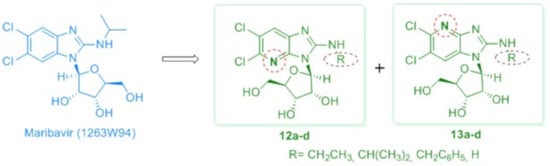
3.2. Synthesis
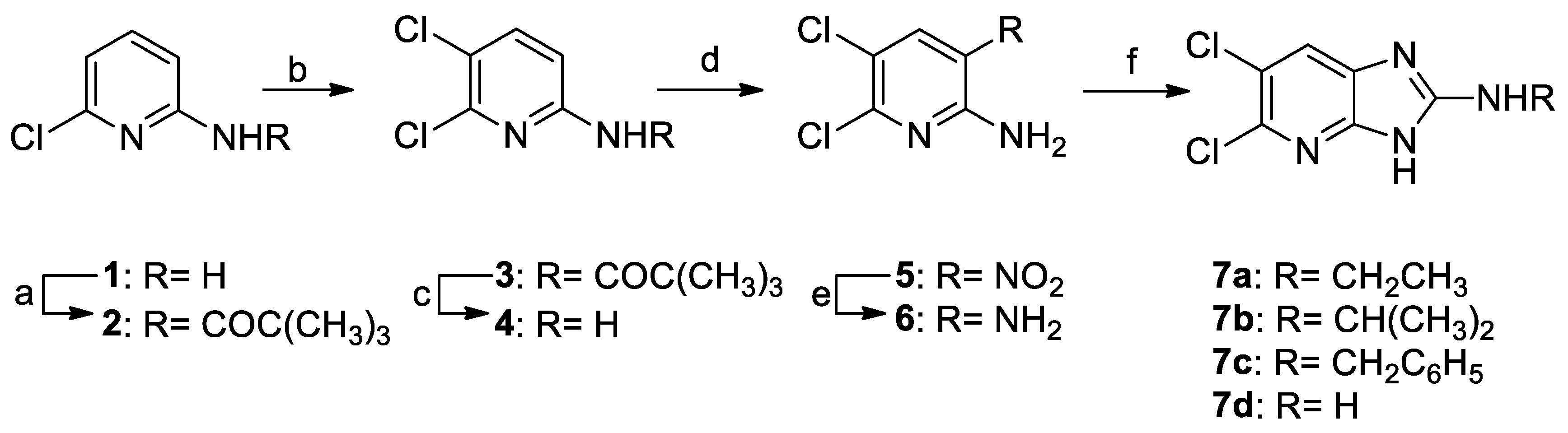
5,6-Dichloro-N-ethyl-3H-imidazo[4,5-b]pyridin-2-amine(7a): Ethylisothiocyanate (1 mL, 11.24 mmol) was added to a solution of diamine 6 (500 mg, 2.81 mmol) in anhydrous THF (10 mL) and the reaction mixture was refluxed under argon for 36 h. The mixture was then cooled to room temperature and HgO (1.22 g, 5.65 mmol) was added, leading to an orange suspension. Refluxing the reaction mixture resulted progressively in a black coloured suspension and within 40 min the reaction was completed. The mixture was filtered warm through a celite pad, which was thoroughly washed with warm MeOH. The solvent was then vacuum-evaporated and the residue was purified by column chromatography using a mixture of CHCl3/MeOH (95/5 to 85/15, v/v) as the eluent, to result in 340 mg of 7a (52% overall yield). Beige solid, mp > 300 °C (dec), (MeOH). 1H-NMR (DMSO-d6, 600MHz) δ 1.16 (t, 3H, CH3, J = 7.2 Hz), 3.33 (m, 2H, CH2), 7.37 (br s, 1H, D2O exchangeable, NH), 7.57 (s, 1H, H-7). 13C-NMR (151 MHz, DMSO-d6) δ 14.87 (CH3), 36.82 (CH2), 116.40 (C-6), 116.58 (C-7, major tautomeric form), 121.73 (C-7, minor tautomeric form), 126.92 (C-7a), 137.46 (C-5), 156.20 (C-3a), 158.92 (C-2). HR-MS (ESI) m/z: Calcd for C8H9Cl2N4: [M + H]+ = 231.0199, found 231.0202.
5,6-Dichloro-N-isopropyl-3H-imidazo[4,5-b]pyridin-2-amine (7b):This compound was prepared by a procedure analogous to that described for 7a, upon reaction of diamine 6 (500 mg, 2.81 mmol) with isopropyl isothiocyanate (0.90 mL, 8.43 mmol). Purification was effected by column chromatography, using a mixture of DCM/EtOAc (70/30 to 20/80, v/v) and then EtOAc/MeOH (99/1 to 97/3, v/v) as eluents. Overall yield 55%. White solid, mp 235–236 °C (acetone). 1H-NMR (600 MHz, DMSO-d6) δ 1.20 (d, 6H, 2xCH3, J = 6.1 Hz), 3.92 (m, 1H, CH), 7.34 (br s, 1H, D2O exchangeable, NH), 7.56 (s, 1H, H-7), 11.06 (br s, 1H, D2O exchangeable, imidazole NH of the major tautomeric form), 11.66 (br s, 1H, D2O exchangeable, imidazole NH of the minor tautomeric form). 13C-NMR (151 MHz, DMSO-d6) δ 22.58 (CH3), 43.97 (CH), 116.37 (C-6), 116.61 (C-7 of the major tautomeric form), 121.62 (C-7 of the minor tautomeric form), 126.86 (C-7a), 137.41 (C-5), 156.22 (C-3a), 158.24 (C-2). HR-MS (ESI) m/z: Calcd for C9H11Cl2N4: [M + H]+ = 245.0355, found 245.0352.
N-Benzyl-5,6-dichloro-3H-imidazo[4,5-b]pyridin-2-amine (7c): This compound was prepared by a procedure analogous to that described for 7a, upon reaction of diamine 6 (500 mg, 2.81 mmol) with benzyl isothiocyanate (0.80 mL, 6.03 mmol). Purification was effected by column chromatography, using a mixture of DCM/EtOAc (90/10 to 50/50, v/v) and then EtOAc/MeOH (99/1 to 90/10, v/v) as eluents. Overall yield 51%. White solid, mp: 277–278 °C (MeOH). 1H-NMR (600 MHz, DMSO-d6) δ 4.55 (d, 2H, CH2, J = 6.0 Hz), 7.24 (t, 1H, phenyl H-4′’, J = 7.2 Hz), 7.32 (t, 2H, phenyl H-3′’, H-5′’, J = 7.6 Hz), 7.36 (d, 2H, phenyl H-2′’, H-6′’, J = 7.3 Hz), 7.60 (s, 1H, H-7), 7.94 (br s, 1H, D2O exchangeable, NH), 11.38 (br s, 1H, D2O exchangeable, imidazole NH). 13C-NMR (151 MHz, DMSO-d6) δ 45.36 (CH2), 116.89 (C-6), 117.05 (C-7), 126.97 (phenyl C-4′’), 127.13 (C-7a), 127.25 (phenyl C-3′’, C-5′’), 128.33 (phenyl C-2′’, C-6′’), 137.50 (C-5), 139.34 (phenyl C-1′’), 155.88 (C-3a), 158.81 (C-2). HR-MS (ESI) m/z: Calcd for C13H11Cl2N4: [M + H]+ = 293.0355, found 293.0358.
