen, L.B.; Boyle, R.W. Unique Diagnostic and Therapeutic Roles of Porphyrins and Phthalocyanines in Photodynamic Therapy, Imaging and Theranostics. Theranostics 2012, 2, 916. [Google Scholar] [CrossRef]
- Dabrowski, J.M.; Pucelik, B.; Regiel-Futyra, A.; Brindell, M.; Mazuryk, O.; Kyzioł, A.; Stochel, G.; Macyk, W.; Arnaut, L.G. Engineering of relevant photodynamic processes through structural modifications of metallotetrapyrrolic photosensitizers. Coord. Chem. Rev. 2016, 325, 67. [Google Scholar] [CrossRef]
- Nyokong, T. Effects of substituents on the photochemical and photophysical properties of main group metal phthalocyanines. Coord. Chem. Rev. 2007, 251, 1707. [Google Scholar] [CrossRef]
- Miller, J.D.; Baron, E.D.; Scull, H.; Hsia, A.; Berlin, J.C.; McCormick, T.; Colussi, V.; Kenney, M.E.; Cooper, K.D.; Oleinick, N.L. Photodynamic therapy with the phthalocyanine photosensitizer Pc 4: The case experience with preclinical mechanistic and early clinical–translational studies. Toxicol. Appl. Pharmcol. 2007, 224, 290. [Google Scholar] [CrossRef]
- Figueira, F.; Pereira, P.M.R.; Silva, S.; Cavaleiro, J.A.S.; Tomé, J.P.C. Porphyrins and Phthalocyanines Decorated with Dendrimers: Synthesis and Biomedical Applications. Current Org. Synth. 2014, 11, 110. [Google Scholar] [CrossRef]
- Venkatram, N.; Rao, D.N.; Giribabu, L.; Rao, S.V. Femtosecond nonlinear optical properties of alkoxy phthalocyanines at 800 nm studied using Z-Scan technique. Chem. Phys. Lett. 2008, 464, 211. [Google Scholar] [CrossRef]
- Makarov, N.S.; Drobizhev, M.; Rebane, A. Two-photon absorption standards in the 550–1600 nm excitation wavelength range. Opt. Express 2008, 16, 4029. [Google Scholar] [CrossRef]
- Drobizhev, M.; Makarov, N.S.; Rebane, A.; de la Torre, G.; Torres, T. Strong Two-Photon Absorption in Push-Pull Phthalocyanines: Role of Resonance Enhancement and Permanent Dipole Moment Change upon Excitation. J. Phys. Chem. C 2008, 112, 848. [Google Scholar] [CrossRef]
- Morisue, M.; Ogawa, K.; Kamada, K.; Ohta, K.; Kobuke, Y. Strong two-photon and three-photon absorptions in the antiparallel dimer of a porphyrin–phthalocyanine tandem. Chem. Commun. 2010, 46, 2121. [Google Scholar] [CrossRef] [PubMed]
- Bolze, F.; Jenni, S.; Sour, A.; Heitz, V. Molecular photosensitisers for two-photon photodynamic therapy. Chem. Commun. 2017, 53, 12857. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, L.-P.; Wu, F.; Zhao, Y. Photosensitizers for Two-Photon Excited Photodynamic Therapy. Adv. Funct. Mater. 2017, 27, 1704079. [Google Scholar] [CrossRef]
- Liu, Z.; Xiong, X.; Li, Y.; Lia, S.; Qin, J. Synthesis, optical properties and singlet oxygen generation of a phthalocyanine derivative containing strong two-photon-absorbing chromophores in the periphery. Photochem. Photobiol. Sci. 2011, 10, 1804. [Google Scholar] [CrossRef] [PubMed]
- Paul-Roth, C.O.; Simonneaux, G. Porphyrins with fluorenyl and fluorenone pendant arms. Tetrahedron Lett. 2006, 47, 3275. [Google Scholar] [CrossRef]
- Paul-Roth, C.O.; Simonneaux, G. Porphyrins with fluorenyl and fluorenone pendant arms as red-light-emitting devices. C.R. Acad. Sci. Ser. IIb Chim. 2006, 9, 1277. [Google Scholar] [CrossRef]
- Li, B.; Li, J.; Fu, Y.; Bo, Z. Porphyrins with Four Monodisperse Oligofluorene Arms as Efficient Red Light-Emitting Materials. J. Am. Chem. Soc. 2004, 126, 3430. [Google Scholar] [CrossRef]
