
BODIPY- and Porphyrin-Based Sensors for Recognition of Amino Acids and Their Derivatives



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
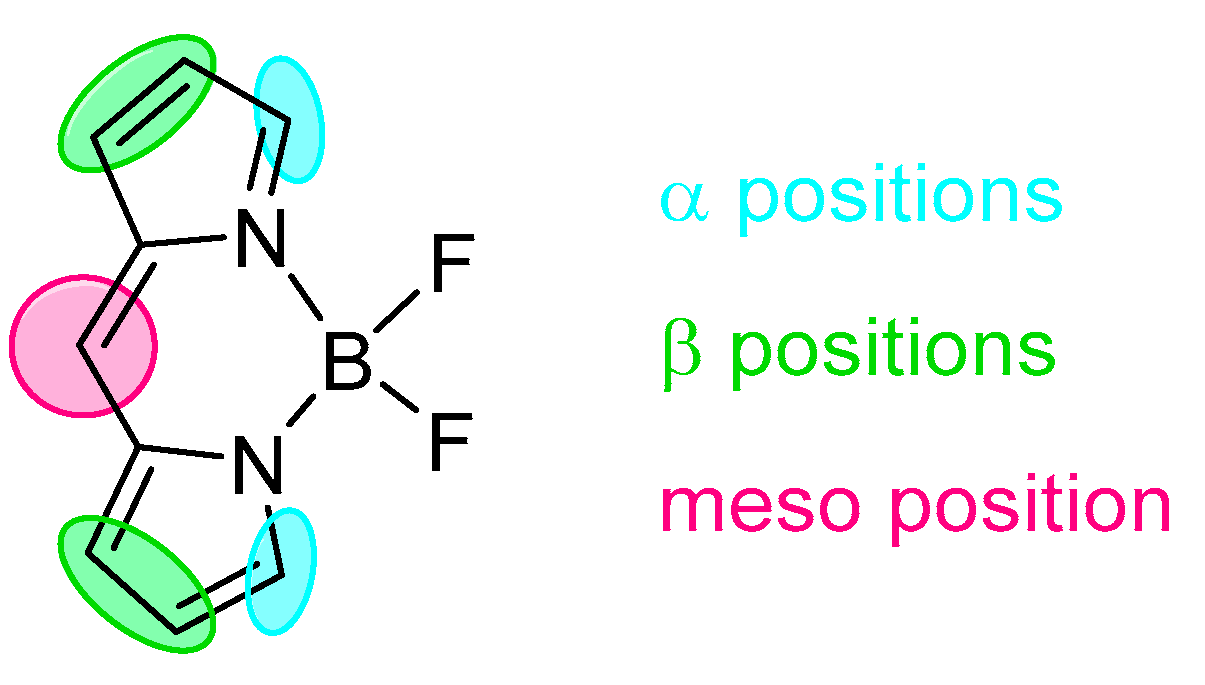{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
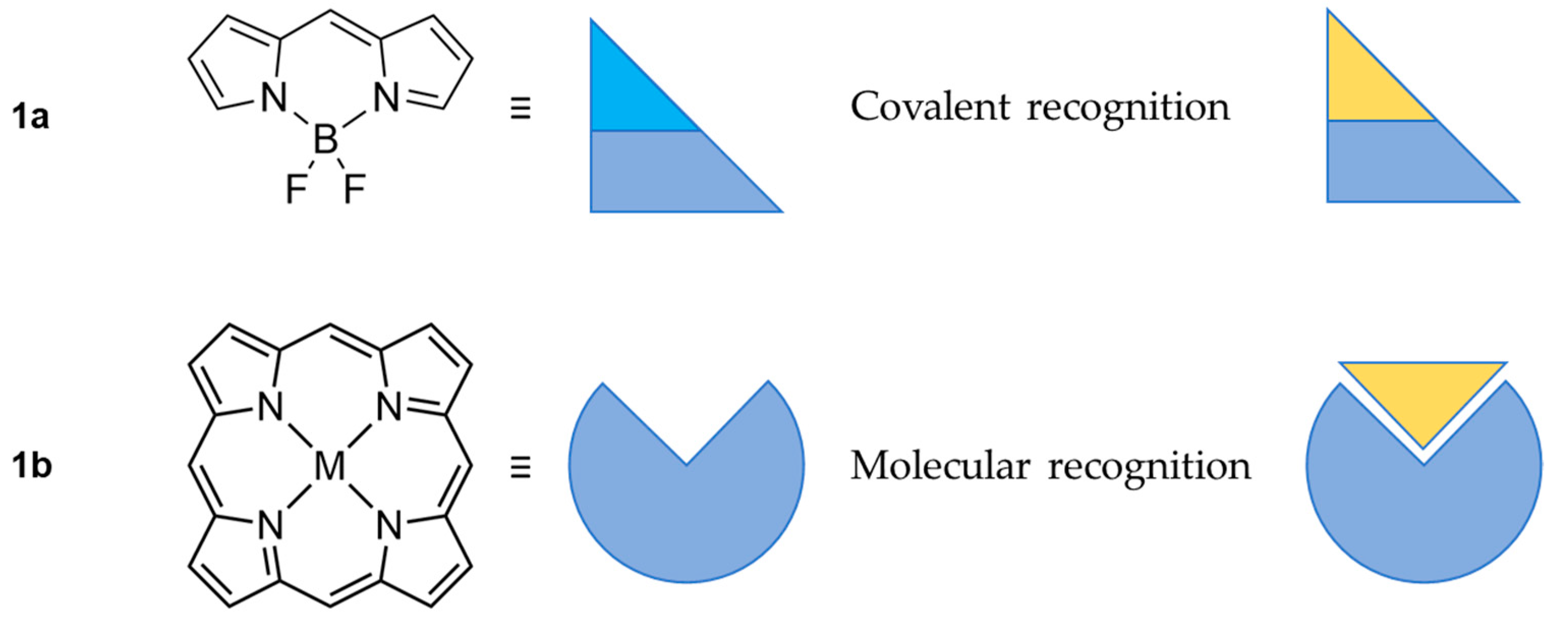
1. Introduction
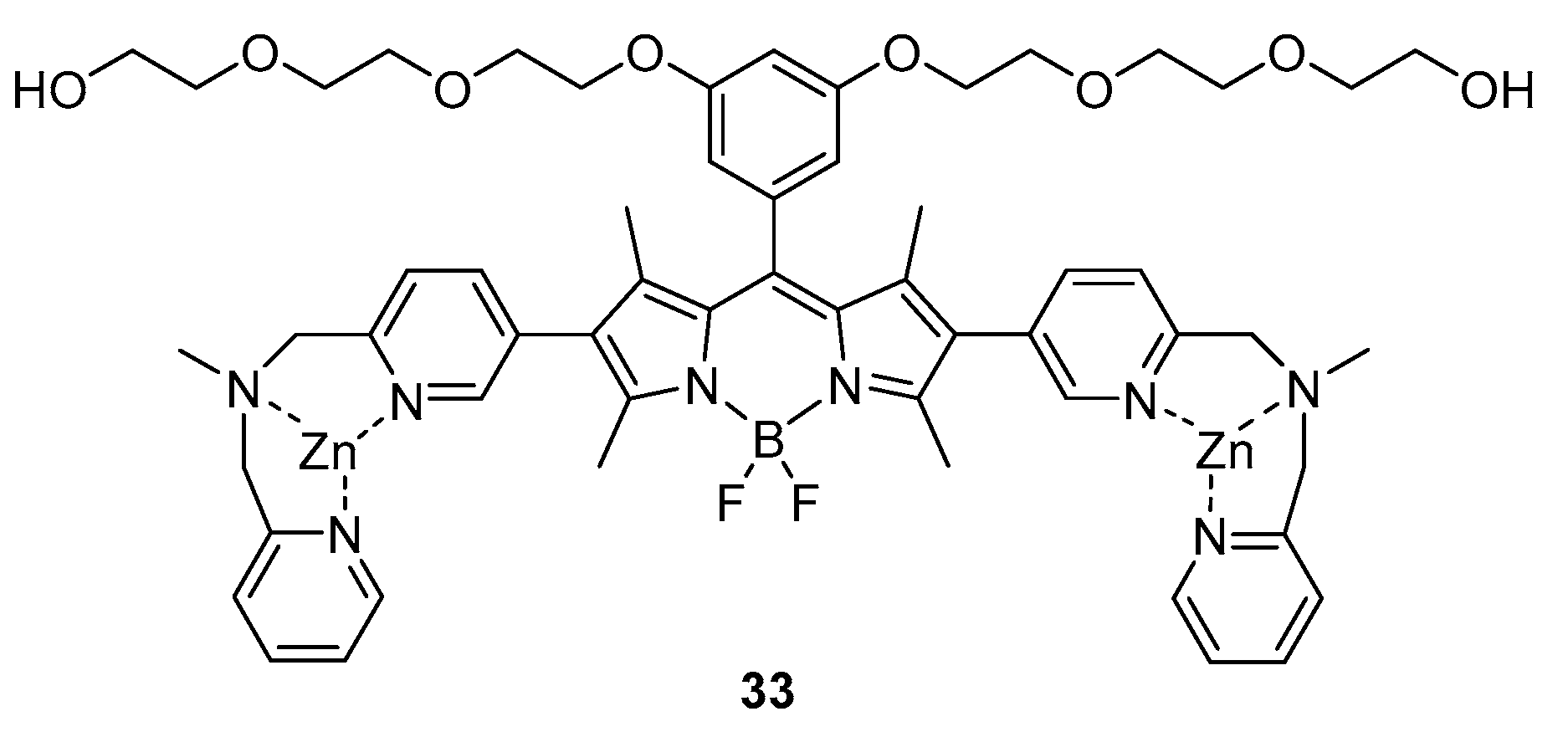
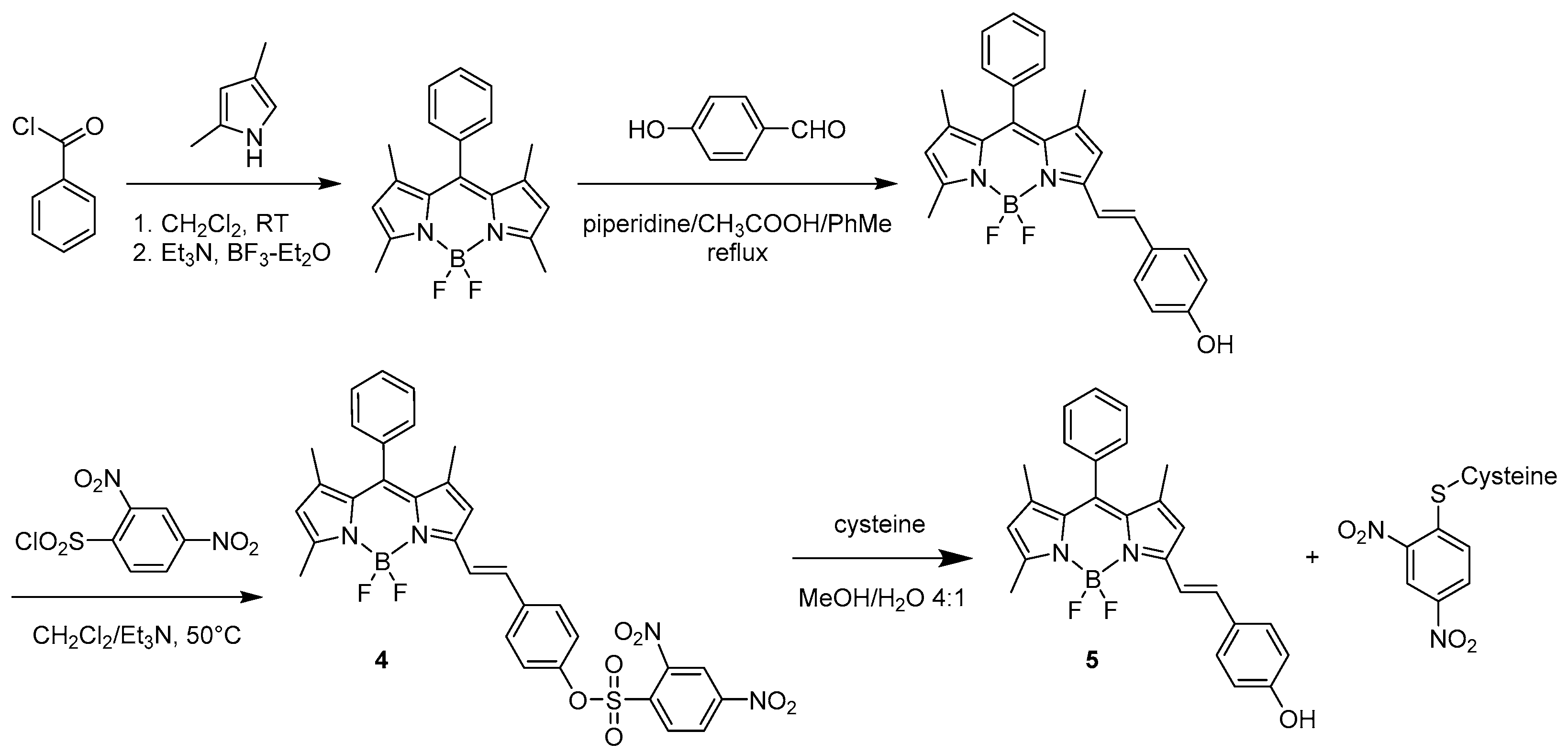
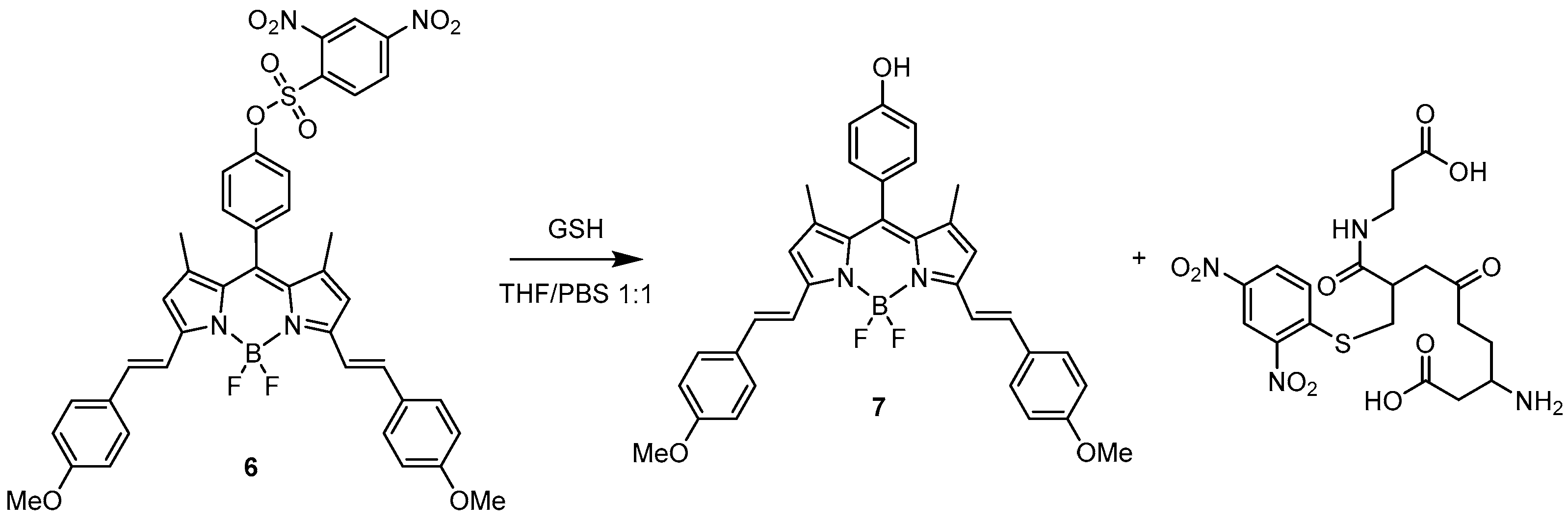
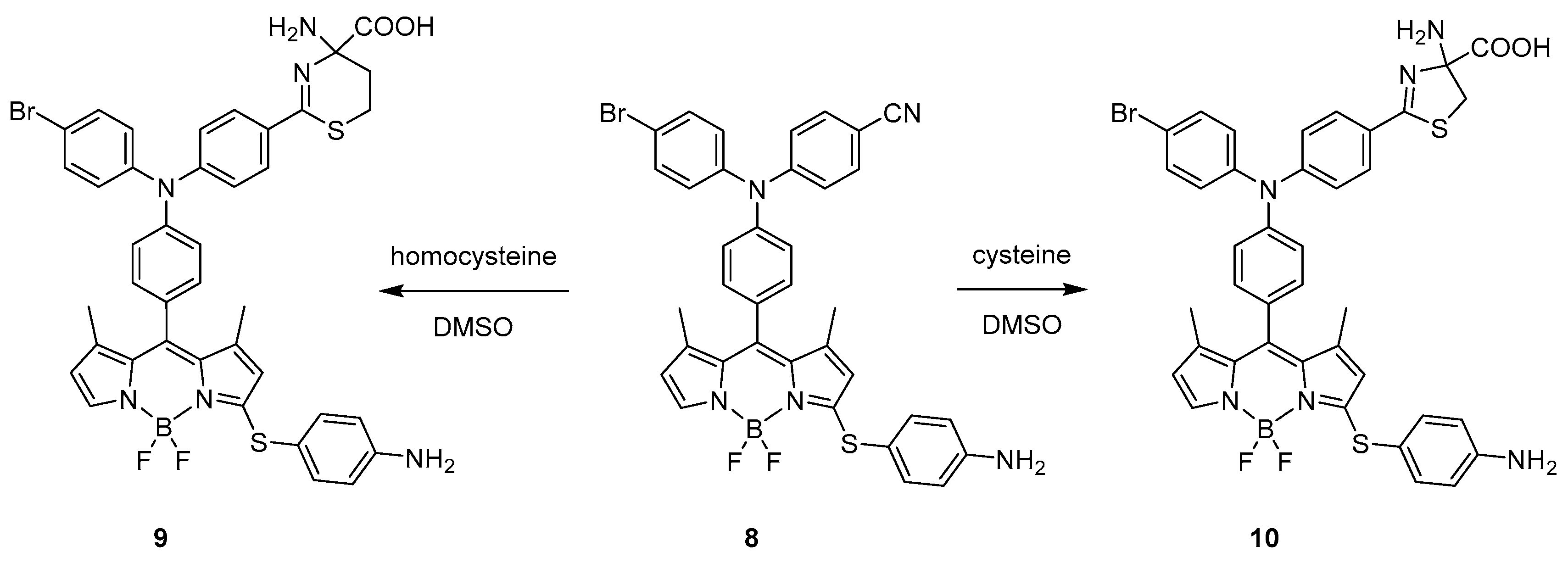
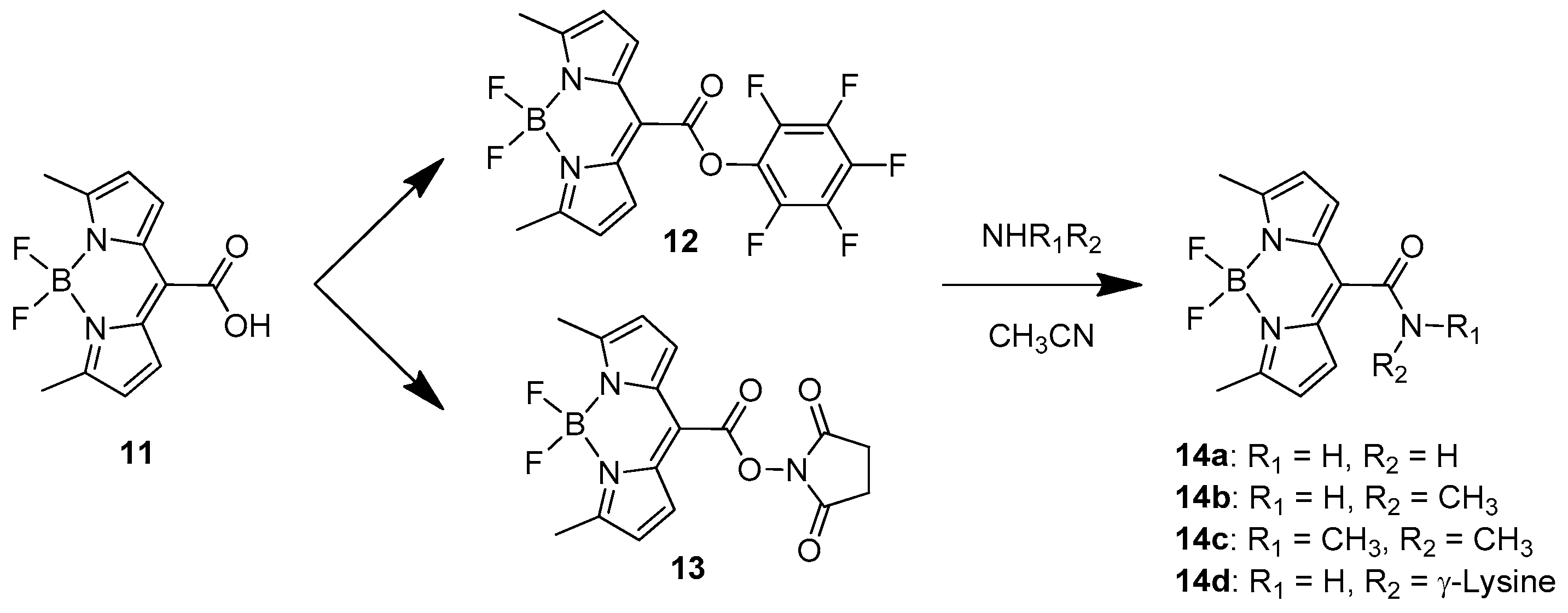
2. BODIPY Probes
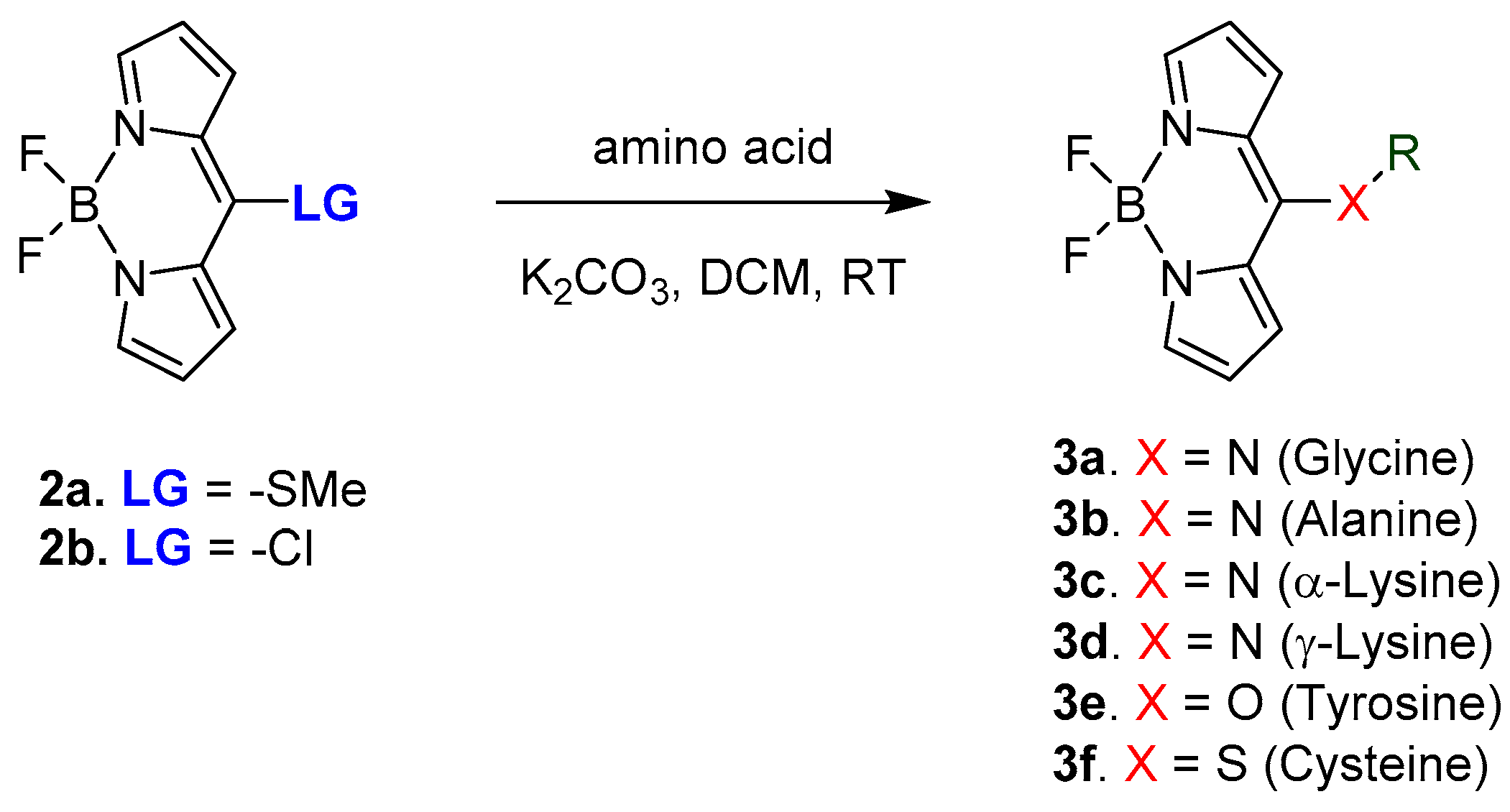
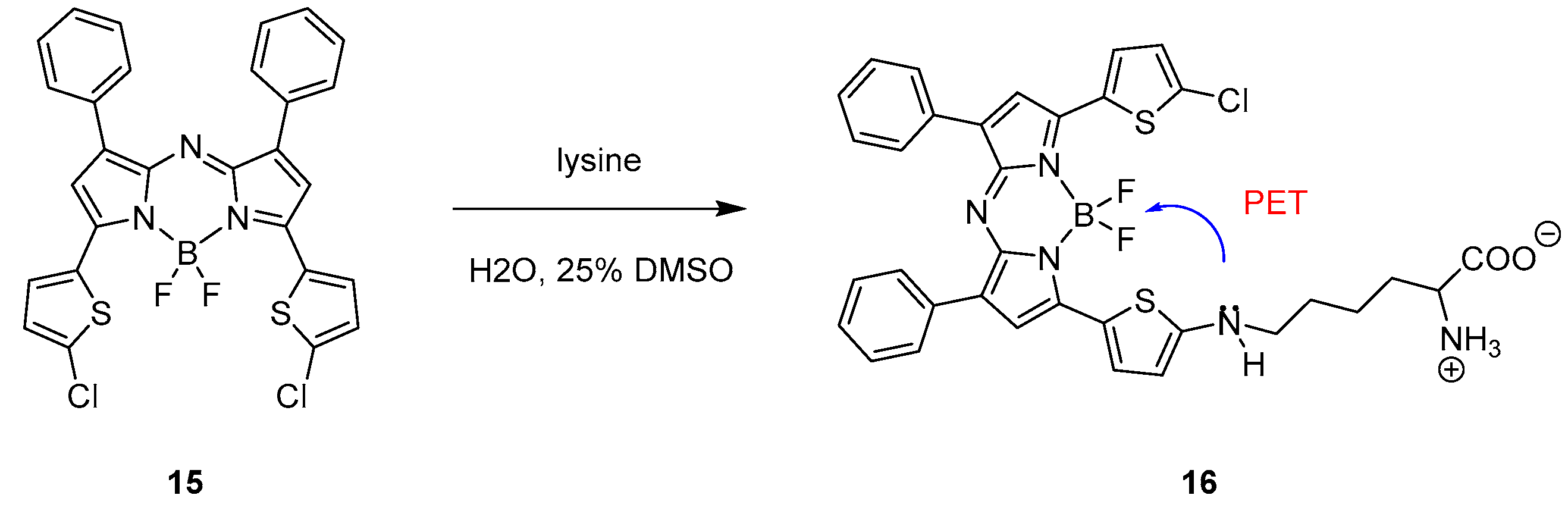
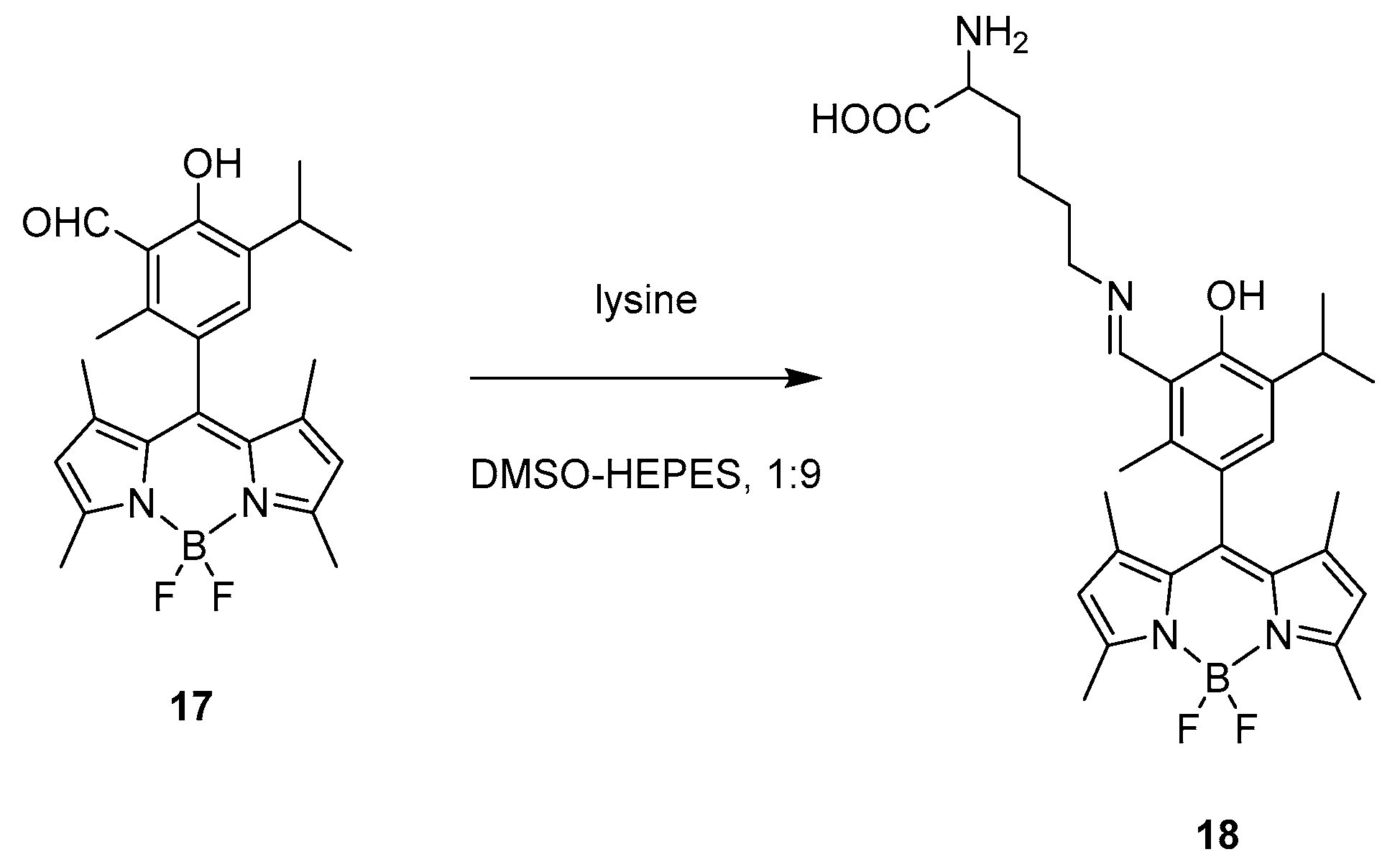
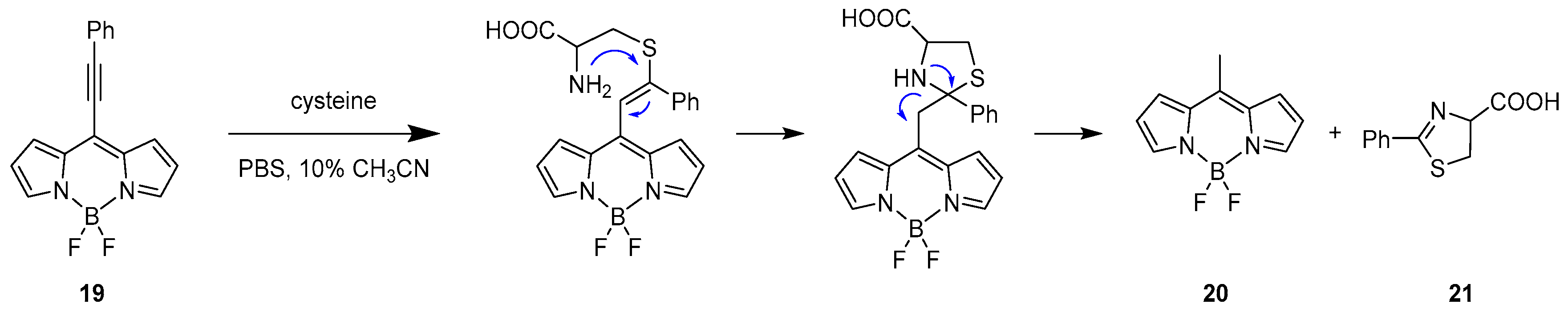
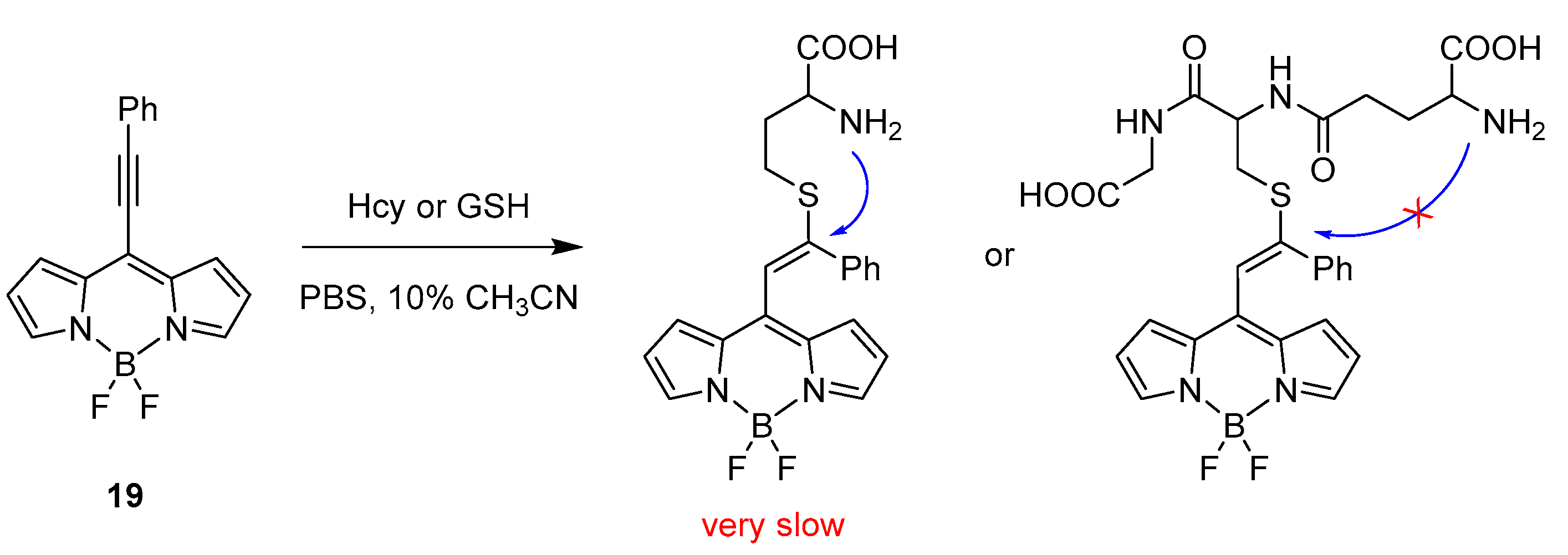
2.1. Covalent Interaction (Recognition)
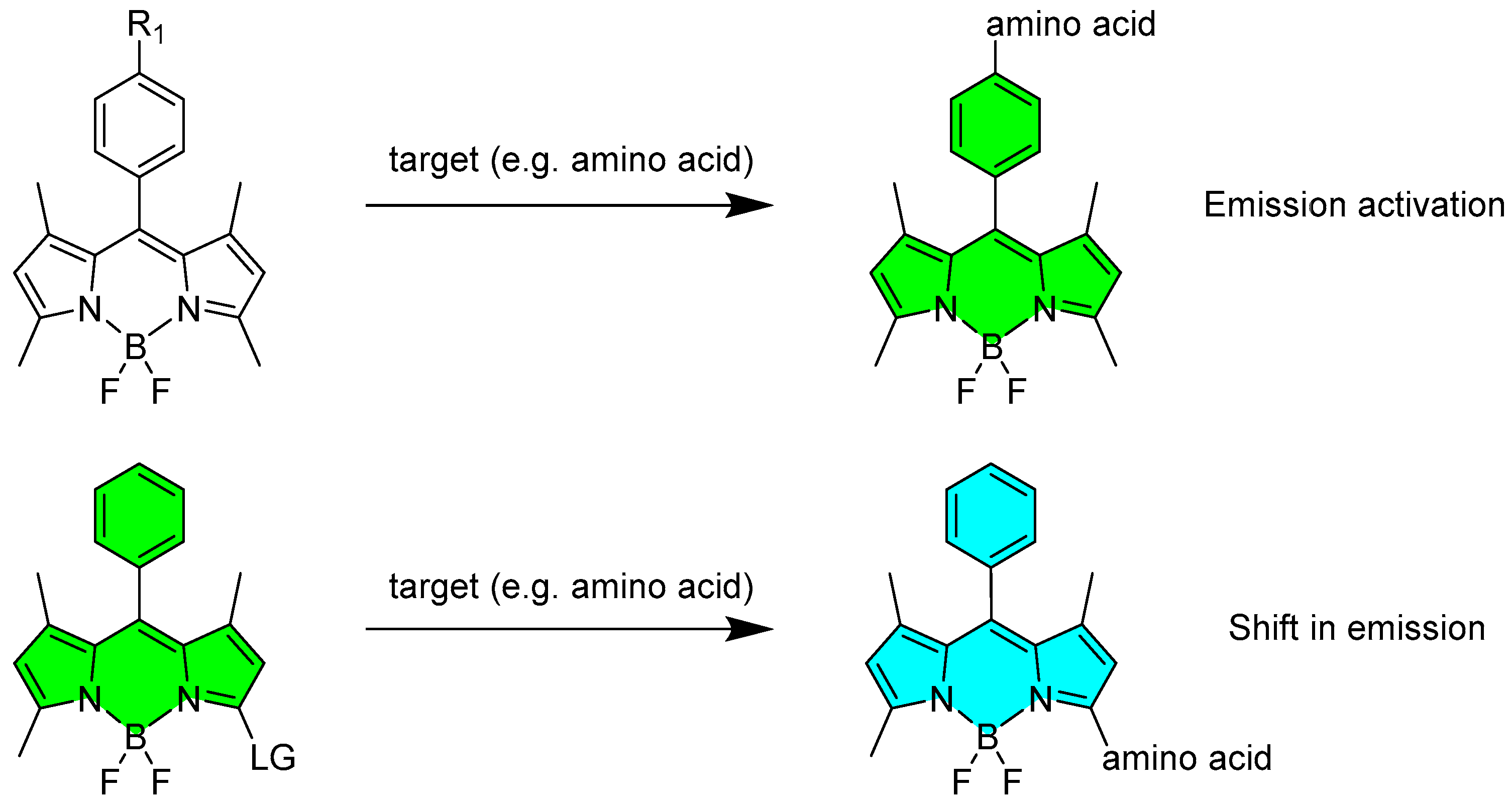
2.1.1. Emission Triggering
2.1.2. Emission Shift
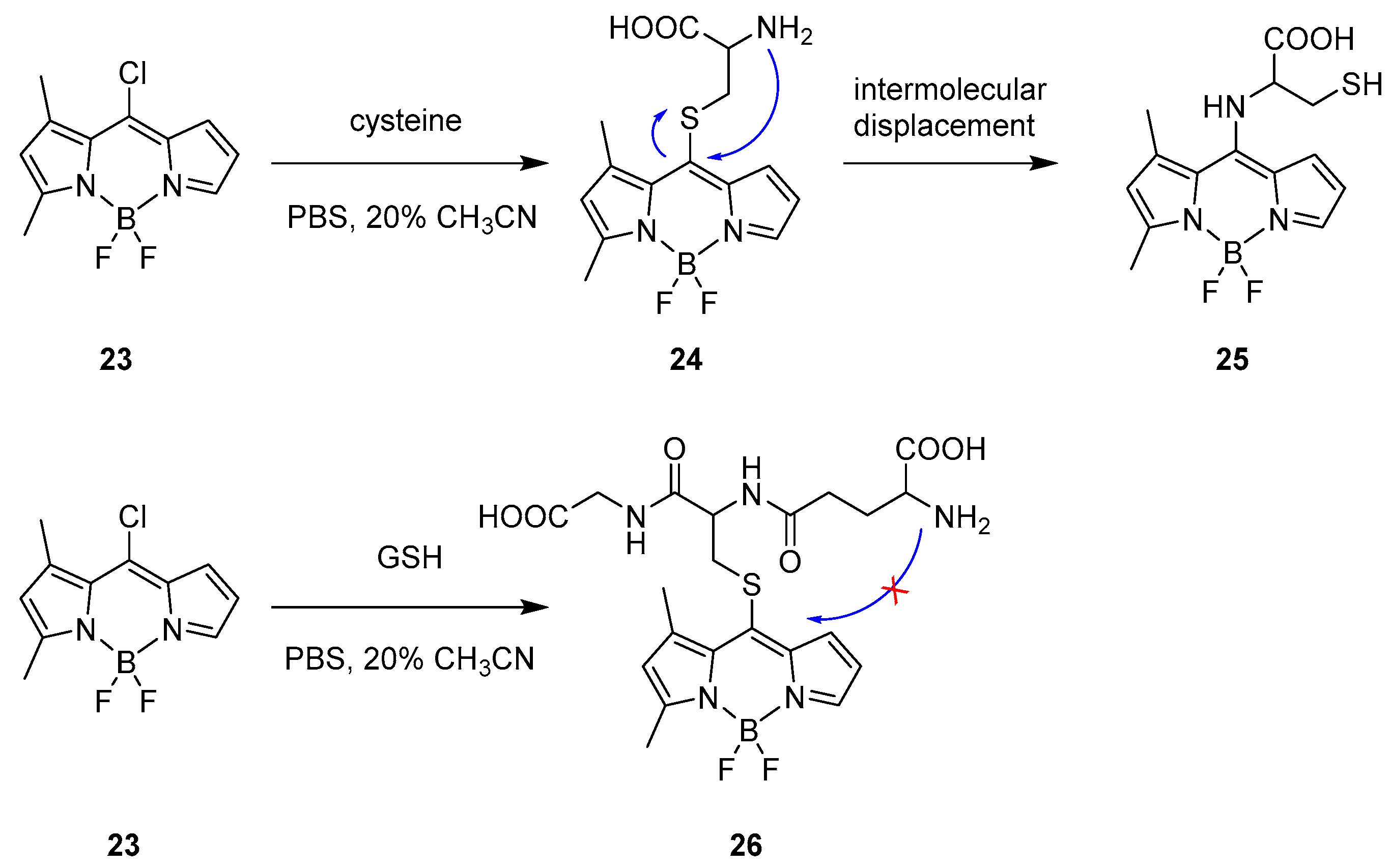
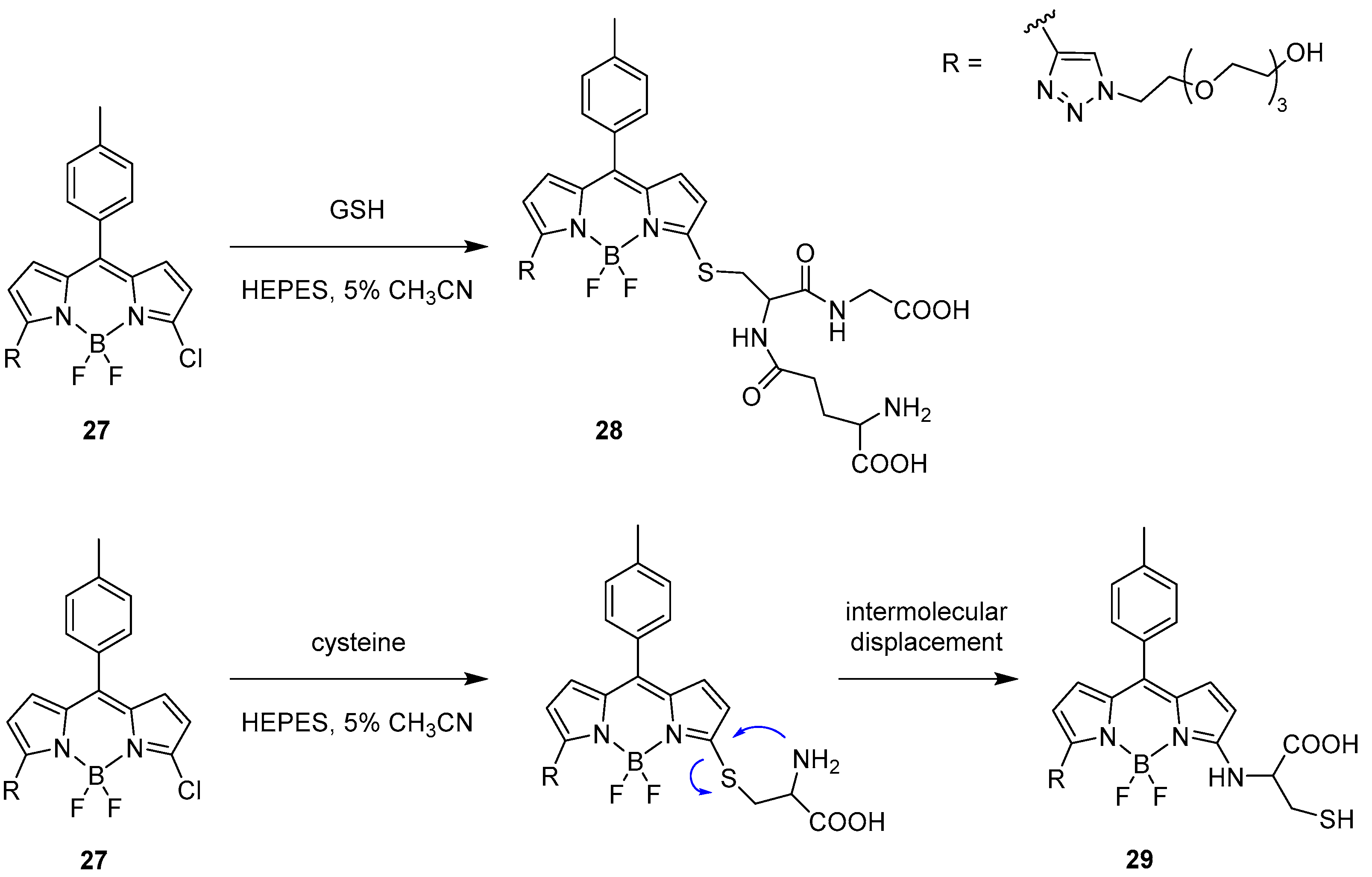
Intermolecular Displacement
2.2. Molecular Recognition (Non-Covalent)
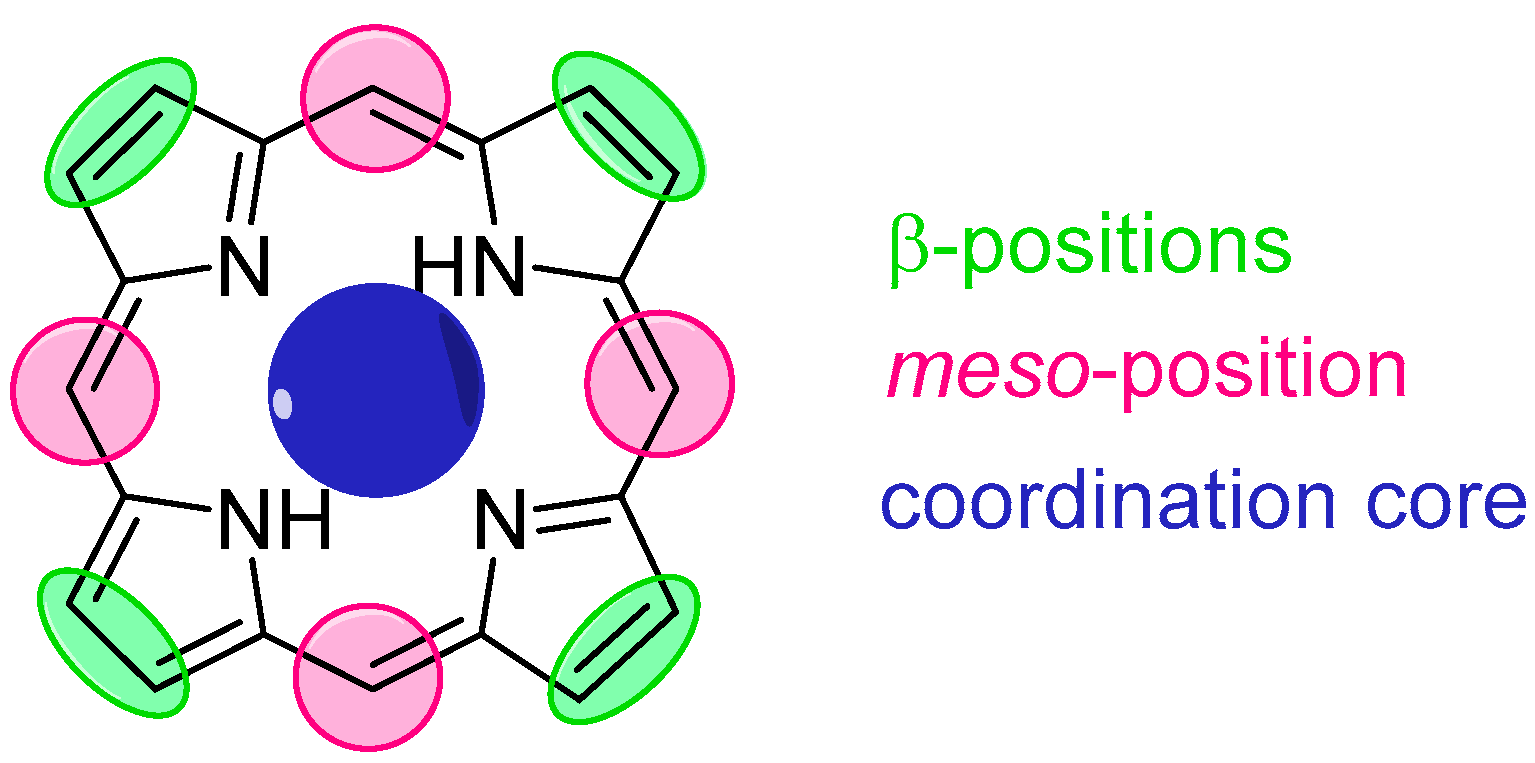
3. Porphyrin Probes
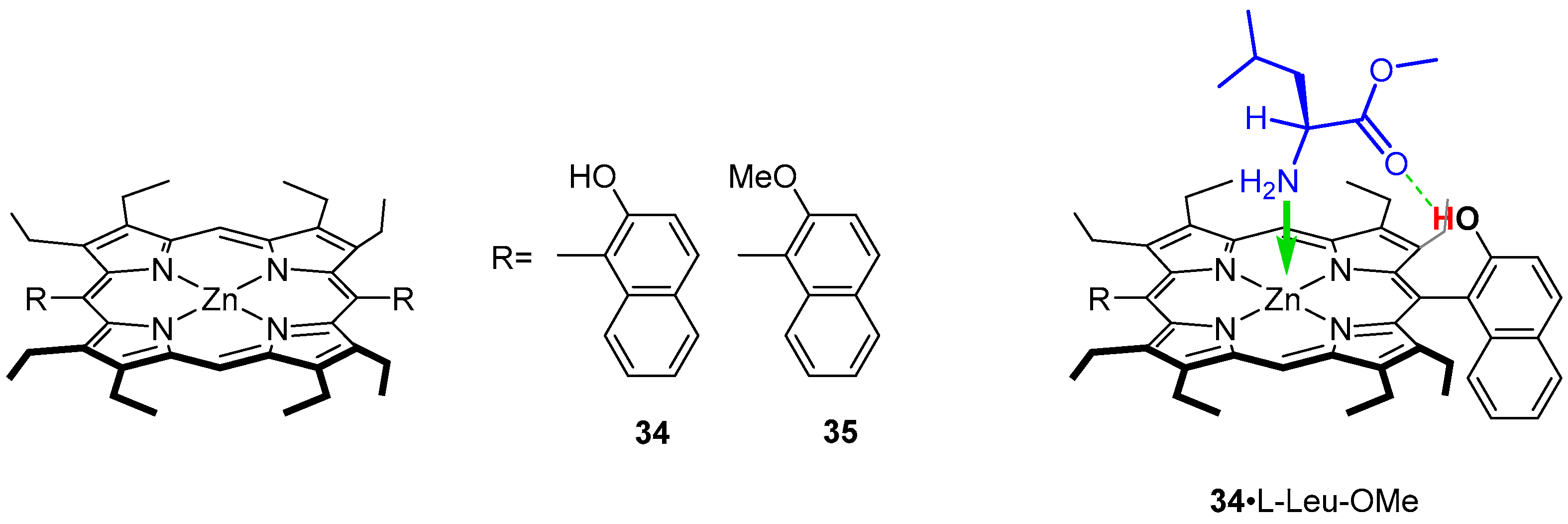
3.1. Monomeric Hosts
3.1.1. Early Reports
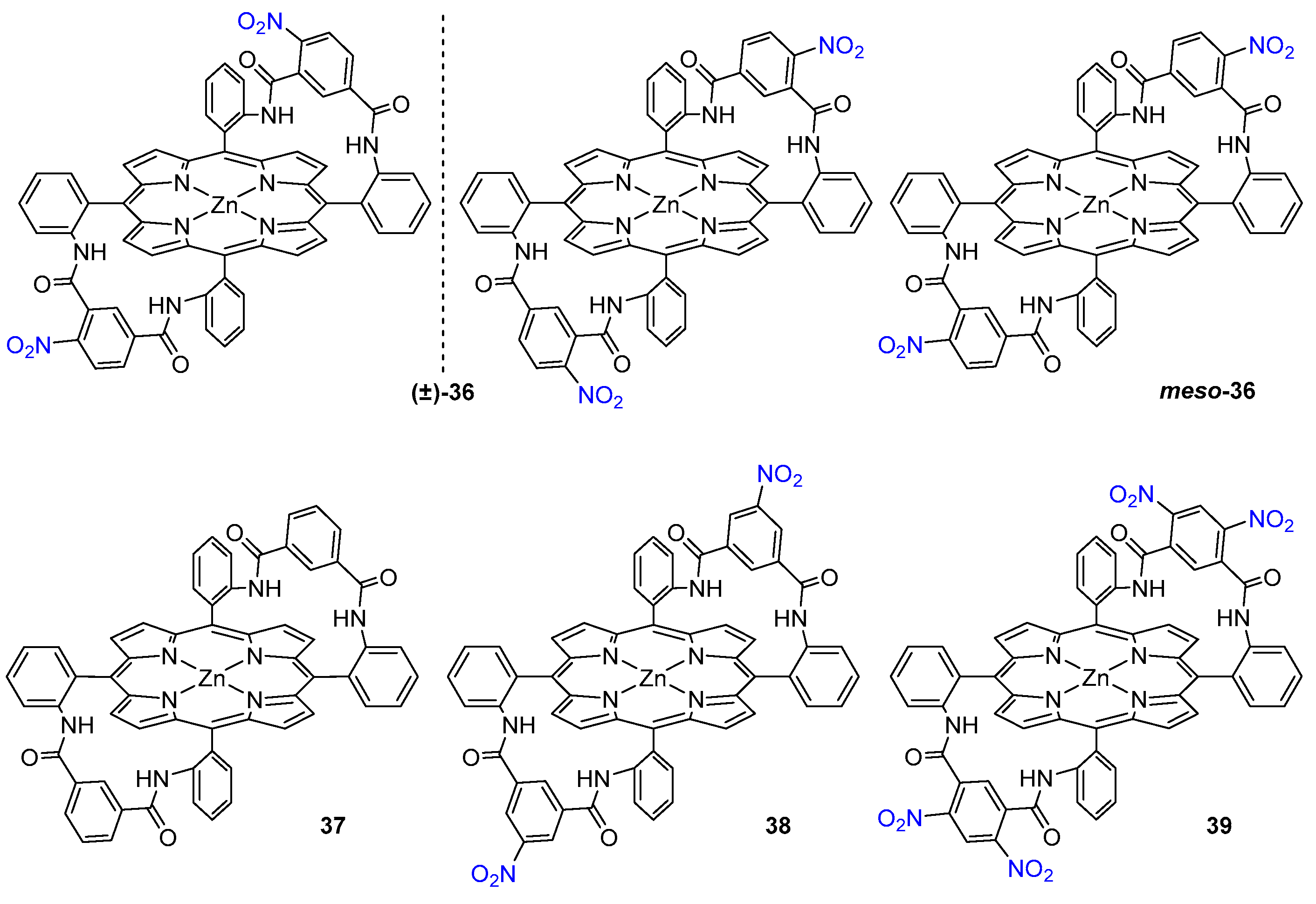
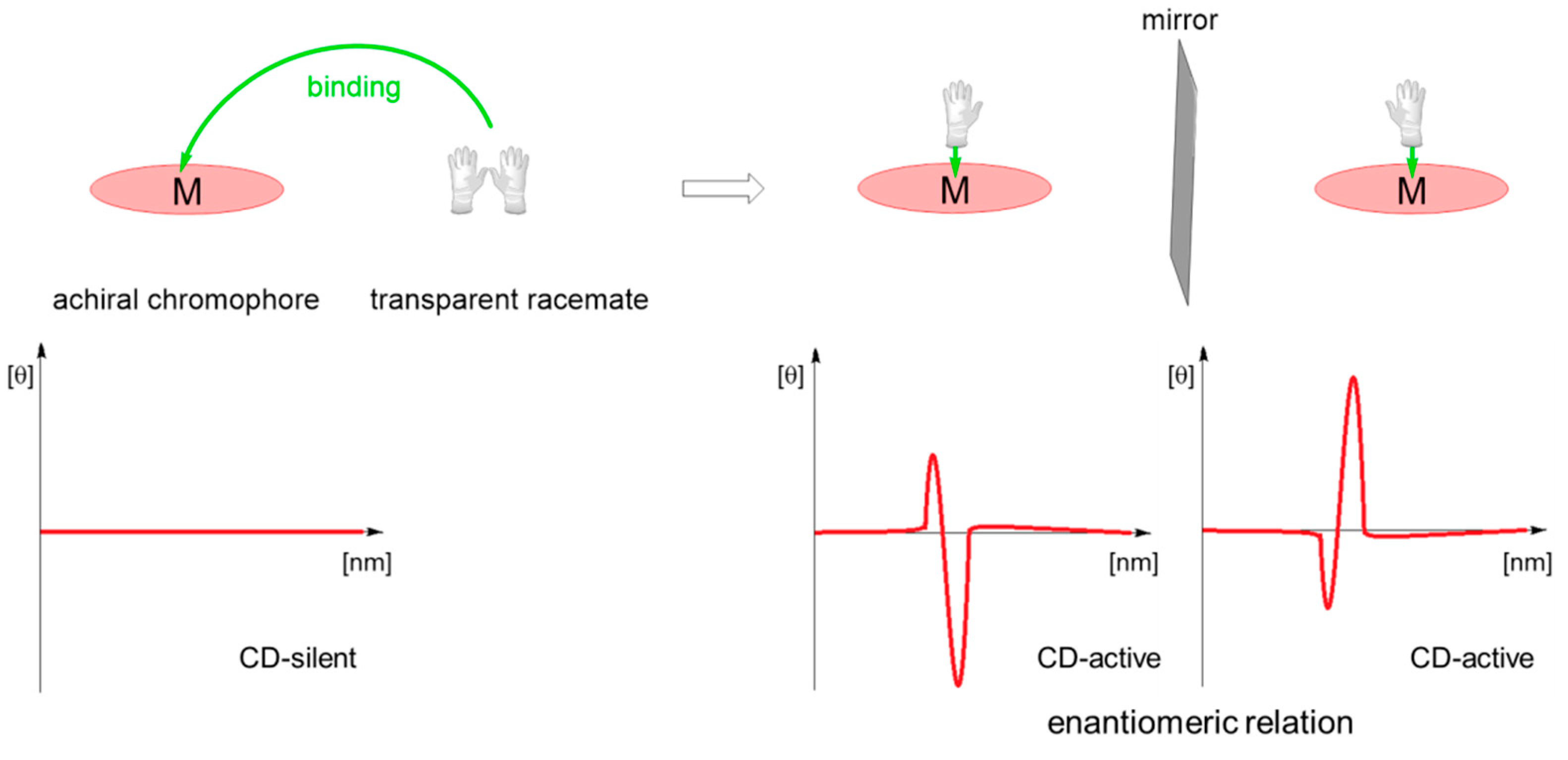
3.1.2. Achiral Hosts
3.1.3. Chiral Hosts
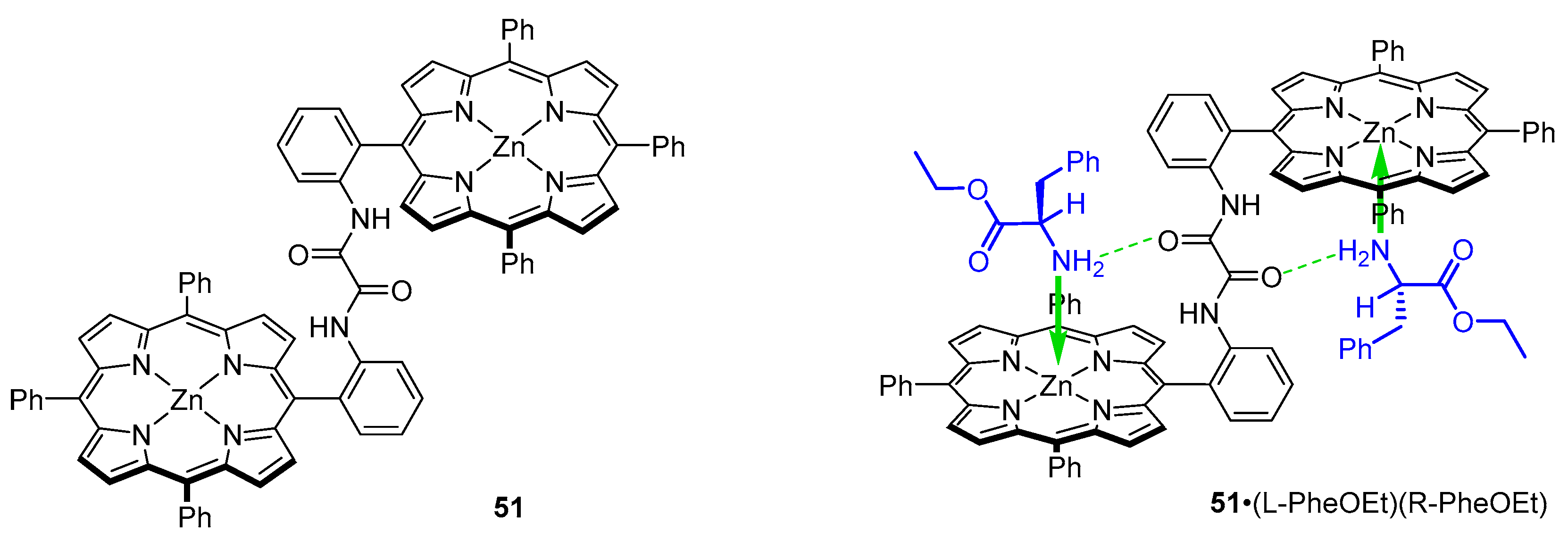
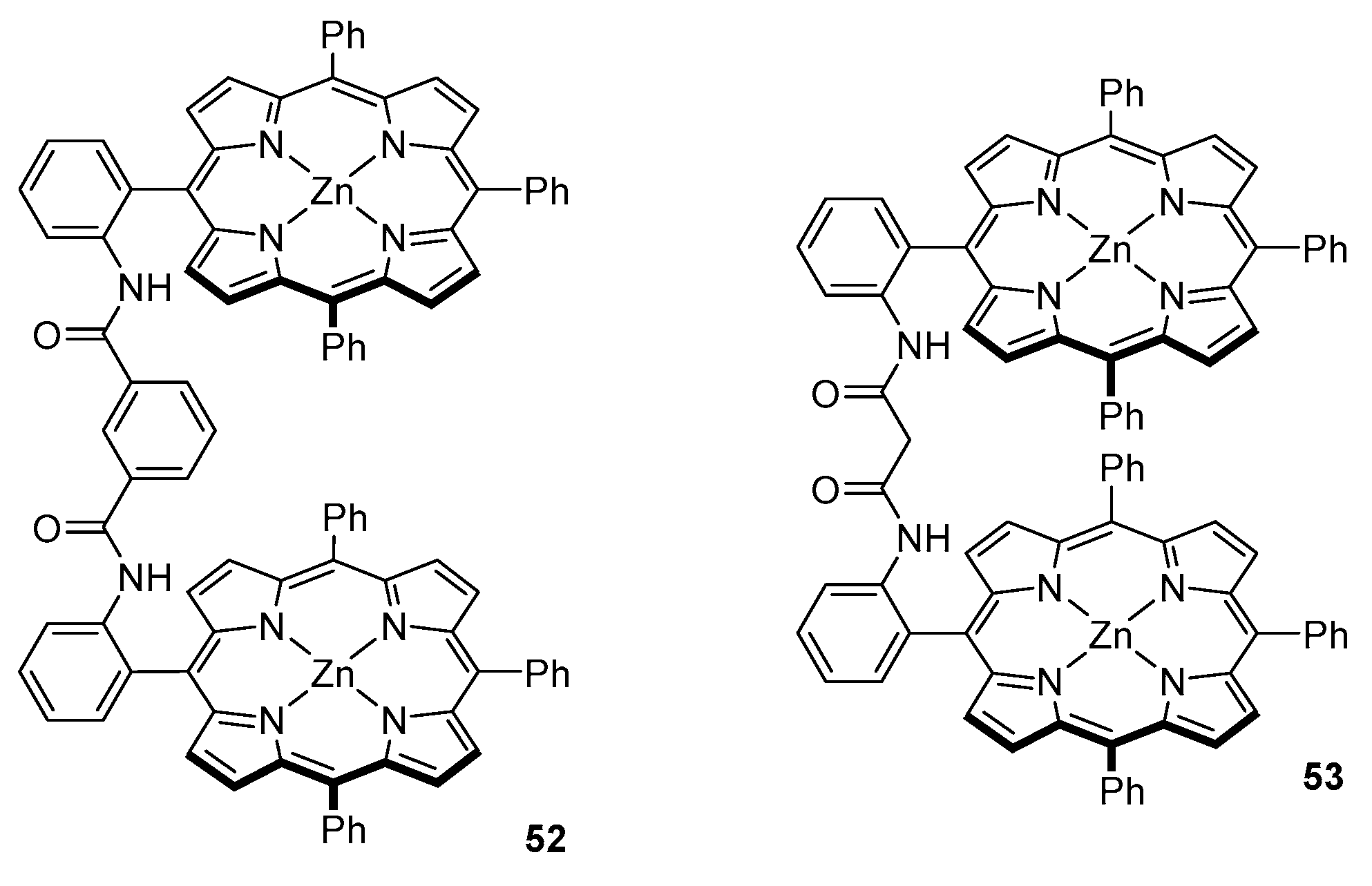
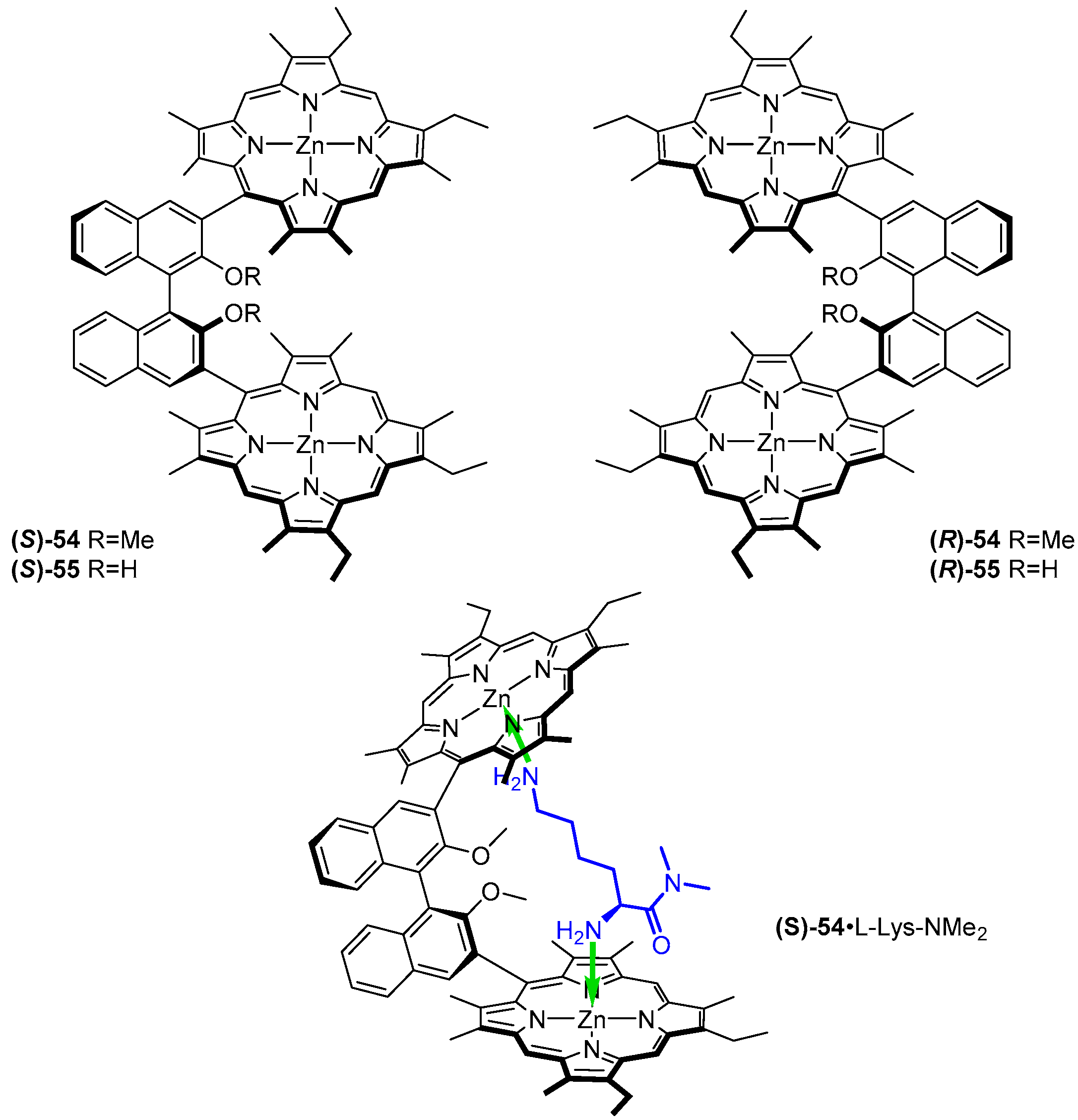
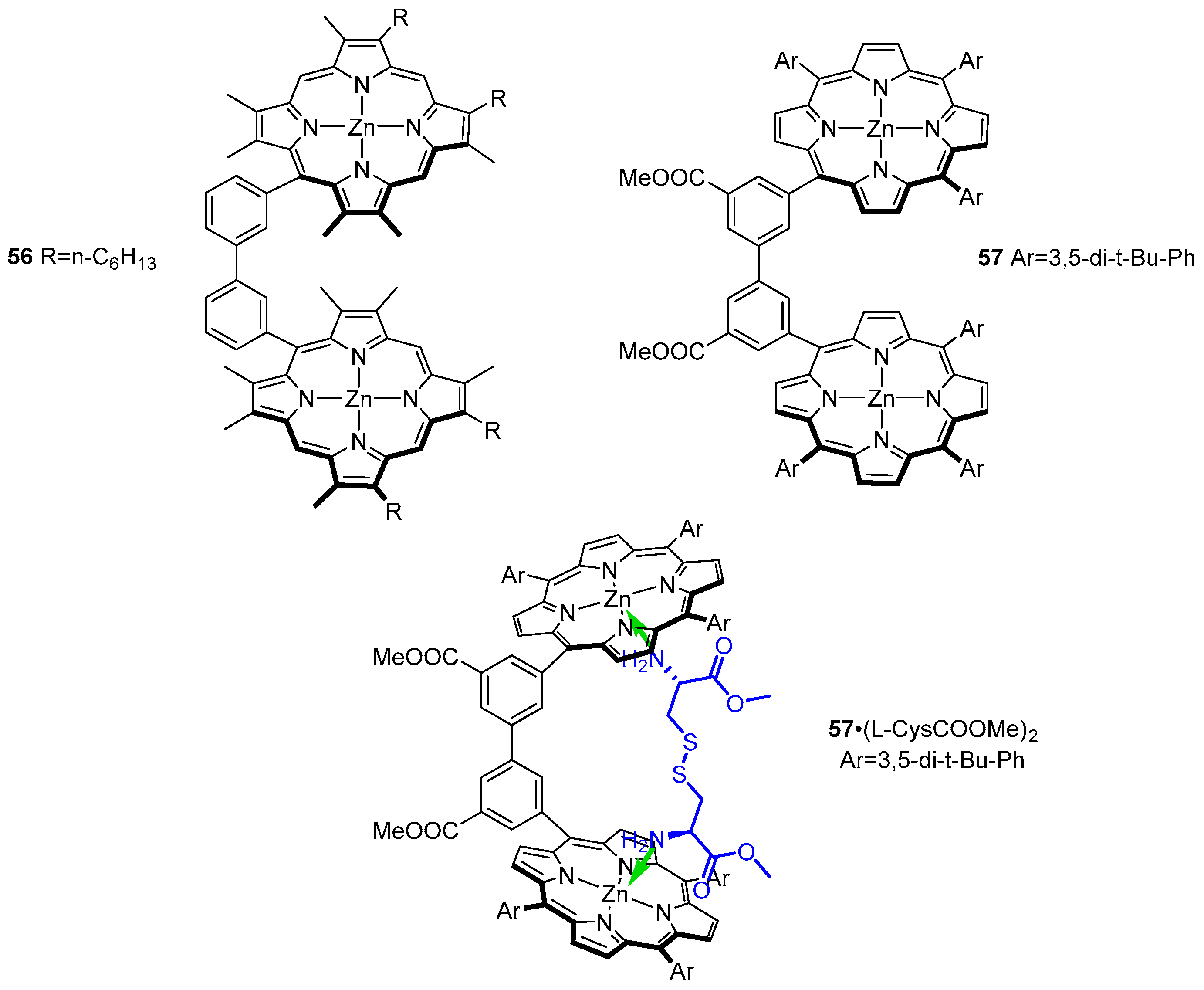
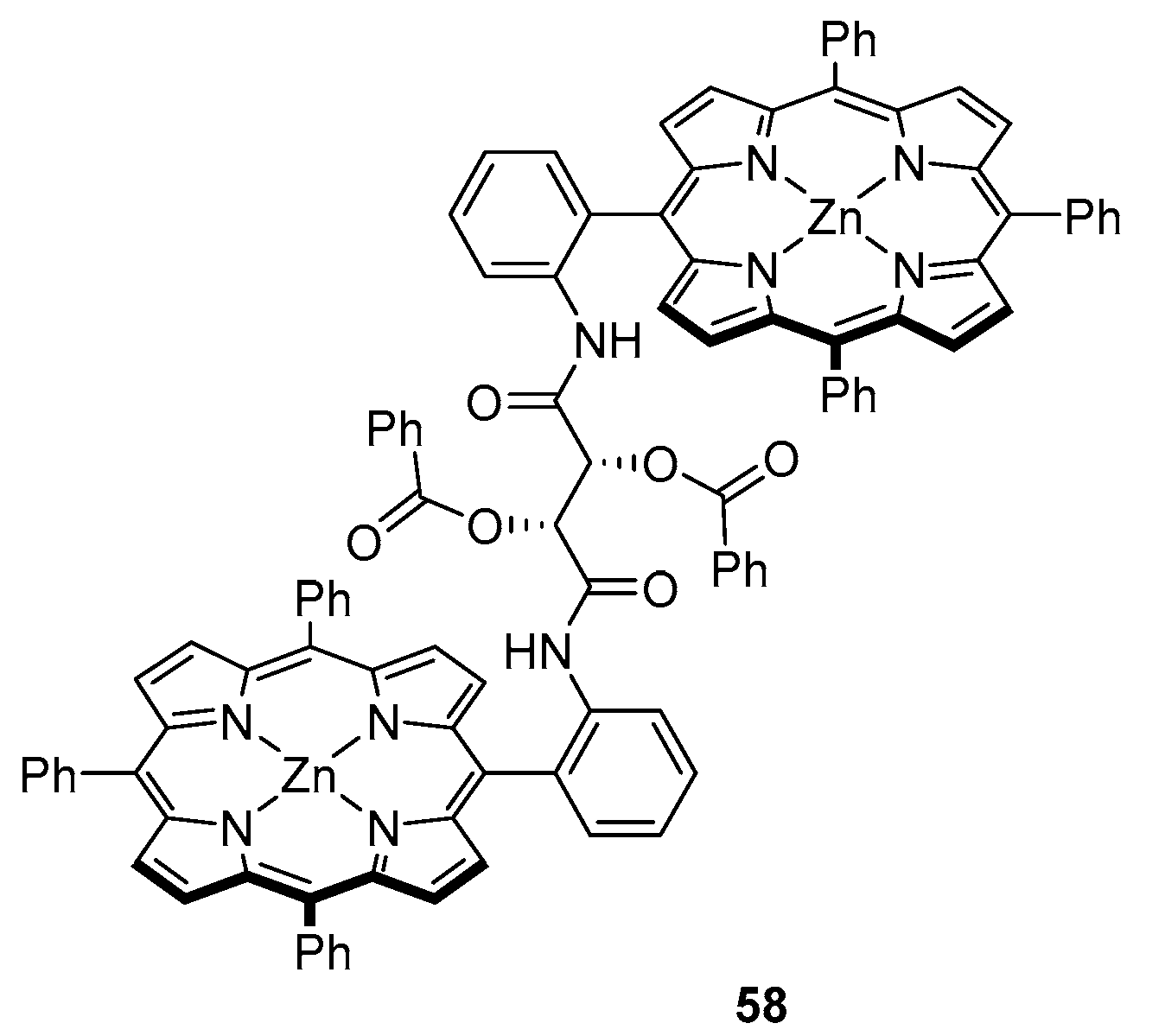
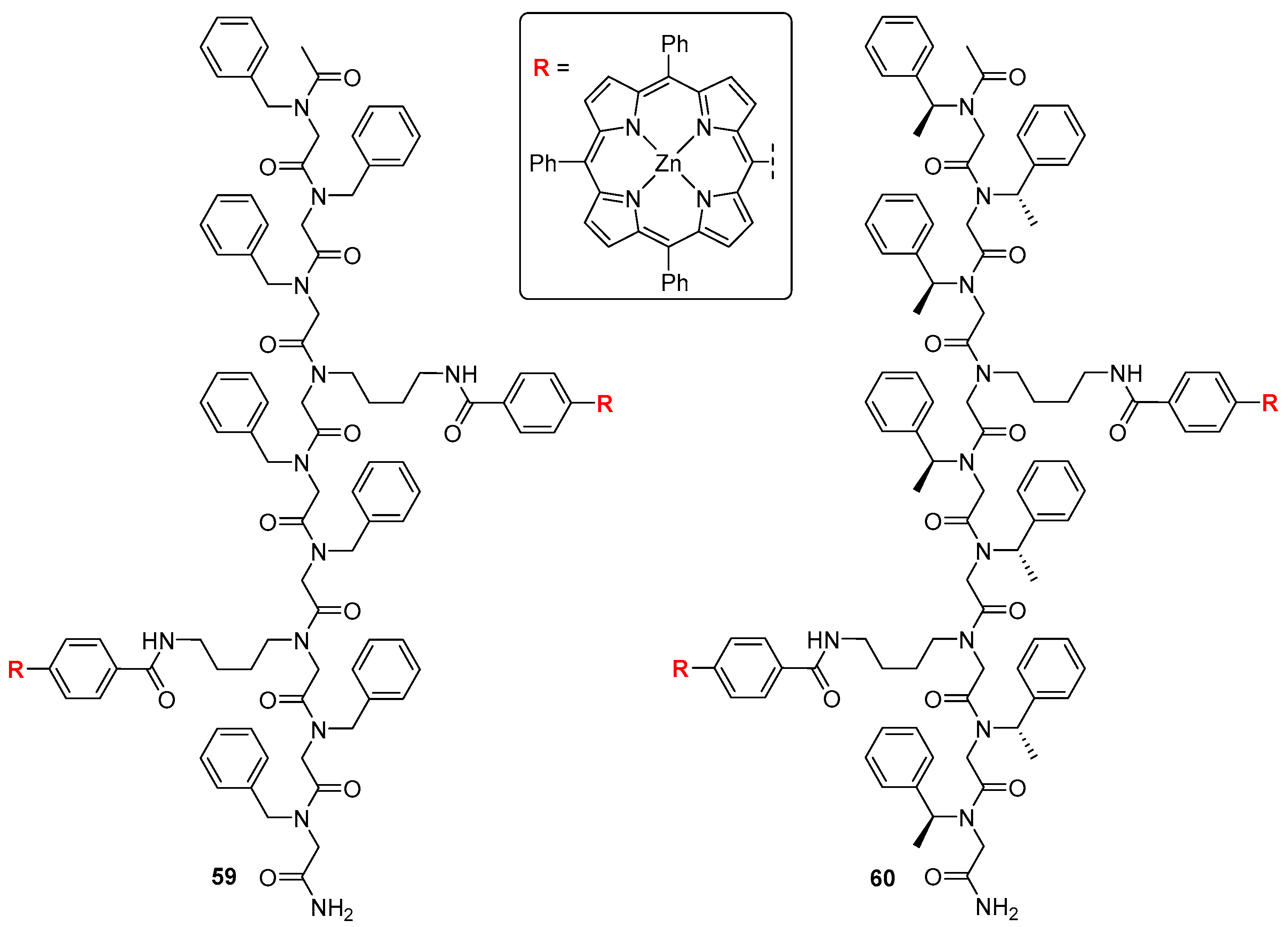
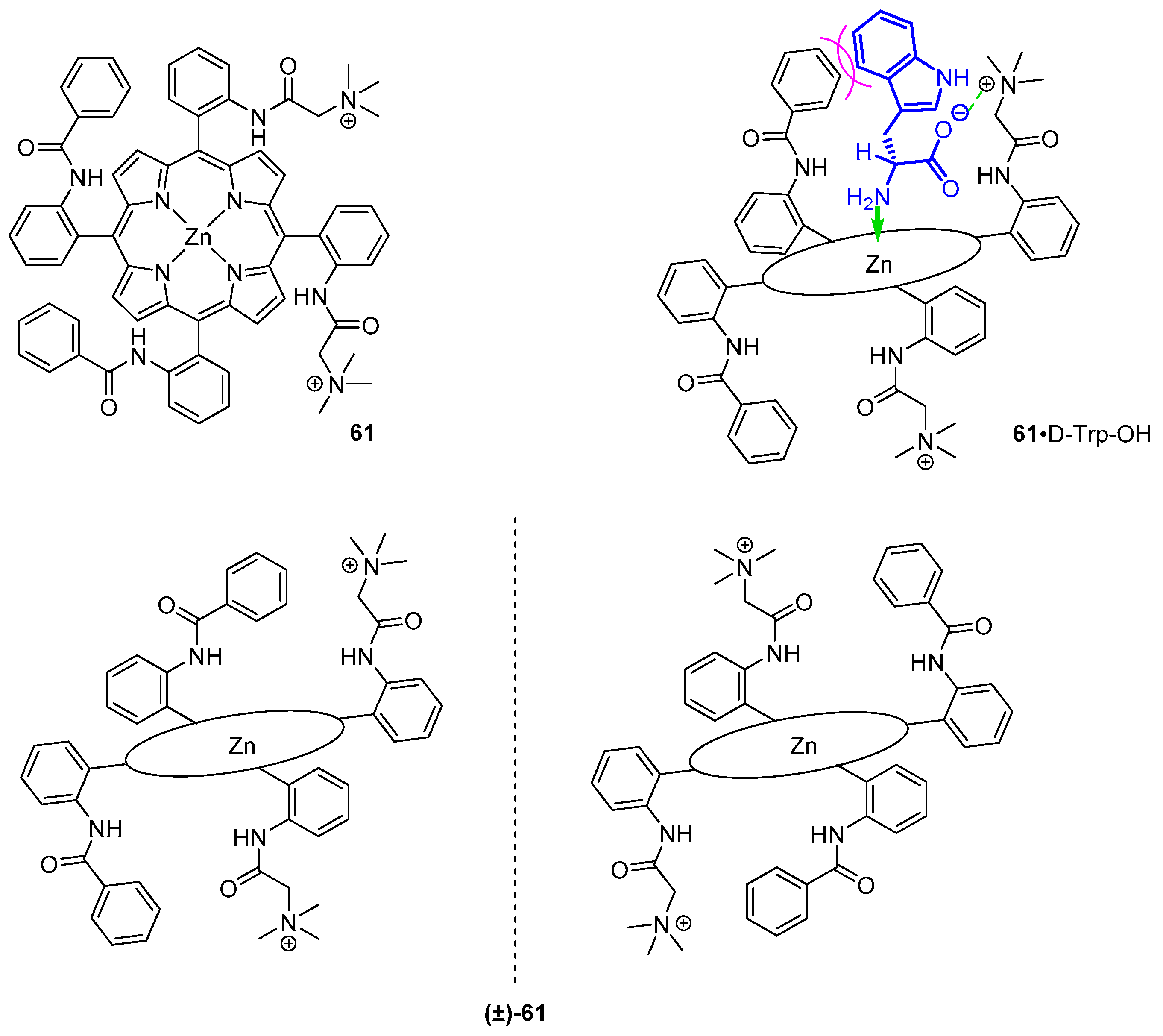
3.2. Dimeric Hosts
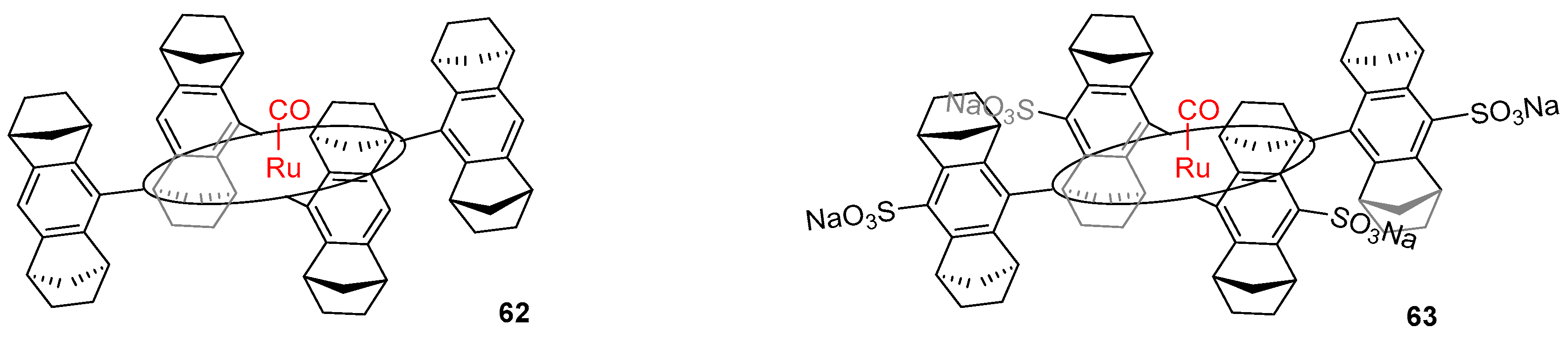
3.3. Aqueous Phase
4. Conclusions
Funding
Conflicts of Interest
References
- Persch, E.; Dumele, O.; Diederich, F. Molecular Recognition in Chemical and Biological Systems. Angew. Chem. Int. Ed. 2015, 54, 3290–3327. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; McCammon, J.A. Molecular Recognition and Ligand Association. Annu. Rev. Phys. Chem. 2013, 64, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Ariga, K.; Ito, H.; Hillab, J.P.; Tsukube, H. Molecular recognition: From solution science to nano/materials technology. Chem. Soc. Rev. 2012, 41, 5800–5835. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kaneko, Y.; Nishibori, E.; Nabeshima, T. Molecular Recognition by Multiple Metal Coordination inside Wavy-Stacked Macrocycles. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef]