5,6-Dichloro-3H-imidazo[4,5-b]pyridin-2-amine (7d): To a suspension of the diamine 6 (570 mg, 3.20 mmol) in MeOH (12 mL) and H2O (12 mL) was added BrCN (1.60 mL, 5M solution in acetonitrile, 8 mmol) and the reaction mixture was stirred at room temperature for 48 h. MeOH was concentrated under vacuo and the pH of the resulting aqueous phase was adjusted to 8 with a saturated NaHCO3 solution. The aqueous layer (150 mL) was extracted with EtOAc (5 × 200 mL) and the combined organic layers were dried (anhydrous Na2SO4) and concentrated to dryness. The residue thus obtained was triturated with diethyl ether to afford 510mg of 7d as an amorphous solid (79%). 1H-NMR (DMSO-d6, 600MHz) δ7.05 (br s, 2H, D2O exchangeable, NH2), 7.58 (s, 1H, H-7). 13C-NMR (151 MHz, DMSO-d6) δ 117.54 (C-6), 118.24 (C-7), 129.57 (C-7a), 136.73 (C-5), 153.60 (C-3a), 158.85 (C-2). HR-MS (ESI) m/z: Calcd for C6H5Cl2N4: [M + H]+ = 202.9886, found 202.9887.
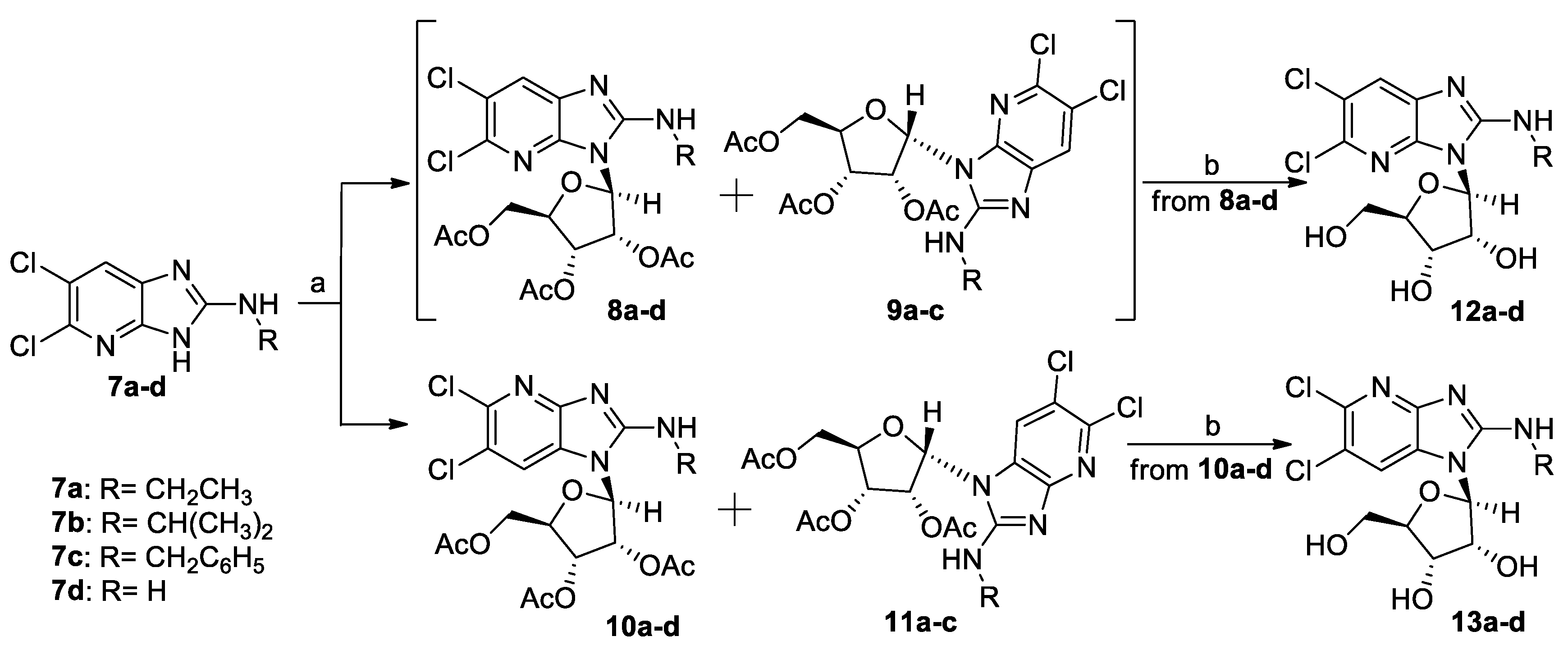
5,6-Dichloro-N-ethyl-3-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-3H-imidazo[4,5-b]pyridin-2-amine (8a), 5,6-dichloro-N-ethyl-3-(2′,3′,5′-tri-O-acetyl-α-d-ribofuranosyl)-3H-imidazo[4,5-b]pyridin-2-amine (9a), 5,6-dichloro-N-ethyl-1-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (10a) and 5,6-dichloro-N-ethyl-1-(2′,3′,5′-tri-O-acetyl-α-d-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (11a): To a suspension of the imidazopyridine 7a (250 mg, 1.08 mmol) in dry CH3CN (10 mL) was added N,O-bis-(trimethylsilyl)acetamide (291 mg, 1.43 mmol) and the reaction mixture was refluxed for 2 h. The suspension was then cooled to room temperature and 1,2,3,5-tetra-O-acetyl-β-d-ribofuranose (413 mg, 1.30 mmol) was added, followed by the dropwise addition of trimethylsilyltriflate (0.3 mL, 1.54 mmol) at 0 °C. The mixture was refluxed for 3 h, the solvent was vacuum evaporated, the residue was dissolved in EtOAc (100 mL) and washed with a saturated NaHCO3 solution (100 mL). The aqueous phase was extracted once more with EtOAc (100 mL) and the combined organic layers were washed with brine (200 mL), dried (Na2SO4) and evaporated to dryness. The resulting oil was purified by column chromatography, using a mixture of CHCl3/MeOH (99/1 to 97/3, v/v) as the eluent, to afford 8a, as a mixture with its corresponding α-anomer 9a (280 mg, total yield 53% for two anomers, 8a:9a (3-β:α) ratio 12:1, as estimated by 1H-NMR), 10a (115 mg, yield 22%) and 11a (27 mg, yield 5%).