- Paul-Roth, C.O.; Williams, J.A.G.; Letessier, J.; Simonneaux, G. New tetra-aryl and bi-aryl porphyrins bearing 5,15-related fluorenyl pendants: The influence of arylation on fluorescence. Tetrahedron Lett. 2007, 48, 4317. [Google Scholar] [CrossRef]
- Paul-Roth, C.O.; Rault-Berthelot, J.; Simonneaux, G. New polymers for catalytic carbene transfer: Electropolymerization of tetrafluorenylporphyrinruthenium carbon monoxide. Tetrahedron 2004, 60, 12169. [Google Scholar] [CrossRef]
- Drouet, S.; Paul-Roth, C.O.; Simonneaux, G. Synthesis and photophysical properties of porphyrins with fluorenyl pendant arms. Tetrahedron 2009, 65, 2975. [Google Scholar] [CrossRef]
- Drouet, S.; Paul-Roth, C.O. Fluorenyl Dendrimer Porphyrins: Synthesis and Photophysical Properties. Tetrahedron 2009, 65, 10693. [Google Scholar] [CrossRef]
- Harth, E.M.; Hecht, S.; Helms, B.; Malmstrom, E.E.; Fréchet, J.M.J.; Hawker, C.J. The Effect of Macromolecular Architecture in Nanomaterials: A Comparison of Site Isolation in Porphyrin Core Dendrimers and Their Isomeric Linear Analogues. J. Am. Chem. Soc. 2002, 124, 3926. [Google Scholar] [CrossRef] [PubMed]
- Mongin, O.; Hugues, V.; Blanchard-Desce, M.; Merhi, A.; Drouet, S.; Yao, D.; Paul-Roth, C. Fluorenyl porphyrins for combined two-photon excited fluorescence and photosensitization. Chem. Phys. Lett. 2015, 625, 151. [Google Scholar] [CrossRef]
- Yao, D.; Zhang, X.; Triadon, A.; Richy, N.; Mongin, O.; Blanchard-Desce, M.; Paul, F.; Paul-Roth, C.O. New Conjugated meso-Tetrafluorenylporphyrin-cored Derivatives as Fluorescent Two-photon Photosensitizers for Singlet Oxygen Generation. Chem. Eur. J. 2017, 23, 2635. [Google Scholar] [CrossRef]
- Lawrence, D.S.; Whitten, D.G. Photochemistry and Photophysical Properties of Novel, Unsymmetrically Substituted Metallophthalocyanines. Photochem. Photobiol. 1996, 64, 923. [Google Scholar] [CrossRef] [PubMed]
- Vincett, P.S.; Voigt, E.M.; Rieckhoff, K.E.J. Phosphorescence and fluorescence of phthalocyanines. Chem. Phys. 1971, 55, 4131. [Google Scholar] [CrossRef]
- Ogunsipe, A.; Maree, D.; Nyokong, T. Solvent effects on the photochemical and fluorescence properties of zinc phthalocyanine derivatives. J. Mol. Struct. 2003, 650, 131. [Google Scholar] [CrossRef]
- Barker, C.A.; Zeng, X.; Bettington, S.; Batsanov, S.A.; Bryce, M.R.; Beeby, A. Porphyrin, Phthalocyanine and Porphyrazine Derivatives with Multifluorenyl Substituents as Efficient Deep-Red Emitters. Chem. Eur. J. 2007, 13, 6710. [Google Scholar] [CrossRef]
- Cabir, B.; Agırtas, M.S.; Duygulu, E.; Yuksel, F. Synthesis of some metallophthalocyanines bearing 9-phenyl-9Hfluoren- 9-yl) oxy functional groups and investigation of their photophysical properties. J. Mol. Struct. 2017, 1142, 194. [Google Scholar] [CrossRef]
- Görlach, B.; Dachtler, M.; Glaser, T.; Albert, K.; Hanack, M. Synthesis and separation of structural isomers of 2(3),9(10),16(17),23(24)-tetrasubstituted phthalocyanines. Chem. Eur. J. 2001, 7, 2459. [Google Scholar] [CrossRef]
- Troisi, A.; Ratner, M.A. Molecular rectification through electric field induced conformational changes. J. Am. Chem. Soc. 2002, 124, 14528. [Google Scholar] [CrossRef] [PubMed]