- Fan, E.; Van Arman, S.A.; Kincaid, S.; Hamilton, A.D. Molecular Recognition: Hydrogen-Bonding Receptors That Function in Highly Competitive Solvents. J. Am. Chem. Soc. 1993, 115, 369–370. [Google Scholar] [CrossRef]
- Yao, H.; Ke, H.; Zhang, X.; Pan, S.-J.; Li, M.-S.; Yang, L.-P.; Schreckenbach, G.; Jiang, W. Molecular Recognition of Hydrophilic Molecules in Water by Combining the Hydrophobic Effect with Hydrogen Bonding. J. Am. Chem. Soc. 2018, 140, 13466–13477. [Google Scholar] [CrossRef]
- Yang, L.; Adam, C.; Nichol, G.S.; Cockroft, S.L. How Much Do van Der Waals Dispersion Forces Contribute to Molecular Recognition in Solution? Nat. Chem. 2013, 5, 1006–1010. [Google Scholar] [CrossRef]
- Muehldorf, A.V.; Van Engen, D.; Warner, J.C.; Hamilton, A.D. Aromatic-Aromatic Interactions in Molecular Recognition: A Family of Artificial Receptors for Thymine That Shows both Face-to-Face and Edge-to-Face Orientations. J. Am. Chem. Soc. 1988, 110, 6561–6562. [Google Scholar] [CrossRef]
- Crowley, J.D.; Goshe, A.J.; Bosnich, B. Molecular Recognition. Electrostatic Effects in Supramolecular Self-Assembly. Chem. Commun. 2003, 9, 392–393. [Google Scholar] [CrossRef]
- Hosoowi, L.; Hong, K.I.; Jang, W.D. Design and Applications of Molecular Probes Containing Porphyrin Derivatives. Coord. Chem. Rev. 2018, 354, 46–73. [Google Scholar] [CrossRef]
- Imai, H.; Munakata, H.; Uemori, Y.; Sakura, N. Chiral Recognition of Amino Acids and Dipeptides by a Water-Soluble Zinc Porphyrin. Inorg. Chem. 2004, 43, 1211–1213. [Google Scholar] [CrossRef]
- Randazzo, R.; Gaeta, M.; Gangemi, C.M.A.; Fragalà, M.E.; Purrello, R.; D’Urso, A. Chiral Recognition of l- and d- Amino Acid by Porphyrin Supramolecular Aggregates. Molecules 2019, 24, 84. [Google Scholar] [CrossRef]
- Huang, X.; Nakanishi, K.; Berova, N. Porphyrins and Metalloporphyrins: Versatile Circular Dichroic Reporter Groups for Structural Studies. Chirality 2000, 12, 237–255. [Google Scholar] [CrossRef]