Data for 10a: Oil, [α]D +30.55 (c=0.475, CHCl3). 1H-NMR (600 MHz, CDCl3) δ 1.32 (t, 3H, CH3, J = 7.1 Hz), 2.04 (s, 3H, CH3CO), 2.17 (s, 3H, CH3CO), 2.20 (s, 3H, CH3CO), 3.59 (m, 1H, CH2), 3.67 (m, 1H, CH2), 4.34 (dd, 1H, H-5′, J5′,4′ = 2.2 Hz, J5′,5′ = 12.7 Hz), 4.41 (m, 1H, H-4′), 4.65 (dd, 1H, H-5′, J5′,4′ = 3.8 Hz, J5′,5′ = 12.7 Hz), 5.31 (dd, 1H, H-3′, J3′,4′ = 3.6 Hz, J3′,2′ = 6.2 Hz), 5.35 (m, 1H, H-2′), 5.98 (d, 1H, H-1′, J1′,2′= 6.5 Hz), 6.13 (br s, 1H, D2O exchangeable, NH), 7.52 (s, 1H, H-7). 13C-NMR (151 MHz, CDCl3) δ 15.03 (CH3), 20.42 (CH3CO), 20.69 (CH3CO), 20.93 (CH3CO), 38.92 (CH2), 62.85 (C-5′), 69.67 (C-3′), 71.61 (C-2′), 81.18 (C-4′), 86.35 (C-1′), 117.71 (C-7), 120.20 (C-6), 125.61 (C-7a), 141.57 (C-5), 153.55 (C-3α), 156.43 (C-2), 169.49 (CO), 169.78 (CO), 170.26 (CO). HR-MS (ESI) m/z: Calcd for C19H22Cl2N4O7Na: [M + Na]+ = 511.0758, found 511.0762.
Data for 11a: Oil, [α]D +61.23 (c=0.684, CHCl3). 1H-NMR (600 MHz, CDCl3) δ 1.28 (t, 3H, CH3, J = 7.2 Hz), 1.82 (s, 3H, CH3CO), 2.11 (s, 3H, CH3CO), 2.15 (s, 3H, CH3CO), 3.57 (m, 2H, CH2), 4.29 (dd, 1H, H-5′, J5′,4′ = 4.9 Hz, J5′,5′ = 12.3 Hz), 4.33 (dd, 1H, H-5′, J5′,4′ = 3.5 Hz, J5′,5′ = 12.3 Hz), 4.66 (m, 1H, H-4′), 5.27 (t, 1H, D2O exchangeable, NH, J = 5.3 Hz), 5.45 (t, 1H, H-3′, J = 5.3 Hz), 5.73 (t, 1H, H-2′, J = 5.1 Hz), 6.19 (d, 1H, H-1′, J1′,2′ = 4.9 Hz), 7.55 (s, 1H, H-7). 13C-NMR (151 MHz, CDCl3) δ 14.96 (CH3), 20.28 (CH3CO), 20.55 (CH3CO), 20.94 (CH3CO), 38.71 (CH2), 63.31 (C-5′), 71.08 (C-3′), 71.55 (C-2′), 78.96 (C-4′), 85.40 (C-1′), 119.37 (C-7), 119.69 (C-6), 125.67 (C-7a), 141.09 (C-5), 154.24 (C-3a), 157.01 (C-2), 169.26 (CO), 169.55 (CO), 170.51 (CO). HR-MS (ESI) m/z: Calcd for C19H22Cl2N4O7Na: [M + Na]+ = 511.0758, found 511.0764.
5,6-Dichloro-N-isopropyl-3-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-3H-imidazo[4,5-b]pyridin-2-amine (8b), 5,6-dichloro-N-isopropyl-3-(2′,3′,5′-tri-O-acetyl-α-d-ribofuranosyl)-3H-imidazo[4,5-b]pyridin-2-amine (9b), 5,6-dichloro-N-isopropyl-1-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (10b) and 5,6-dichloro-N-isopropyl-1-(2′,3′,5′-tri-O-acetyl-α-D-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (11b): These derivatives were prepared by a procedure analogous to that described for 8a,9a,10a,11a, starting from imidazopyridine 7b (280 mg, 1.14 mmol). Purification was effected by column chromatography, using a mixture of CHCl3/MeOH (99.5/0.5 to 98/2, v/v) as the eluent, to afford 8b, as a mixture with its corresponding α-anomer 9b (350 mg, 61% total yield for two anomers, 8b:9b (3-β:α) ratio 24:1, as estimated by 1H-NMR), 10b (140 mg, 24% yield) and 11b (50 mg, 9% yield).
Data for 10b: Beige solid, mp: 165-166 °C (EtOAc/n-pentane). [α]D +37.77 (c=0.495, CHCl3). 1H NMR (600 MHz, CDCl3) δ 1.30 (d, 3H, CH3, J = 6.5 Hz), 1.31 (d, 3H, CH3, J = 6.5 Hz), 2.04 (s, 3H, CH3CO), 2.15 (s, 3H, CH3CO), 2.19 (s, 3H, CH3CO), 4.27 (m, 1H, CH), 4.32 (dd, 1H, H-5′, J5′,4′= 2.2 Hz, J5′,5′ = 12.7 Hz), 4.39 (m, 1H, H-4′), 4.62 (dd, 1H, H-5′, J5′,4′ = 4.0 Hz, J5′,5′ = 12.7 Hz), 5.30 (dd, 1H, H-3′, J3′,4′ = 3.6 Hz, J3′,2′ = 6.1 Hz), 5.33 (m, 1H, H-2′), 5.47 (br s, 1H, D2O exchangeable, NH), 5.86 (d, 1H, H-1′, J1′,2′ = 6.7 Hz), 7.53 (s, 1H, H-7). 13C-NMR (151 MHz, CDCl3) δ 20.41 (CH3CO), 20.67 (CH3CO), 21.04 (CH3CO), 22.76 (CH3), 23.03 (CH3), 46.04 (CH), 62.80 (C-5′), 69.59 (C-3′), 71.87 (C-2′), 81.06 (C-4′), 86.26 (C-1′), 117.74 (C-7), 119.70 (C-6), 125.49 (C-7α), 141.38 (C-5), 154.37 (C-3α), 156.10 (C-2), 169.44 (CO), 169.72 (CO), 170.29 (CO). HR-MS (ESI) m/z: Calcd for C20H24Cl2N4O7Na: [M + Na]+ = 525.0914, found 525.0917.