- Maertens, C.; Detrembleur, C.; Dubois, P.; Jérôme, R.; Boutton, C.; Persoons, A.; Kogej, T.; Bredas, J.L. Structure-Second-Order Polarizability Relationship in Chromophores Incorporating a Spacer: A Joint Experimental and Theoretical Study. Chem. Eur. J. 1999, 5, 369. [Google Scholar] [CrossRef]
- Shorygin, P.P.; Ya, B.K. Conjugation and the periodic system of the elements. Russ. Chem. Rev. 1991, 60, 1. [Google Scholar] [CrossRef]
- Haruhiko, T.; Shojiro, S.; Shojiro, O.; Shinsaku, S. Synthesis of phthalocyanines from phthalonitrile with organic strong bases. Chem. Lett. 1980, 9, 1277. [Google Scholar]
- Touaiti, S.; Hajri, A.; Kahouech, M.S.; Khiari, J.; Jamoussi, B. Synthesis and characterization of new Zn-phtalocyanine-based semi-conducting materials. Arab. J. Chem. 2017, 10, 1553. [Google Scholar] [CrossRef]
- George, R.D.; Snow, A.W. Synthesis of 3-nitrophthalonitrile and tetra-α-substituted phthalocyanines. J. Heterocycl. Chem. 1995, 32, 495. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes, and Bromopyridines. Tetrahedron Lett. 1975, 50, 4467. [Google Scholar] [CrossRef]
- Yao, D.; Zhang, X.; Mongin, O.; Paul, F.; Paul-Roth, C.O. Synthesis and Characterization of New Conjugated Fluorenyl-Porphyrin Dendrimers for Optics. Chem. Eur. J. 2016, 22, 5583. [Google Scholar] [CrossRef]
- Haruhiko, T.; Shojiro, S.; Shinsaku, S. Synthesis of metallophthalocyanines from phthalonitrile with strong organic bases. Chem. Lett. 1983, 12, 313. [Google Scholar]
- Durmu, M.; Yexsilot, S. Separation and mesogenic properties of tetraalkoxy-substituted phthalocyanine isomers. New J. Chem. 2006, 30, 675. [Google Scholar] [CrossRef]
- Kobayashi, N.; Ogata, H.; Nonaka, N.; Luk’yanets, E.A. Effect of peripheral substitution on the electronic absorption and fluorescence spectra of metal-free and zinc phthalocyanines. Chem. Eur. J. 2003, 9, 5123. [Google Scholar] [CrossRef]
- Fernandez, D.A.; Awruch, J.; Dicelio, L.E. Photophysical and Aggregation Studies of t-Butyl-Substituted Zn Phthalocyanines. Photochem. Photobiol. 1996, 63, 784. [Google Scholar] [CrossRef]
- In principle four different regioisomers, two of them being centrosymmetric, can be formed in various ratios during the condensation reaction of their mono-functionalized phthalonitrile precursors [32,42]. A nearly statistical ratio (1:1:2:4) between them is usually expected for β-substituted derivatives [42].
- Fox, J.M.; Katz, T.J.; Elshocht, S.V.; Verbiest, T.; Kauranen, M.; Persoons, A.; Thongpanchang, T.; Krauss, T.; Brus, L. Synthesis, self-assembly, and nonlinear optical properties of conjugated helical metal phthalocyanine derivatives. J. Am. Chem. Soc. 1999, 121, 3453. [Google Scholar] [CrossRef]
- de la Escosura, A.; Martínez-Díaz, M.V.; Thordarson, P.; Rowan, A.E.; Nolte, R.J.M.; Torres, T. Donor-Acceptor Phthalocyanine Nanoaggregates. J. Am. Chem. Soc. 2003, 125, 12300. [Google Scholar] [CrossRef] [PubMed]
- Gouloumis, A.; González-Rodríguez, D.; Vázquez, P.; Torres, T.; Liu, S.; Echegoyen, L.; Ramey, J.; Hug, G.L.; Guldi, D.M. Control Over Charge Separation in Phthalocyanine-Anthraquinone Conjugates as a Function of the Aggregation Status. J. Am. Chem. Soc. 2006, 128, 12674. [Google Scholar] [CrossRef]
- Piechocki, C.; Simon, J. Synthesis of a polar discogen. A new type of discotic mesophase. J. Chem. Soc. Chem. Commun. 1985, 259–260. [Google Scholar] [CrossRef]