- Davis, S.J.; Ikemizu, S.; Evans, E.J.; Fugger, L.; Bakker, T.R.; van der Merwe, P.A. The Nature of Molecular Recognition by T Cells. Nat. Immunol. 2003, 4, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Weerapana, E.; Wang, C.; Simon, G.M.; Richter, F.; Khare, S.; Dillon, M.B.D.; Bachovchin, D.A.; Mowen, K.; Baker, D.; Cravatt, B.F. Quantitative reactivity profiling predicts functional cysteine in proteomes. Nature 2010, 468, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Zhouab, Y.; Yoon, J. Recent progress in fluorescent and colorimetric chemosensors for detection of amino acids. Chem. Soc. Rev. 2012, 41, 52–67. [Google Scholar] [CrossRef]
- Cheng, Z.; Kuru, E.; Sachdeva, A.; Vendrell, M. Fluorescent amino acids as versatile building blocks for chemical biology. Nat. Rev. Chem. 2020, 4, 275–290. [Google Scholar] [CrossRef]
- Kaur, P.; Singh, K. Recent Advances in the Application of BODIPY in Bioimaging and Chemosensing. J. Mater. Chem. C 2019, 7, 11361–11405. [Google Scholar] [CrossRef]
- Jameson, E.E.; Cunliffe, J.M.; Neubig, R.R.; Sunahara, R.K.; Kennedy, R.T. Detection of G Proteins by Affinity Probe Capillary Electrophoresis Using a Fluorescently Labeled GTP Analogue. Anal. Chem. 2003, 75, 4297–4304. [Google Scholar] [CrossRef]
- Ojida, A.; Sakamoto, T.; Inoue, M.; Fujishima, S.; Lippens, G.; Hamachi, I. Fluorescent BODIPY-Based Zn(II) Complex as a Molecular Probe for Selective Detection of Neurofibrillary Tangles in the Brains of Alzheimer’s Disease Patients. J. Am. Chem. Soc. 2009, 131, 6543–6548. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, N.; Ji, X.; Tao, Y.; Wang, J.; Zaho, W. BODIPY-Based Fluorescent Probes for Biothiols. Chem. Eur. J. 2020, 26, 4172–4192. [Google Scholar] [CrossRef] [PubMed]
- Farinone, M.; Cybińska, J.; Pawlicki, M. A Controlled Blue-Shift in Meso-Nitrogen Aryl Fused DIPY and BODIPY Skeletons. Org. Chem. Front. 2019, 6, 2825–2832. [Google Scholar] [CrossRef]
- Farinone, M.; Cybinska, J.; Pawlicki, M. BODIPY-Amino Acid Conjugates—Tuning the Optical Response with a Meso-Heteroatom. Org. Chem. Front. 2020, 7, 2391–2398. [Google Scholar] [CrossRef]
- Sørensen, M.L.H.; Vosch, T.; Laursen, B.W.; Hansen, T. Spectral Shifts of BODIPY Derivatives: A Simple Continuous Model. Photochem. Photobiol. Sci. 2019, 18, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Loudet, A.; Burgess, K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef]