Data for 11b: Oil, [α]D +57.75 (c = 0.561, CHCl3). 1H NMR (400 MHz, CDCl3) δ 1.25 (d, 3H, CH3, J = 6.5 Hz), 1.26 (d, 3H, CH3, J = 6.5 Hz), 1.81 (s, 3H, CH3CO), 2.09 (s, 3H, CH3CO), 2.14 (s, 3H, CH3CO), 4.21 (m, 1H, CH), 4.26 (dd, 1H, H-5′, J5′.4′ = 5.2 Hz, J5′,5′ = 12.5 Hz), 4.32 (dd, 1H, H-5′, J5′,4′ = 3.3 Hz, J5′,5′ = 12.3 Hz), 4.65 (m, 1H, H-4′), 5.16 (d, 1H, D2O exchangeable, NH, J = 7.0 Hz), 5.42 (t, 1H, H-3′, J = 5.4 Hz), 5.74 (t, 1H, H-2′, J = 5.0 Hz), 6.23 (d, 1H, H-1′, J1′,2′ = 4.7 Hz), 7.55 (s, 1H, H-7). 13C-NMR (151 MHz, CDCl3) δ 20.26 (CH3CO), 20.53 (CH3CO), 20.88 (CH3CO), 22.67 (CH3), 23.00 (CH3), 45.87 (CH), 63.29 (C-5′), 70.98 (C-3′), 71.48 (C-2′), 78.84 (C-4′), 85.35 (C-1′), 119.32 (C-7), 119.43 (C-6), 125.58 (C-7a), 140.92 (C-5), 154.52 (C-3a), 156.45 (C-2), 169.20 (CO), 169.55 (CO), 170.49 (CO). HR-MS (ESI) m/z: Calcd for C20H24Cl2N4O7Na: [M + Na]+ = 525.0914, found 525.0917.
N-Benzyl-5,6-dichloro-3-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-3H-imidazo[4,5-b]pyridin-2-amine (8c), N-benzyl-5,6-dichloro-3-(2′,3′,5′-tri-O-acetyl-α-d-ribofuranosyl)-3H-imidazo[4,5-b]pyridin-2-amine (9c), N-benzyl-5,6-dichloro-1-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (10c) and N-benzyl-5,6-dichloro-1-(2′,3′,5′-tri-O-acetyl-α-d-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (11c): These derivatives were prepared by a procedure analogous to that described for 8a,9a,10a,11a, starting from imidazopyridine 7c (350 mg, 1.20mmol). Purification was effected by column chromatography, using a mixture of cyclohexane/EtOAc (70/30 to 20/80, v/v) as the eluent, to afford 8c, as a mixture with its corresponding α-anomer 9c (350 mg, 53% total yield for two anomers, 8c:9c (3-β:α) ratio 12:1, as estimated by 1H-NMR), 10c (100 mg, crude) and 11c (40 mg, 6% yield). Fractions containing 10c were pooled and subjected to column chromatography eluted with CHCl3/MeOH (99.5/0.5, v/v), yielding 80 mg of pure 10c (12% yield).
Data for 10c: Oil, [α]D +46.29 (c = 0.337, CHCl3). 1H NMR (600 MHz, CDCl3) δ 1.91 (s, 3H, CH3CO), 1.99 (s, 3H, CH3CO), 2.15 (s, 3H, CH3CO), 4.21 (dd, 1H, H-5′, J5′,4′ = 2.0 Hz, J5′,5′ = 12.6 Hz), 4.37 (m, 1H, H-4′), 4.57 (dd, 1H, H-5′, J5′,4′ = 3.4 Hz, J5′,5′ = 12.6 Hz), 4.72 (dd, 1H, CH2, JCH2,NH = 5.2 Hz, JCH2,CH2 = 14.8 Hz), 4.82 (dd, 1H, CH2, JCH2,NH = 5.9 Hz, JCH2,CH2 = 14.8 Hz), 5.30 (dd, 1H, H-3′, J3′,4′ = 3.1 Hz, J3′,2′ = 6.1 Hz), 5.37 (m, 1H, H-2′), 5.87 (d, 1H, H-1′, J1′,2′ = 7.6 Hz), 5.88 (br s, 1H, D2O exchangeable, NH), 7.27 (m, 1H, phenyl H-4′’), 7.32 (t, 2H, phenyl H-3′’, H-5′’, J = 7.5 Hz), 7.37 (d, 2H, phenyl H-2′’, H-6′’, J = 7.4 Hz), 7.52 (s, 1H, H-7). 13C-NMR (151 MHz, CDCl3) δ 20.34 (CH3CO), 20.52 (CH3CO), 20.70 (CH3CO), 47.62 (CH2), 62.91 (C-5′), 69.83 (C-3′), 71.64 (C-2′), 81.41 (C-4′), 86.04 (C-1′), 117.40 (C-7), 120.01 (C-6), 126.03 (C-7a), 127.89 (phenyl C-2′’, C-6′’), 128.01 (phenyl C-4′’), 128.96 (phenyl C-3′’, C-5′’), 137.97 (phenyl C-1′’), 141.60 (C-5), 154.26 (C-3a), 156.81 (C-2), 169.27 (CO), 169.71 (CO), 170.16 (CO). HR-MS (ESI) m/z: Calcd for C24H24Cl2N4O7Na: [M + Na]+ = 573.0914, found 573.0919.