- Terekhov, D.S.; Nolan, K.J.M.; McArthur, C.R.; Leznoff, C.C. Synthesis of 2,3,9,10,16,17,23,24-octaalkynylphthalocyanines and the effects of concentration and temperature on their 1H NMR spectra. J. Org. Chem. 1996, 61, 3034. [Google Scholar] [CrossRef]
- Chen, M.J.; Rathke, J.W. Dimeric aggregates of five-coordinated methyl(phthalocyaninato)Rh(III): 1H NMR evidence for staggered and slipped cofacial dimers. J. Porph. Phthalocyan. 2001, 5, 528. [Google Scholar] [CrossRef]
- Edwards, L.; Gouterman, M. Vapor Absorption Spectra and Stability: Phthalocyanines. J. Mol. Spectro. 1970, 33, 292. [Google Scholar] [CrossRef]
- van Leeuwen, M.; Beeby, A.; Ashworth, S.H. The photochemistry and photophysics of a series of non-peripherally substituted zinc phthalocyanines. Photochem. Photobiol. Sci. 2010, 9, 370. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, M.; Beeby, A.; Fernandes, I.; Ashworth, S.H. The photochemistry and photophysics of a series of alpha octa(alkyl-substituted) silicon, zinc and palladium phthalocyanines. Photochem. Photobiol. Sci. 2014, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Lavis, L.D.; Raines, R.T. Bright Ideas for Chemical Biology. ACS Chem. Bio 2008, 3, 142. [Google Scholar] [CrossRef] [PubMed]
- Ogunsipe, A.; Chen, J.-Y.; Nyokong, T. Photophysical and photochemical studies of zinc(II) phthalocyanine derivatives-effects of substituents and solvents. New J. Chem. 2004, 28, 822. [Google Scholar] [CrossRef]
- Xu, C.; Webb, W.W. Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm. J. Opt. Soc. Am. B 1996, 13, 481. [Google Scholar] [CrossRef]
- Drobizhev, M.; Makarov, S.; Stepanenko, Y.; Rebane, A. Near-infrared two-photon absorption in phthalocyanines: Enhancement of lowest gerade-gerade transition by symmetrical electron-accepting substitution. J. Chem. Phys. 2006, 124, 224701. [Google Scholar] [CrossRef]
- Ricciardi, G.; Rosa, A.; Baerends, E.J. Ground and Excited States of Zinc Phthalocyanine Studied by Density Functional Methods. J. Phys. Chem. A 2000, 105, 5242. [Google Scholar] [CrossRef]
- Zagal, J.H.; Gulppi, M.A.; Cardenas-Jiron, G. Metal-centered redox chemistry of substituted cobalt phthalocyanines adsorbed on graphite and correlations with MO calculations and Hammett parameters. Electrocatalytic reduction of a disulfide. Polyhedron 2000, 19, 2255. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameter. Chem. Rev. 1991, 91, 165. [Google Scholar] [CrossRef]
- Happer, D.A.R.; Wright, G.J. The variation of substituent resonance effects with electron demand. J. Chem. Soc. Perkin Trans. 1979, 694–698. [Google Scholar] [CrossRef]
- Pawlicki, M.; Collins, H.A.; Denning, R.G.; Anderson, H.L. Two-Photon Absorption and the Design of Two-Photon Dyes. Angew. Chem. Int. Ed. 2009, 48, 3244. [Google Scholar] [CrossRef] [PubMed]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 3rd ed.; Pergamon Press: Oxford, UK, 1988. [Google Scholar]
- Devi, C.L.; Yesudas, K.; Makarov, N.S.; Rao, V.J.; Bhanuprakash, K.; Perry, J.W. Fluorenylethynylpyrene derivatives with strong two-photon absorption: Influence of substituents on optical properties. J. Mater. Chem. C 2015, 3, 3730. [Google Scholar] [CrossRef]
- Demas, N.; Crosby, G.A. Measurement of photoluminescence quantum yields. J. Phys. Chem. 1971, 75, 991. [Google Scholar]