- Bañuelos, J. BODIPY Dye, the Most Versatile Fluorophore Ever? Chem. Rec. 2016, 16, 335–348. [Google Scholar] [CrossRef]
- Prasannan, D.; Arunkumar, C. A “Turn-on-and-off” PH Sensitive BODIPY Fluorescent Probe for Imaging E. Coli Cells. New J. Chem. 2018, 42, 3473–3482. [Google Scholar] [CrossRef]
- Bricks, J.L.; Kovalchuk, A.; Trieflinger, C.; Nofz, M.; Bueschel, M.; Tolmachev, A.I.; Daub, J.; Rurack, K. On the Development of Sensor Molecules that Display FeIII-amplified Fluorescence. J. Am. Chem. Soc. 2005, 127, 13522–13529. [Google Scholar] [CrossRef]
- Zhanga, J.; Pana, F.; Jina, Y.; Wanga, N.; Hea, J.; Zhanga, W.; Zhao, W. A BODIPY-based dual-responsive turn-on fluorescent probe for NO and nitrite. Dyes Pigment. 2018, 155, 276–283. [Google Scholar] [CrossRef]
- Shao, J.; Guo, H.; Ji, S.; Zhao, J. Styryl-BODIPY Based Red-Emitting Fluorescent OFF–ON Molecular Probe for Specific Detection of Cysteine. Biosens. Bioelectron. 2011, 26, 3012–3017. [Google Scholar] [CrossRef]
- Wang, C.; Xia, X.; Luo, J.; Qian, Y. A Novel Near-Infrared Styryl-BODIPY Fluorescent Probe for Discrimination of GSH and Its Application in Living Cells. Dyes Pigment. 2018, 152, 85–92. [Google Scholar] [CrossRef]
- Wang, Q.; Wei, X.; Li, C.; Xie, Y. A Novel P-Aminophenylthio- and Cyano- Substituted BODIPY as a Fluorescence Turn-on Probe for Distinguishing Cysteine and Homocysteine from Glutathione. Dyes Pigment. 2018, 148, 212–218. [Google Scholar] [CrossRef]
- Leen, V.; Yuan, P.; Wang, L.; Boens, N.; Dehaen, W. Synthesis of Meso-Halogenated BODIPYs and Access to Meso-Substituted Analogues. Org. Lett. 2012, 14, 6150–6153. [Google Scholar] [CrossRef]
- Jeon, S.; Kim, T.; Jin, H.; Lee, U.; Bae, J.; Bouffard, J.; Kim, Y. Amine-Reactive Activated Esters of Meso-CarboxyBODIPY: Fluorogenic Assays and Labeling of Amines, Amino Acids, and Proteins. J. Am. Chem. Soc. 2020, 142, 9231–9239. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Yue, S.; Jia, L.; Li, S.; Li, C.; Li, Q.; Xiao, L. NIR Fluorescent AzaBODIPY-Based Probe for the Specific Detection of l-Lysine. Chem. Sel. 2018, 3, 7581–7585. [Google Scholar] [CrossRef]
- Adhikari, S.; Ghosh, A.; Mandal, S.; Guria, S.; Banerjee, P.P.; Chatterjeec, A.; Das, D. Colorimetric and fluorescence probe for the detection of nano-molar lysine in aqueous medium. Org. Biomol. Chem. 2016, 14, 10688–10694. [Google Scholar] [CrossRef]