Data for 11c: Oil, [α]D +53.17 (c = 0.600, CHCl3). 1H NMR (600 MHz, CDCl3) δ 1.72 (s, 3H, CH3CO), 2.00 (s, 3H, CH3CO), 2.12 (s, 3H, CH3CO), 4.27 (dd, 1H, H-5′, J5′,4′ = 4.8 Hz, J5′,5′ = 12.4 Hz), 4.30 (dd, 1H, H-5′, J5′,4′ = 3.5 Hz, J5′,5′ = 12.3 Hz), 4.62 (m, 1H, H-4′), 4.69 (dd, 1H, CH2, JCH2,NH = 5.0 Hz, JCH2,CH2 = 14.5 Hz), 4.76 (dd, 1H, CH2, JCH2,NH = 5.5 Hz, JCH2,CH2 = 14.5 Hz), 5.44 (t, 1H, H-3′, J = 5.2 Hz), 5.60 (br s, 1H, D2O exchangeable, NH), 5.71 (t, 1H, H-2′, J = 5.2 Hz), 6.19 (d, 1H, H-1′, J1′,2′ = 5.0 Hz), 7.28 (m, 1H, phenyl H-4′’), 7.32 (m, 2H, phenyl H-3′’, H-5′’), 7.40 (m, 2H, phenyl H-2′’, H-6′’), 7.53 (s, 1H, H-7). 13C-NMR (151 MHz, CDCl3) δ 20.08 (CH3CO), 20.40 (CH3CO), 20.94 (CH3CO), 47.73 (CH2), 63.24 (C-5′), 71.03 (C-3′), 71.39 (C-2′), 79.20 (C-4′), 85.46 (C-1′), 118.91 (C-7), 119.87 (C-6), 125.89 (C-7a), 128.02 (phenyl C-4′’), 128.23 (phenyl C-2′’, C-6′’), 128.90 (phenyl C-3′’, C-5′’), 138.03 (phenyl C-1′’), 141.44 (C-5), 154.31 (C-3a), 157.13 (C-2), 169.33 (CO), 169.53 (CO), 170.39 (CO). HR-MS (ESI) m/z: Calcd for C24H24Cl2N4O7Na: [M + Na]+ = 573.0914, found 573.0920.
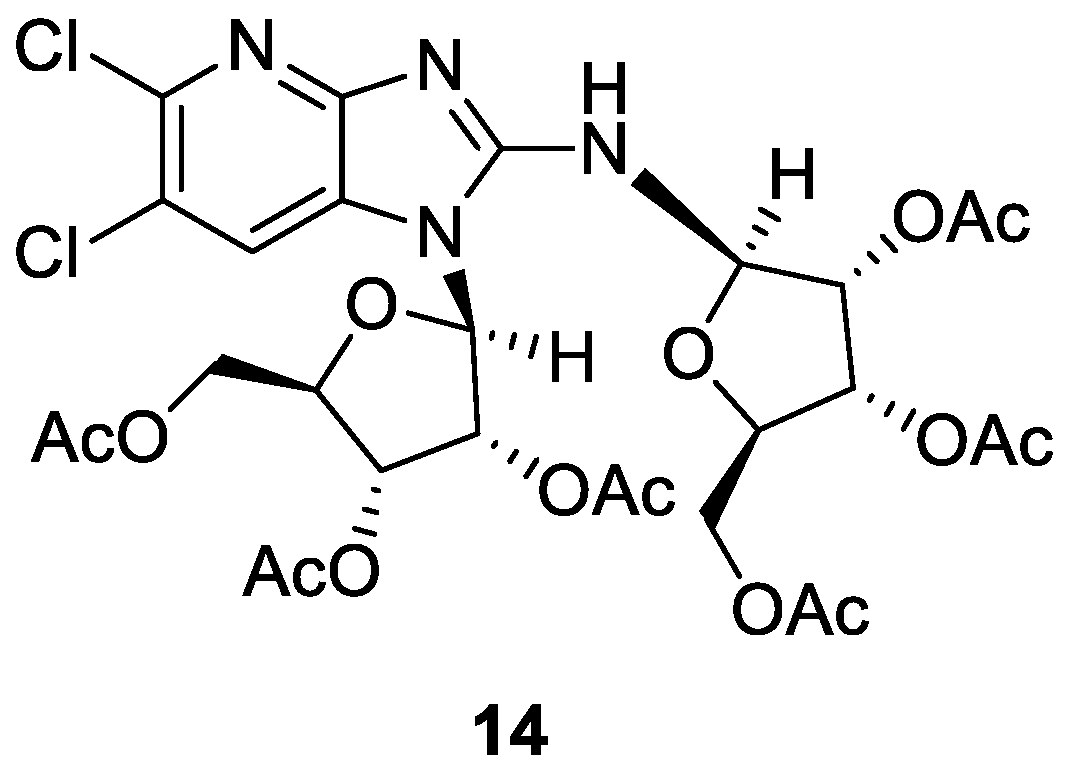
5,6-Dichloro-N,1-bis-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (14), 5,6-dichloro-3-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-3H-imidazo[4,5-b]pyridin-2-amine (8d) and 5,6-dichloro-1-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (10d): These derivatives were prepared by a procedure analogous to that described for 8a and 10a, starting from 7d (245 mg, 1.21 mmol). Purification was effected by column chromatography, using a mixture of CHCl3/MeOH (99.5/0.5 to 95.5/4.5, v/v) as the eluent, to afford 14 (300 mg, 35% yield), 8d (150 mg, 27% yield) and 10d (40 mg, 7% yield).
Data for 14: Oil, [α]D −25.60 (c=0.703, CHCl3). 1H NMR (600 MHz, CDCl3) δ 2.07 (s, 3H, CH3CO), 2.07 (s, 3H, CH3CO), 2.11 (s, 3H, CH3CO), 2.13 (s, 3H, CH3CO), 2.14 (s, 3H, CH3CO), 2.23 (s, 3H, CH3CO), 4.30–4.33 (m, 3H, 2 × H-4′, 1 × H-5′), 4.36 (dd, 1H, H-5′, J5′,4′ = 5.1 Hz, J5′,5′ = 12.1 Hz), 4.42 (dd, 1H, H-5′, J5′,4′ = 3.4 Hz, J5′,5′ = 12.1 Hz), 4.50 (m, 1H, H-5′), 5.44 (dd, 1H, H-3′, J3′,4′ = 3.6 Hz, J3′,2′ = 6.2 Hz), 5.56 (br s, 1H, H-2′), 5.71 (br s, 1H, H-3′), 5.97 (br s, 1H, H-1′), 6.03 (br s, 1H, H-2′), 6.07 (br s, 1H, H-1′), 7.39 (s,1H, H-7). 13C-NMR (151 MHz, CDCl3) δ 20.50 (CH3CO), 20.60 (CH3CO), 20.68 (CH3CO), 20.69 (CH3CO), 20.90 (CH3CO), 21.06 (CH3CO), 63.35 (2xC-5′), 70.04, 70.10, 70.49, 70.77 (2 × C-2′, 2 × C-3′), 79.73 (C-4′), 80.33 (C-4′), 85.06 (C-1′), 85.13 (C-1′), 117.37 (C-7), 122.62, 123.67 (C-6 and C-7a), 139.26, 142.47 (C-3a, C-5), 149.92 (C-2), 169.67 (3 ×CO), 169.76 (CO), 170.37 (CO), 170.65 (CO). HR-MS (ESI) m/z: Calcd for C28H32Cl2N4O14Na: [M + Na]+ = 741.1184, found 741.1185.