- Eaton, G.R. Reference Materials for Fluorescence Measurment. Pure Appl. Chem. 1988, 60, 1107. [Google Scholar] [CrossRef]
- Werts, M.H.V.; Nerambourg, N.; Pélégry, D.; Le Grand, Y.; Blanchard-Desce, M. Action cross sections of two-photon excited luminescence of some Eu(III) and Tb(III) complexes. Photochem. Photobiol. Sci. 2005, 4, 531. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local-density-functional theories. J. Chem. Phys. 1993, 98, 1372. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | λabs Fluorenyl (nm) | λabs N and B (nm) | λabs Q (nm) | λmax (M−1·cm−1) | λem (nm) | Ф Fa | λmax.ФFb | ФΔc |
|---|---|---|---|---|---|---|---|---|
| ZnPc | - | 343 | 602, 666 | 234,000 | 671, 741 | 0.26 d | 60840 | 0.61 |
| ZnTOFPc1 | 276 | 303, 350 | 611, 677 | - | 687, 758 | 0.29 | - | - |
| ZnTOFPc2 | 276 | 306, 351 | 611, 677 | 215,000 | 688, 758 | 0.33 | 70950 | 0.57 |
| H2TOFPc2 | 276 | 306, 343, 387 | 606, 644, 667, 703 | 136,000 | 709, 790 | 0.43 | 58480 | - |
| ZnTOFPc3 | 270 | 307, 353 | 628, 698 | 270,000 | 706, 779 | 0.23 | 62100 | 0.60 |
| ZnTOFPc4 | 309 (sh), 328, 344 | 609, 675 | 291,000 | 683, 753 | 0.37 | 107670 | 0.54 | |
| H2TOFPc4 | 310 (sh), 327, 344 | 604, 637, 665, 699 | 175,000 | 705, 786 | 0.47 | 82250 | - | |
| H2TOFP e | 304 | 423 | 516, 551, 590, 647 | 213,000 | 663, 728 | 0.10 | 21300 | - |
| H2OOFP e | 304 | 423 | 516, 551, 592, 653 | 245,300 | 652, 721 | 0.13 | 31889 | 0.64 |
| Compounds | λ2PAmax2 (nm) | σ2max2 a (GM) | σ2max2.ФFb (GM) | λ2PAmax1 (nm) | σ2max1 a (GM) | σ2max1.ФFb (GM) |
|---|---|---|---|---|---|---|
| ZnPc b | 820 | 200 | 52 | 880 | 150 c | 14 |
| ZnTOFPc2 | 830 | 330 | 109 | 880 | 160 | 53 |
| H2TOFPc2 | 840 | 350 | 150 | 880 | 130 | 56 |
| ZnTOFPc3 | 860 | 160 | 37 | 880 | 140 | 32 |
| ZnTOFPc4 | 830 | 510 | 189 | 880 | 240 | 89 |
| H2TOFPc4 | 840 | 360 | 169 | 880 | 200 | 94 |
| H2OOFP d | 790 | 45 | 5 | - | - | - |
| H2SOFP d | 790 | 75 | 11 | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abid, S.; Ben Hassine, S.; Richy, N.; Camerel, F.; Jamoussi, B.; Blanchard-Desce, M.; Mongin, O.; Paul, F.; Paul-Roth, C.O. Phthalocyanine-Cored Fluorophores with Fluorene-Containing Peripheral Two-Photon Antennae as Photosensitizers for Singlet Oxygen Generation. Molecules 2020, 25, 239. https://doi.org/10.3390/molecules25020239
Abid S, Ben Hassine S, Richy N, Camerel F, Jamoussi B, Blanchard-Desce M, Mongin O, Paul F, Paul-Roth CO. Phthalocyanine-Cored Fluorophores with Fluorene-Containing Peripheral Two-Photon Antennae as Photosensitizers for Singlet Oxygen Generation. Molecules. 2020; 25(2):239. https://doi.org/10.3390/molecules25020239
Chicago/Turabian StyleAbid, Seifallah, Sarra Ben Hassine, Nicolas Richy, Franck Camerel, Bassem Jamoussi, Mireille Blanchard-Desce, Olivier Mongin, Frédéric Paul, and Christine O. Paul-Roth. 2020. "Phthalocyanine-Cored Fluorophores with Fluorene-Containing Peripheral Two-Photon Antennae as Photosensitizers for Singlet Oxygen Generation" Molecules 25, no. 2: 239. https://doi.org/10.3390/molecules25020239
APA StyleAbid, S., Ben Hassine, S., Richy, N., Camerel, F., Jamoussi, B., Blanchard-Desce, M., Mongin, O., Paul, F., & Paul-Roth, C. O. (2020). Phthalocyanine-Cored Fluorophores with Fluorene-Containing Peripheral Two-Photon Antennae as Photosensitizers for Singlet Oxygen Generation. Molecules, 25(2), 239. https://doi.org/10.3390/molecules25020239