- Liu, Y.; Lv, X.; Hou, M.; Shi, Y.; Guo, W. Selective Fluorescence Detection of Cysteine over Homocysteine and Glutathione Based on a Cysteine-Triggered Dual Michael Addition/Retro-Aza-Aldol Cascade Reaction. Anal. Chem. 2015, 87, 11475–11483. [Google Scholar] [CrossRef]
- Avellanal-Zaballa, E.; Ramos-Torres, Á.; Prieto-Castañeda, A.; García-Garrido, F.; Bañuelos, J.; Agarrabeitia, A.R.; Ortiz, M.J. A BODIPY-Based Fluorescent Sensor for Amino Acids Bearing Thiol. Proceedings 2019, 41, 18. [Google Scholar] [CrossRef]
- Wang, N.; Wang, Y.; Gao, J.; Ji, X.; He, J.; Zhang, J.; Zhao, W. A Ratiometric Fluorescent BODIPY-Based Probe for Rapid and Highly Sensitive Detection of Cysteine in Human Plasma. Analyst 2018, 143, 5728–5735. [Google Scholar] [CrossRef]
- Niu, L.; Guan, Y.; Chen, Y.; Wu, L.; Tung, C.; Yang, Q. BODIPY-Based Ratiometric Fluorescent Sensor for Highly Selective Detection of Glutathione over Cysteine and Homocysteine. J. Am. Chem. Soc. 2012, 134, 18928–18931. [Google Scholar] [CrossRef]
- Guria, S.; Ghosh, A.; Manna, K.; Pal, A.; Adhikary, A.; Adhikari, S. Rapid detection of aspartic acid in water by BODIPY-Based fluorescent probe: Live-cell imaging and DFT studies. Dyes Pigment. 2019, 168, 111–122. [Google Scholar] [CrossRef]
- Bastug, E.; Kursunlu, A.N.; Guler, E. A fluorescent clever macrocycle: Deca-bodipy bearing a pillar [5]arene and its selective binding of asparagine in half-aqueous medium. J. Lumin 2020, 225, 117343. [Google Scholar] [CrossRef]
- Mizutani, T.; Wada, K.; Kitagawa, S. Molecular Recognition of Amines and Amino Esters by Zinc Porphyrin Receptors: Binding Mechanisms and Solvent Effects. J. Org. Chem. 2000, 65, 6097–6106. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Yotsukura, M.; Noji, M.; Takanami, T. Bis(Zinc Porphyrin) as a CD-Sensitive Bidentate Host Molecule: Direct Determination of Absolute Configuration of Mono-Alcohols. Chem. Commun. 2015, 51, 11068–11071. [Google Scholar] [CrossRef] [PubMed]
- Borovkov, V.V.; Lintuluoto, J.M.; Inoue, Y. Supramolecular Chirogenesis in Zinc Porphyrins: Mechanism, Role of Guest Structure, and Application for the Absolute Configuration Determination. J. Am. Chem. Soc. 2001, 123, 2979–2989. [Google Scholar] [CrossRef]