Data for 8d: Oil, [α]D +52.57 (c=0.675, CHCl3). 1H NMR (600 MHz, CDCl3) δ 2.03 (s, 3H, CH3CO), 2.11 (s, 3H, CH3CO), 2.16 (s, 3H, CH3CO), 4.35 (dd, 1H, H-5′, J5′,4′ = 2.7 Hz, J5′,5′ = 12.2 Hz), 4.38 (m, 1H, H-4′), 4.59 (dd, 1H, H-5′, J5′,4′ = 4.0 Hz, J5′,5′ = 12.2 Hz), 5.50 (dd, 1H, H-3′, J3′,4′ = 4.3 Hz, J3′,2′ = 6.2 Hz), 5.75 (m, 1H, H-2′), 5.87 (br s, 2H, D2O exchangeable, NH2), 6.23 (d, 1H, H-1′, J1′,2′ = 6.5 Hz), 7.63 (s, 1H, H-7). 13C-NMR (151 MHz, CDCl3) δ 20.48 (CH3CO), 20.70 (CH3CO), 20.83 (CH3CO), 63.24 (C-5′), 70.27 (C-3′), 71.19 (C-2′), 80.73 (C-4′), 85.04 (C-1′), 123.94 (C-6), 125.17 (C-7), 135.02, 138.38 (C-5 and C-7a), 144.67 (C-3a), 155.30 (C-2), 169.85 (CO), 169.86 (CO), 170.24 (CO). HR-MS (ESI) m/z: Calcd for C17H18Cl2N4O7Na: [M + Na]+ = 483.0445, found 483.0449.
Data for 10d: Oil, [α]D +22.70 (c = 0.185, CHCl3). 1H NMR (600 MHz, CDCl3) δ 2.02 (s, 3H, CH3CO), 2.16 (s, 3H, CH3CO), 2.35 (s, 3H, CH3CO), 4.31 (dd, 1H, H-5′, J5′,4′ = 2.1 Hz, J5′,5′ = 12.5 Hz), 4.43 (m, 1H, H-4′), 4.65 (dd, 1H, H-5′, J5′,4′ = 1.9 Hz, J5′,5′ = 12.5 Hz), 5.39 (dd, 1H, H-3′, J3′,4′ = 3.2 Hz, J3′,2′ = 6.2 Hz), 5.47 (m, 1H, H-2′), 5.89 (d, 1H, H-1′, J1′,2′ = 7.5 Hz), 7.47 (br s, 2H, D2O exchangeable, NH2), 7.52 (s, 1H, H-7). 13C-NMR (151 MHz, CDCl3) δ 20.36 (CH3CO), 20.65 (CH3CO), 21.23 (CH3CO), 63.01 (C-5′), 70.07 (C-3′), 71.20 (C-2′), 81.20 (C-4′), 86.11 (C-1′), 117.54 (C-7), 119.80 (C-6), 125.35 (C-7a), 141.36 (C-5), 153.77 (C-3a), 157.58 (C-2), 169.30 (CO), 169.71 (CO), 170.43 (CO). HR-MS (ESI) m/z: Calcd for C17H18Cl2N4O7Na: [M + Na]+ = 483.0445, found 483.0440.
5,6-Dichloro-N-ethyl-3-(β-d-ribofuranosyl)-3H-imidazo[4,5-b]pyridin-2-amine (12a): A mixture of 8a and 9a (80 mg, 0.16 mmol) was treated for 18 h with a saturated methanolic ammonia solution (15 mL). Upon evaporation of the solvent, the residue was purified by column chromatography using a mixture of EtOAc/MeOH: 99/1 (v/v), as the eluent and the desired product was recrystallized from methanol to provide pure 12a (35 mg, 59% yield). White solid, mp: 191–192 °C (MeOH). [α]D +31.88 (c = 0.367, MeOH). 1H NMR (600 MHz, DMSO-d6) δ 1.19 (t, 3H, CH3, J = 7.2 Hz), 3.41 (m, 2H, CH2, overlapping with water of DMSO-d6), 3.66 (m, 2H, H-5′), 3.99 (m, 1H, H-4′), 4.12 (dd, 1H, H-3′, J3′,4′ = 1.5 Hz, J3′,2′ = 5.2 Hz), 4.59 (dd, 1H, H-2′, J2′,3′ = 5.2 Hz, J2′,1′ = 7.3 Hz), 5.05–5.55 (br s, 2H, D2O exchangeable, 2 × OH), 5.75 (br s, 1H, D2O exchangeable, OH), 5.96 (d, 1H, H-1′, J1′,2′ = 7.5 Hz), 7.64 (br s, 1H, D2O exchangeable, NH), 7.78 (s, 1H, H-7). 13C-NMR (151 MHz, DMSO-d6) δ 14.50 (CH3), 37.08 (CH2), 61.42 (C-5′), 70.29 (C-2′), 71.04 (C-3′), 85.74 (C-4′), 86.37 (C-1′), 121.03 (C-6), 122.82 (C-7), 134.65, 136.07 (C-5 and C-7a), 145.96 (C-3a), 155.48 (C-2). HR-MS (ESI) m/z: Calcd for C13H17Cl2N4O4: [M + H]+ = 363.0621, found 363.0615.
5,6-Dichloro-N-isopropyl-3-(β-d-ribofuranosyl)-3H-imidazo[4,5-b]pyridin-2-amine (12b): This compound was prepared by a procedure analogous to that described for 12a starting from 8b (90 mg, 0.18mmol, containing also the corresponding α-anomer 9b). Purification was effected by column chromatography using a mixture of DCM/MeOH: 98/2 to 95/5 (v/v) as the eluent, to afford 12b (55 mg, 81% yield). White solid, mp: 127–128 °C (EtOAc/n-pentane). [α]D +30.77 (c = 0.494, MeOH). 1H NMR (600 MHz, DMSO-d6) δ 1.21 (d, 3H, CH3, J = 6.5 Hz), 1.22 (d, 3H, CH3, J = 6.5 Hz), 3.65 (dd, 1H, H-5′, J5′,4′ = 2.8 Hz, J5′,5′ = 11.7 Hz), 3.68 (dd, 1H, H-5′, J5′,4′ = 2.0 Hz, J5′,5′ = 11.7 Hz), 3.99 (m, 1H, H-4′), 4.11–4.18 (m, 2H, H-3′, CH), 4.56 (dd, 1H, H-2′, J2′,3′ = 5.4 Hz, J2′,1′ = 7.6 Hz), 5.02–5.52 (br s, 2H, D2O exchangeable, 2 × OH), 5.72 (br s, 1H, D2O exchangeable, OH), 5.97 (d, 1H, H-1′, J1′,2′ = 7.6 Hz), 7.44 (d, 1H, D2O exchangeable, NH, J = 7.5 Hz), 7.78 (s, 1H, H-7). 13C-NMR (151 MHz, DMSO-d6) δ 22.23 (CH3), 22.37 (CH3), 44.36 (CH), 61.42 (C-5′), 70.25 (C-2′), 71.03 (C-3′), 85.71 (C-4′), 86.26 (C-1′), 121.00 (C-6), 122.72 (C-7), 134.60, 135.99 (C-5 and C-7a), 145.91 (C-3a), 154.84 (C-2). HR-MS (ESI) m/z: Calcd for C14H19Cl2N4O4: [M + H]+ = 377.0778, found 377.0786.
N-Benzyl-5,6-dichloro-3-(β-d-ribofuranosyl)-3H-imidazo[4,5-b]pyridin-2-amine (12c): This compound was prepared by a procedure analogous to that described for 12a starting from 8c (130 mg, 0.23 mmol, containing also the corresponding α-anomer 9c). Purification was effected by column chromatography using a mixture of DCM/MeOH: 98/2 to 90/10 (v/v) to afford an anomeric mixture β/α, in a 12/1 ratio (80 mg). A second column chromatography was performed (DCM/MeOH: 97.5/2.5 to 90/10, v/v) and the fractions pooled (30 mg) were enriched in the desired β-anomer 12c (β/αratio: 17/1). From this mixture, pure 12c (12 mg) was obtained by semi-preparative HPLC, eluted with H2O (+0.2% Acetic Acid)/ACN (70/30 to 60/40, v/v, over a period of 40 min); tR=23.96 min. Oil, [α]D +6.44 (c=0.652, MeOH). 1H NMR (600 MHz, DMSO-d6) δ 3.67 (br s, 2H, H-5′), 4.02 (m, 1H, H-4′), 4.14 (m, 1H, H-3′), 4.58 (dd, 1H, CH2, JCH2,NH = 5.8 Hz, JCH2,CH2 = 15.6 Hz), 4.63 (dd, 1H, CH2, JCH2,NH = 6.4 Hz, JCH2,CH2 = 15.6 Hz), 4.67 (m, 1H, H-2′), 5.22 (d, 1H, D2O exchangeable, OH-3′, J = 4.1 Hz), 5.40 (d, 1H, D2O exchangeable, OH-2′, J = 6.3 Hz), 5.72 (t, 1H, D2O exchangeable, OH-5′, J = 4.3 Hz), 6.01 (d, 1H, H-1′, J1′,2′ = 7.5 Hz), 7.23 (t, 1H, phenyl H-4′’, J = 7.1 Hz), 7.32 (t, 2H, phenyl H-3′’, H-5′’, J = 7.6 Hz), 7.35 (d, 2H, phenyl H-2′’, H-6′’, J = 7.2 Hz), 7.78 (s, 1H, H-7), 8.25 (t, 1H, D2O exchangeable, NH, J = 6.1 Hz). 13C-NMR (151 MHz, DMSO-d6) δ 45.14 (CH2), 61.45 (C-5′), 70.53 (C-2′), 71.05 (C-3′), 85.80 (C-4′), 86.46 (C-1′), 121.08 (C-6), 123.09 (C-7), 126.79 (phenyl C-4′’), 126.99 (phenyl C-2′’, C-6′’), 128.26 (phenyl C-3′’, C-5′’), 134.73, 136.10 (C-5 and C-7a), 139.25 (phenyl C-1′’), 146.01 (C-3a), 155.69 (C-2). HR-MS (ESI) m/z: Calcd for C18H17Cl2N4O4: [M − H]− = 423.0632, found 423.0632.
5,6-Dichloro-3-(β-d-ribofuranosyl)-3H-imidazo[4,5-b]pyridin-2-amine (12d): This compound was prepared by a procedure analogous to that described for 12a, starting from 8d (100 mg, 0.22 mmol). Purification was effected by column chromatography using a mixture of DCM/MeOH: 97/3 to 90/10 (v/v) as the eluent, to result in 12d (70 mg, 96% yield). Beige solid, mp: 142–143 °C (toluene). [α]D +16.50 (c = 0.594, MeOH). 1HNMR (600 MHz, DMSO-d6) δ 3.64 (brs, 2H, H-5′), 3.98 (m, 1H, H-4′), 4.12 (m, 1H, H-3′), 4.62 (m, 1H, H-2′), 5.20 (br s, 1H, D2O exchangeable, OH-3′), 5.35 (brs, 1H, D2O exchangeable, OH-2′), 5.58 (brs, 1H, D2O exchangeable, OH-5′), 5.94 (d, 1H, H-1′, J1′,2′ = 7.4 Hz), 7.39 (brs, 2H, D2O exchangeable, NH2), 7.71 (s, 1H, H-7). 13C-NMR (151 MHz, DMSO-d6) δ 61.44 (C-5′), 70.31 (C-2′), 70.95 (C-3′), 85.73 (C-4′), 86.37 (C-1′), 121.12 (C-6), 122.58 (C-7), 134.39, 136.37 (C-5 and C-7a), 145.71 (C-3a), 156.45 (C-2). HR-MS (ESI) m/z: Calcd for C11H13Cl2N4O4: [M + H]+ = 335.0308, found 335.0317.
5,6-Dichloro-N-ethyl-1-(β-d-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (13a): This compound was prepared by a procedure analogous to that described for 12a starting from 10a (50 mg, 0.10 mmol). Purification was effected by column chromatography using a mixture of DCM/MeOH: 98/2 to 85/15 (v/v) as the eluent to afford 13a (30 mg, 81% yield). White solid, mp: 226-227 °C (MeOH). [α]D +47.23 (c = 0.271, MeOH). 1HNMR (600 MHz, DMSO-d6) δ 1.18 (t, 3H, CH3, J = 7.2 Hz), 3.41 (m, 2H, CH2, overlapping with water of DMSO-d6), 3.66 (dd, 1H, H-5′, J5′,4′ = 2.0 Hz, J5′,5′ = 11.8 Hz), 3.71 (m, 1H, H-5′), 4.00 (m, 1H, H-4′), 4.08 (dd, 1H, H-3′, J3′,4′ = 1.6 Hz, J3′,2′ = 5.5 Hz), 4.28 (m, 1H, H-2′), 5.22–5.37 (brs, 2H, D2O exchangeable, 2 × OH), 5.73 (brs, 1H, D2O exchangeable, OH), 5.77 (d, 1H, H-1′, J1′,2′ = 7.7 Hz), 7.61 (t, 1H, D2O exchangeable, NH, J = 5.3 Hz), 8.04 (s, 1H, H-7). 13C-NMR (151 MHz, DMSO-d6) δ 14.55 (CH3), 37.52 (CH2), 61.09 (C-5′), 70.26 (C-3′), 71.49 (C-2′), 86.00 (C-4′), 87.55 (C-1′), 117.00 (C-6), 118.13 (C-7), 126.68 (C-7a), 138.26 (C-5), 154.74 (C-3a), 157.22 (C-2). HR-MS (ESI) m/z: Calcd for C13H17Cl2N4O4: [M + H]+ = 363.0621, found 363.0630.