- Allenmark, S. Induced Circular Dichroism by Chiral Molecular Interaction. Chirality 2003, 15, 409–422. [Google Scholar] [CrossRef]
- Mizutani, T.; Ema, T.; Yoshida, T.; Kuroda, Y.; Ogoshi, H. Recognition of.Alpha.-Amino Acid Esters by Zinc Porphyrin Derivatives via Coordination and Hydrogen Bonding Interactions. Evidence for Two-Point Fixation from Thermodynamic and Induced Circular Dichroism Spectroscopic Studies. Inorg. Chem. 1993, 32, 2072–2077. [Google Scholar] [CrossRef]
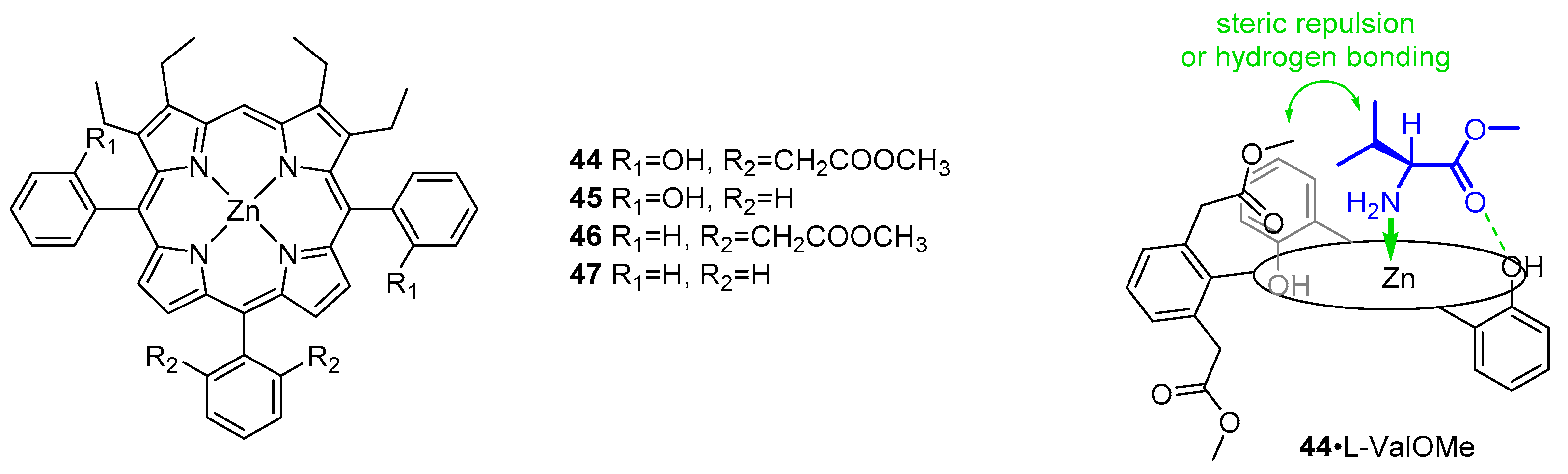
- Kuroda, Y.; Kato, Y.; Higashioji, T.; Hasegawa, J.; Kawanami, S.; Takahashi, M.; Shiraishi, N.; Tanabe, K.; Ogoshi, H. Chiral Amino Acid Recognition by a Porphyrin-Based Artificial Receptor. J. Am. Chem. Soc. 1995, 117, 10950–10958. [Google Scholar] [CrossRef]
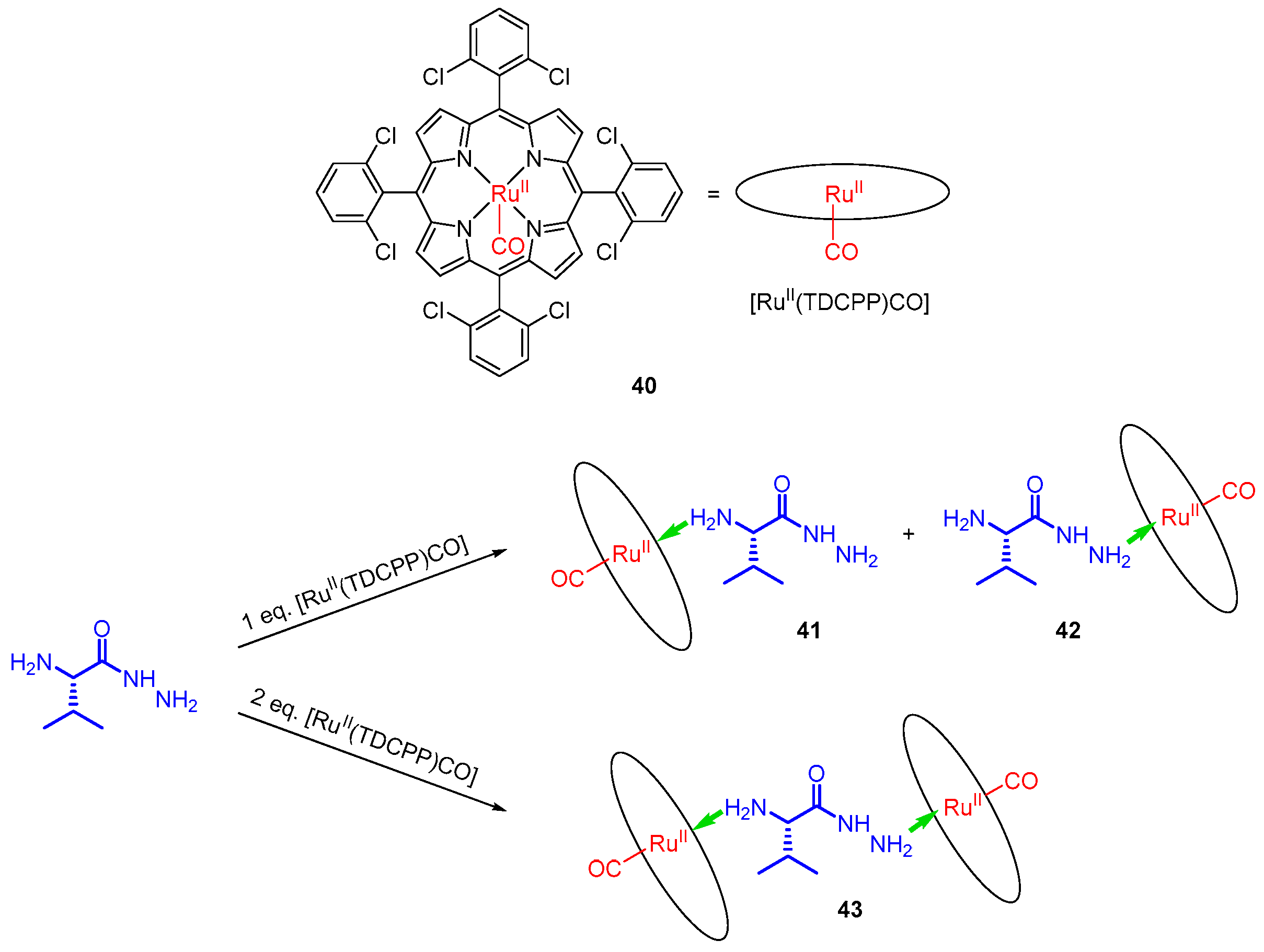
- Liang, Q.-F.; Liu, J.-J.; Chen, J. Sandwich Structure of a Ruthenium Porphyrin and an Amino Acid Hydrazide for Probing Molecular Chirality by Circular Dichroism. Tetrahedron Lett. 2011, 52, 3987–3991. [Google Scholar] [CrossRef]
- Mizutani, T.; Ema, T.; Tomita, T.; Kuroda, Y.; Ogoshi, H. Design and Synthesis of a Trifunctional Chiral Porphyrin with C2 Symmetry as a Chiral Recognition Host for Amino Acid Esters. J. Am. Chem. Soc. 1994, 116, 4240–4250. [Google Scholar] [CrossRef]
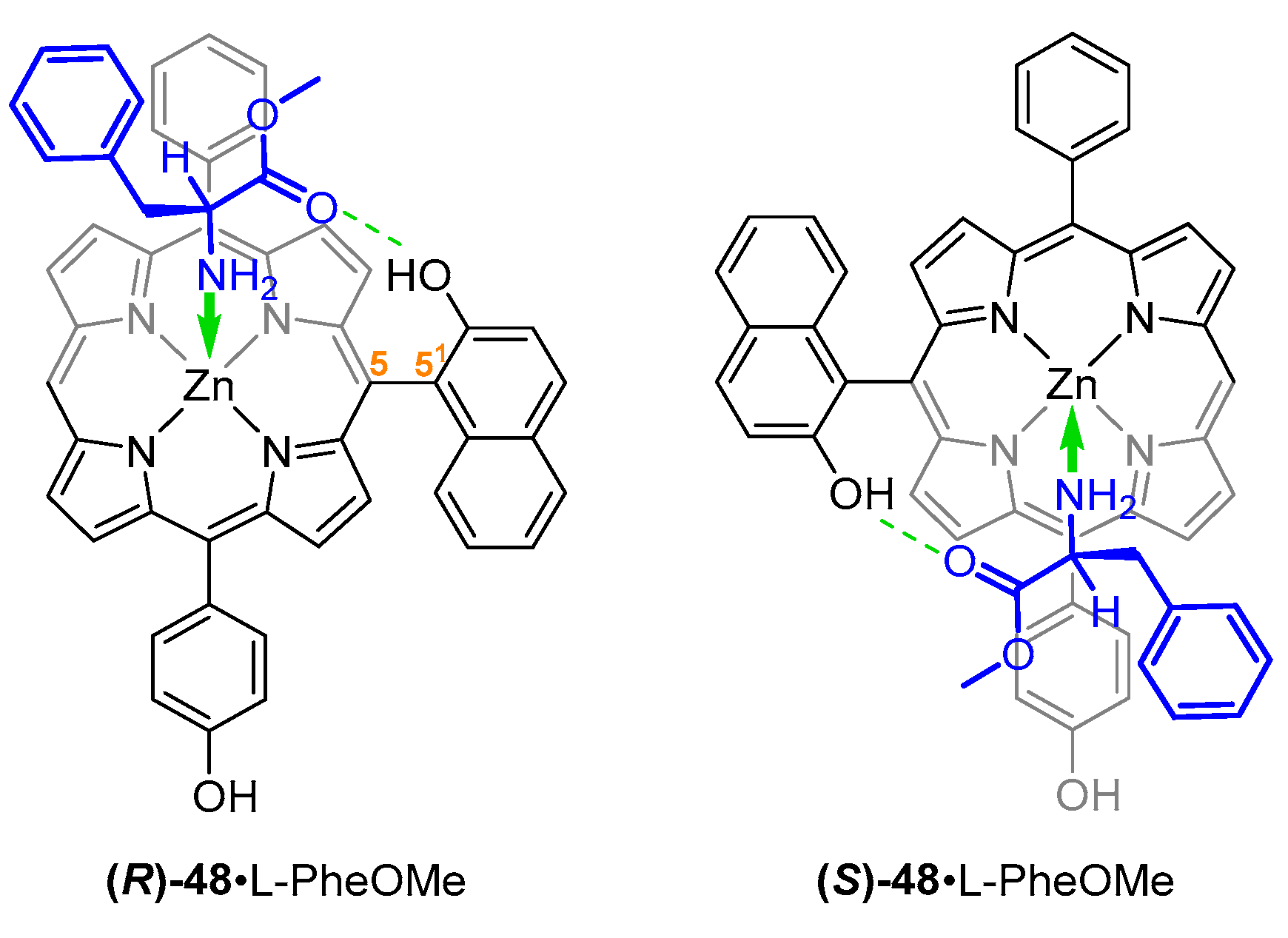
- Yang, L.; Zhou, Y.; Zhu, M.; Zhao, L.; Wei, L.; Bian, Y. Stereochemistry and Solid-State Structure of an Intrinsically Chiral Meso-Patterned Porphyrin: Case Study by NMR and Single-Crystal X-Ray Diffraction Analysis. J. Org. Chem. 2013, 78, 9949–9955. [Google Scholar] [CrossRef]
- Valderrey, V.; Aragay, G.; Ballester, P. Porphyrin Tweezer Receptors: Binding Studies, Conformational Properties and Applications. Coord. Chem. Rev. 2014; 258 259, 137–156. [Google Scholar] [CrossRef]
- Chen, C.W.; Whitlock, H.W. Molecular Tweezers: A Simple Model of Bifunctional Intercalation. J. Am. Chem. Soc. 1978, 100, 4921–4922. [Google Scholar] [CrossRef]
- Sanders, J.K.M.; Bampos, N.; Clyde-Watson, Z.E.; Darling, S.L.; Hawley, J.C.; Kim, H.-J.; Mak, C.C.; Webb, S.J. Axial Coordination Chemistry of Metalloporphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: London, UK, 2000; Volume 3, pp. 30–31. [Google Scholar]