5,6-Dichloro-N-isopropyl-1-(β-d-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (13b):This compound was prepared by a procedure analogous to that described for 12a starting from 10b (90 mg, 0.18 mmol). Purification was effected by column chromatography, using a mixture of DCM/MeOH: 97/3 to 88/12 (v/v) as the eluent, to afford 13b (65 mg, 96% yield). White solid, mp: 191–192 °C (EtOAc/n-pentane). [α]D +45.80 (c = 0.559, MeOH). 1H NMR (600 MHz, DMSO-d6) δ 1.22 (d, 6H, 2 × CH3, J = 6.6 Hz), 3.65 (ddd, 1H, H-5′, J5′,4′ = 2.5 Hz, J5′,OH = 4.4 Hz, J5′,5′ = 11.9 Hz), 3.71 (m, 1H, H-5′), 4.00 (m, 1H, H-4′), 4.08 (m, 1H, H-3′), 4.11 (m, 1H, CH), 4.27 (m, 1H, H-2′), 5.27 (d, 1H, D2O exchangeable, OH-3′, J = 3.9 Hz), 5.30 (d, 1H, D2O exchangeable, OH-2′, J = 7.6 Hz), 5.70 (t, 1H, D2O exchangeable, OH-5′, J = 4.4 Hz), 5.78 (d, 1H, H-1′, J1′,2′ = 7.8 Hz), 7.37 (d, 1H, D2O exchangeable, NH, J = 7.7 Hz), 8.02 (s, 1H, H-7). 13C-NMR (151 MHz, DMSO-d6) δ 22.23 (CH3), 22.33 (CH3), 44.87 (CH), 61.11 (C-5′), 70.26 (C-3′), 71.49 (C-2′), 86.02 (C-4′), 87.53 (C-1′), 116.90 (C-6), 117.96 (C-7), 126.73 (C-7a), 138.22 (C-5), 154.76 (C-3a), 156.63 (C-2). HR-MS (ESI) m/z: Calcd for C14H19Cl2N4O4: [M + H]+ = 377.0778, found 377.0785.
N-Benzyl-5,6-dichloro-1-(β-d-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (13c): This compound was prepared by a procedure analogous to that described for 12a starting from 10c (60 mg, 0.11 mmol). Purification was effected by column chromatography, using a mixture of DCM/MeOH: 97.5/2.5 to 85/15 (v/v) as the eluent to afford 13c (40 mg, 90% yield).White solid, mp: 136-137 °C (toluene). [α]D +17.37 (c = 0.570, MeOH). 1H NMR (600 MHz, DMSO-d6) δ3.66 (dd, 1H, H-5′, J5′,4′ = 2.5 Hz, J5′,5′ = 11.9 Hz), 3.72 (dd, 1H, H-5′, J5′,4′ = 1.8 Hz, J5′,5′ = 11.9 Hz), 4.03 (m, 1H, H-4′), 4.09 (dd, 1H, H-3′, J3′,4′ = 1.6 Hz, J3′,2′ = 5.4 Hz), 4.32 (dd, 1H, H-2′, J2′,3′ = 5.5 Hz, J2′,1′ = 7.8 Hz), 4.59 (d, 2H, CH2, JCH2,NH = 6.1 Hz), 5.07–5.57 (br s, 2H, D2O exchangeable, 2 × OH), 5.77 (br s, 1H, D2O exchangeable, OH), 5.83 (d, 1H, H-1′, J1′,2′ = 7.8 Hz), 7.24 (t, 1H, phenyl H-4′’, J = 7.2 Hz), 7.32 (t, 2H, phenyl H-3′’, H-5′’, J = 7.6 Hz), 7.36 (d, 2H, phenyl H-2′’, H-6′’, J = 7.1 Hz), 8.09 (s, 1H, H-7), 8.26 (t, 1H, D2O exchangeable, NH, JNH,CH2 = 6.1 Hz). 13C-NMR (151 MHz, DMSO-d6) δ 45.71 (CH2), 61.11 (C-5′), 70.26 (C-3′), 71.67 (C-2′), 86.10 (C-4′), 87.55 (C-1′), 117.22 (C-6), 118.50 (C-7), 126.64 (C-7a), 126.87 (phenyl C-4′), 127.21 (phenyl C-2′’, C-6′’), 128.26 (phenyl C-3′’, C-5′’), 138.40 (C-5), 139.24 (phenyl C-1′’), 154.43 (C-3a), 157.31 (C-2). HR-MS (ESI) m/z: Calcd for C18H19Cl2N4O4: [M + H]+ = 425.0778, found 425.0784.
5,6-Dichloro-1-(β-d-ribofuranosyl)-1H-imidazo[4,5-b]pyridin-2-amine (13d): This compound was prepared by a procedure analogous to that described for 12a starting from 10d (40 mg, 0.09 mmol). Purification was effected by column chromatography, using a mixture of DCM/MeOH: 97/3 to 88/12 (v/v) as the eluent, to give 13d (25 mg, 86% yield). Beige solid, mp: 194–195 °C (toluene). [α]D +19.44 (c = 0.360, MeOH). 1H NMR (600 MHz, DMSO-d6) δ 3.64 (m, 1H, H-5′), 3.70 (m, 1H, H-5′), 3.99 (m, 1H, H-4′), 4.08 (dd, 1H, H-3′, J3′,4′ = 1.5 Hz, J3′,2′ = 5.5 Hz), 4.29 (m, 1H, H-2′), 5.25 (br s, 1H, D2O exchangeable, OH-3′), 5.30 (br s, 1H, D2O exchangeable, OH-2′), 5.60 (br s, 1H, D2O exchangeable, OH-5′), 5.76 (d, 1H, H-1′, J1′,2′ = 7.7 Hz), 7.45 (br s, 2H, D2O exchangeable, NH2), 8.08 (s, 1H, H-7). 13C-NMR (151 MHz, DMSO-d6) δ 61.01 (C-5′), 70.07 (C-3′), 71.56 (C-2′), 85.96 (C-4′), 87.60 (C-1′), 116.70 (C-6), 118.29 (C-7), 126.13 (C-7a), 138.41 (C-5), 154.85 (C-3a), 158.06 (C-2). HR-MS (ESI) m/z: Calcd for C11H13Cl2N4O4: [M + H]+ = 335.0308, found 335.0318.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