- Borovkov, V.V.; Hembury, G.A.; Yamamoto, N.; Inoue, Y. Supramolecular Chirogenesis in Zinc Porphyrins: Investigation of Zinc-Freebase Bis-Porphyrin, New Mechanistic Insights, Extension of Sensing Abilities, and Solvent Effect. J. Phys. Chem. A 2003, 107, 8677–8686. [Google Scholar] [CrossRef]
- Jiang, J.; Feng, Z.; Liu, B.; Hu, C.; Wang, Y. Chiral Recognition of Amino Acid Esters by a Novel Oxalic Amide-Linked Bisporphyrin. Dalton Trans. 2013, 42, 7651–7659. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Fang, X.; Liu, B.; Hu, C. M-Phthalic Diamide-Linked Zinc Bisporphyrinate: Spontaneous Resolution of Its Crystals and Its Application in Chiral Recognition of Amino Acid Esters. Inorg. Chem. 2014, 53, 3298–3306. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Jiang, J.; Fang, X.; Hu, C. Absolute Configurational Assignments of Amino Acid Esters by a CD-Sensitive Malonamide-Linked Zinc Bisporphyrinate Host. Chin. J. Chem. 2014, 32, 797–802. [Google Scholar] [CrossRef]
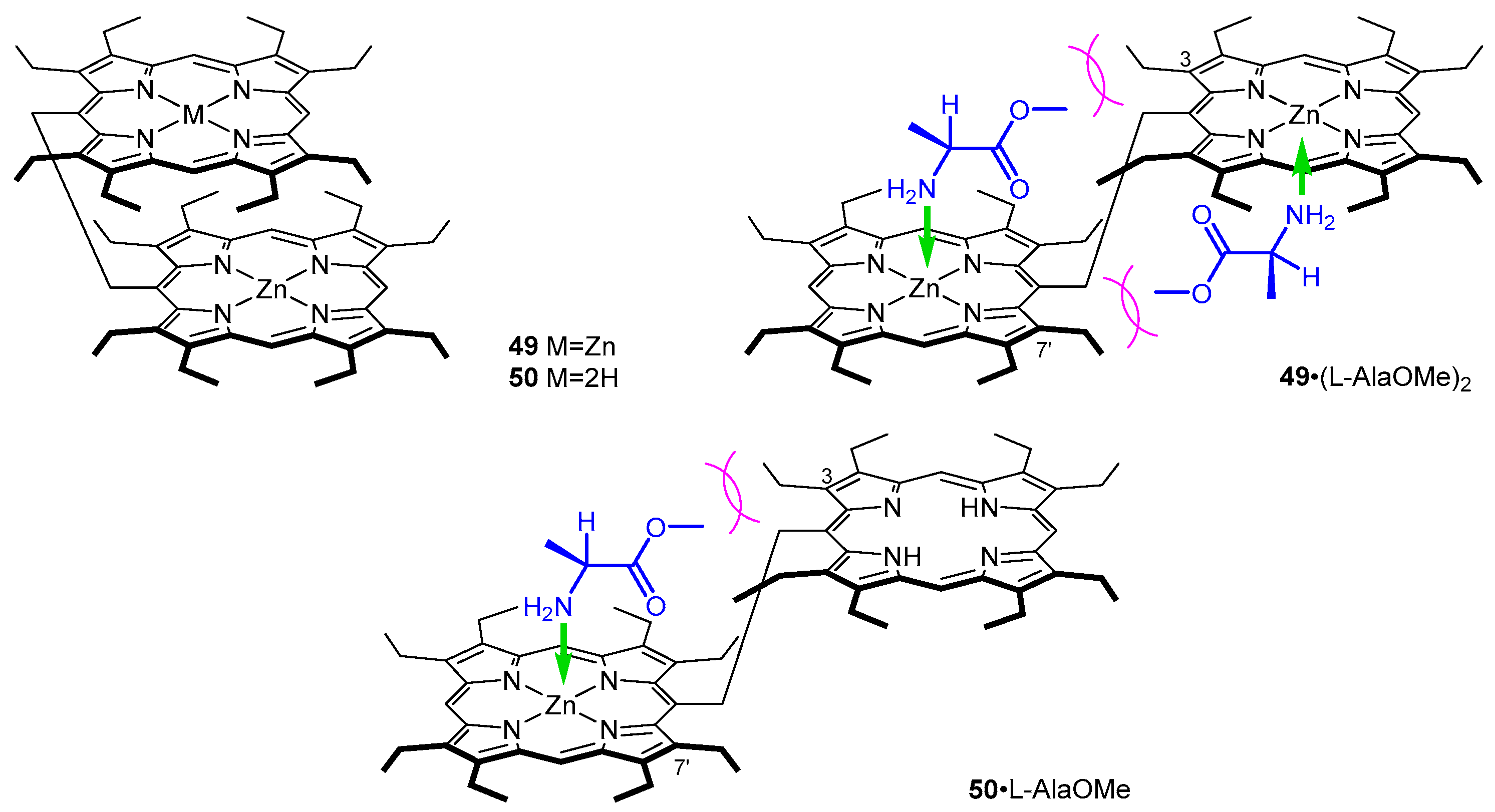
- Hayashi, T.; Aya, T.; Nonoguchi, M.; Mizutani, T.; Hisaeda, Y.; Kitagawa, S.; Ogoshi, H. Chiral Recognition and Chiral Sensing Using Zinc Porphyrin Dimers. Tetrahedron 2002, 58, 2803–2811. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.; Hu, C.; Wang, Y. Enantioselectivity of a Tartaric Acid Amide Linked Zinc Bisporphyrinate towards Amino Acid Esters. Dyes Pigment. 2020, 176, 108223. [Google Scholar] [CrossRef]
- Zuckermann, R.N.; Kerr, J.M.; Kent, S.B.H.; Moos, W.H. Efficient Method for the Preparation of Peptoids by Submonomer Solid-Phase Synthesis. J. Am. Chem. Soc. 1992, 114, 10646–10647. [Google Scholar] [CrossRef]
- Kang, B.; Yang, W.; Lee, S.; Mukherjee, S.; Forstater, J.; Kim, H.; Goh, B.; Kim, T.-Y.; Voelz, V.A.; Pang, Y.; et al. Precisely Tuneable Energy Transfer System Using Peptoid Helix-Based Molecular Scaffold. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Yang, W.; Kang, B.; Voelz, V.A.; Seo, J. Control of Porphyrin Interactions via Structural Changes of a Peptoid Scaffold. Org. Biomol. Chem. 2017, 15, 9670–9679. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Chung, S.; Ahn, Y.D.; Lee, J.; Seo, J. Porphyrin–Peptoid Conjugates: Face-to-Face Display of Porphyrins on Peptoid Helices. Org. Lett. 2013, 15, 1670–1673. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kang, B.; Seo, J. Metalloporphyrin Dimers Bridged by a Peptoid Helix: Host-Guest Interaction and Chiral Recognition. Molecules 2018, 23, 2741. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Wada, K.; Kitagawa, S. Porphyrin Receptors for Amines, Amino Acids, and Oligopeptides in Water. J. Am. Chem. Soc. 1999, 121, 11425–11431. [Google Scholar] [CrossRef]
- Imai, H.; Misawa, K.; Munakata, H.; Uemori, Y. Water-Soluble Zinc Porphyrins as Receptors for Amino Carboxylates. Chem. Lett. 2001, 30, 688–689. [Google Scholar] [CrossRef]
- Nicolas, I.; Chevance, S.; Maux, P.L.; Simonneaux, G. Chiral Recognition of Amines and Amino Acid Derivatives by Optically Active Ruthenium Halterman Porphyrins in Organic Solvents and Water. Tetrahedron Asymmetry 2010, 21, 1788–1792. [Google Scholar] [CrossRef]
- Maux, P.L.; Bahri, H.; Simonneaux, G. Molecular Recognition of Racemic Phosphines by a Chiral Ruthenium Porphyrin. J. Chem. Soc. Chem. Commun. 1991, 1350–1352. [Google Scholar] [CrossRef]
- Galardon, E.; Le Maux, P.; Bondon, A.; Simonneaux, G. Chiral Recognition of Amino Esters by a Ruthenium Porphyrin Complex: Kinetics of the Exchange Process Determined by 1H NMR. Tetrahedron Asymmetry 1999, 10, 4203–4210. [Google Scholar] [CrossRef]



































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farinone, M.; Urbańska, K.; Pawlicki, M. BODIPY- and Porphyrin-Based Sensors for Recognition of Amino Acids and Their Derivatives. Molecules 2020, 25, 4523. https://doi.org/10.3390/molecules25194523
Farinone M, Urbańska K, Pawlicki M. BODIPY- and Porphyrin-Based Sensors for Recognition of Amino Acids and Their Derivatives. Molecules. 2020; 25(19):4523. https://doi.org/10.3390/molecules25194523
Chicago/Turabian StyleFarinone, Marco, Karolina Urbańska, and Miłosz Pawlicki. 2020. "BODIPY- and Porphyrin-Based Sensors for Recognition of Amino Acids and Their Derivatives" Molecules 25, no. 19: 4523. https://doi.org/10.3390/molecules25194523
APA StyleFarinone, M., Urbańska, K., & Pawlicki, M. (2020). BODIPY- and Porphyrin-Based Sensors for Recognition of Amino Acids and Their Derivatives. Molecules, 25(19), 4523. https://doi.org/10.3390/molecules25194523