
Structure-Function Analyses of Human Bitter Taste Receptors—Where Do We Stand?


Abstract
:
1. Introduction
2. General Features of TAS2Rs
3. Different Approaches to Investigate TAS2Rs
3.1. Obtaining Experimental Structures
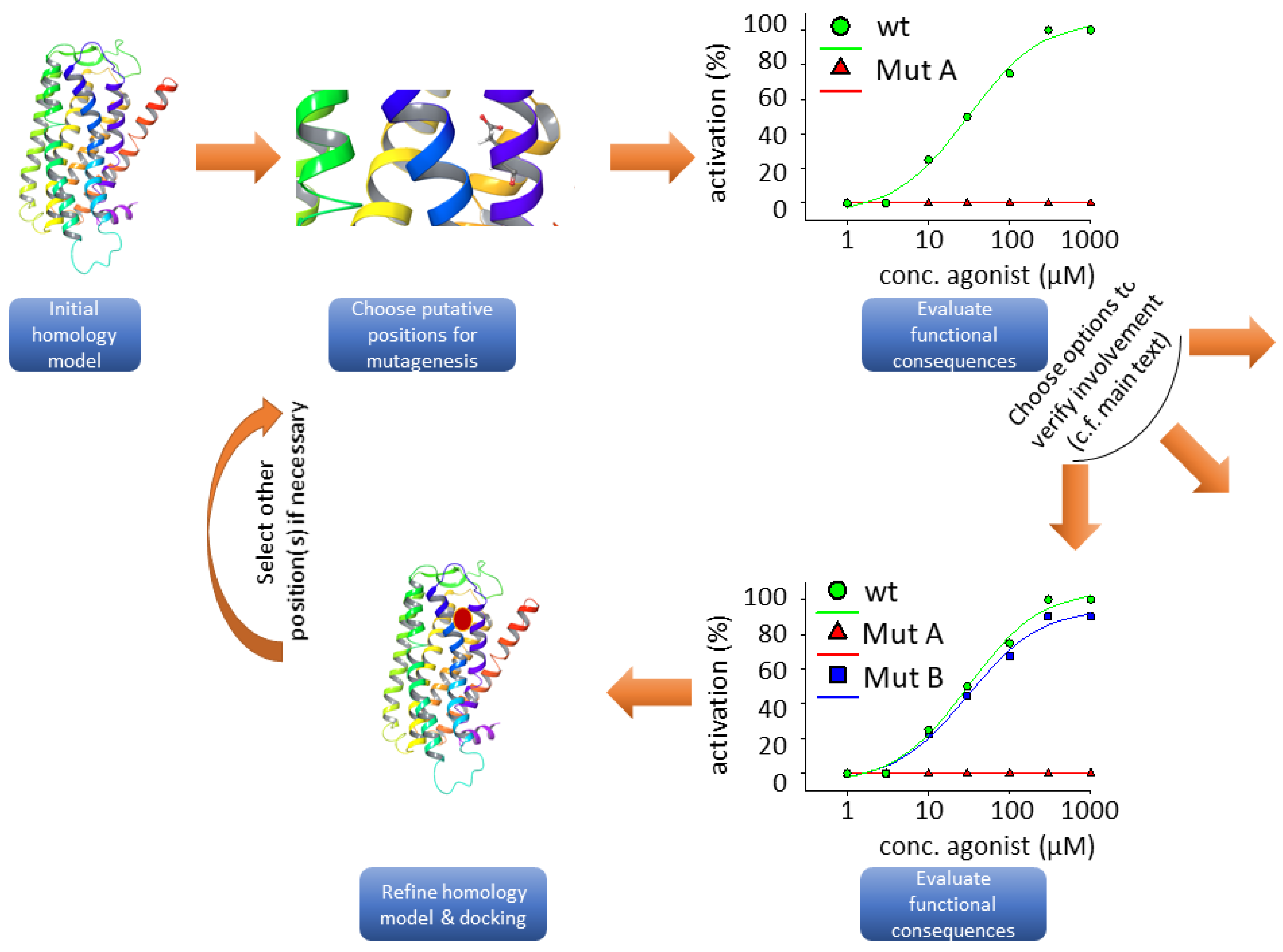
3.2. Homology Modeling
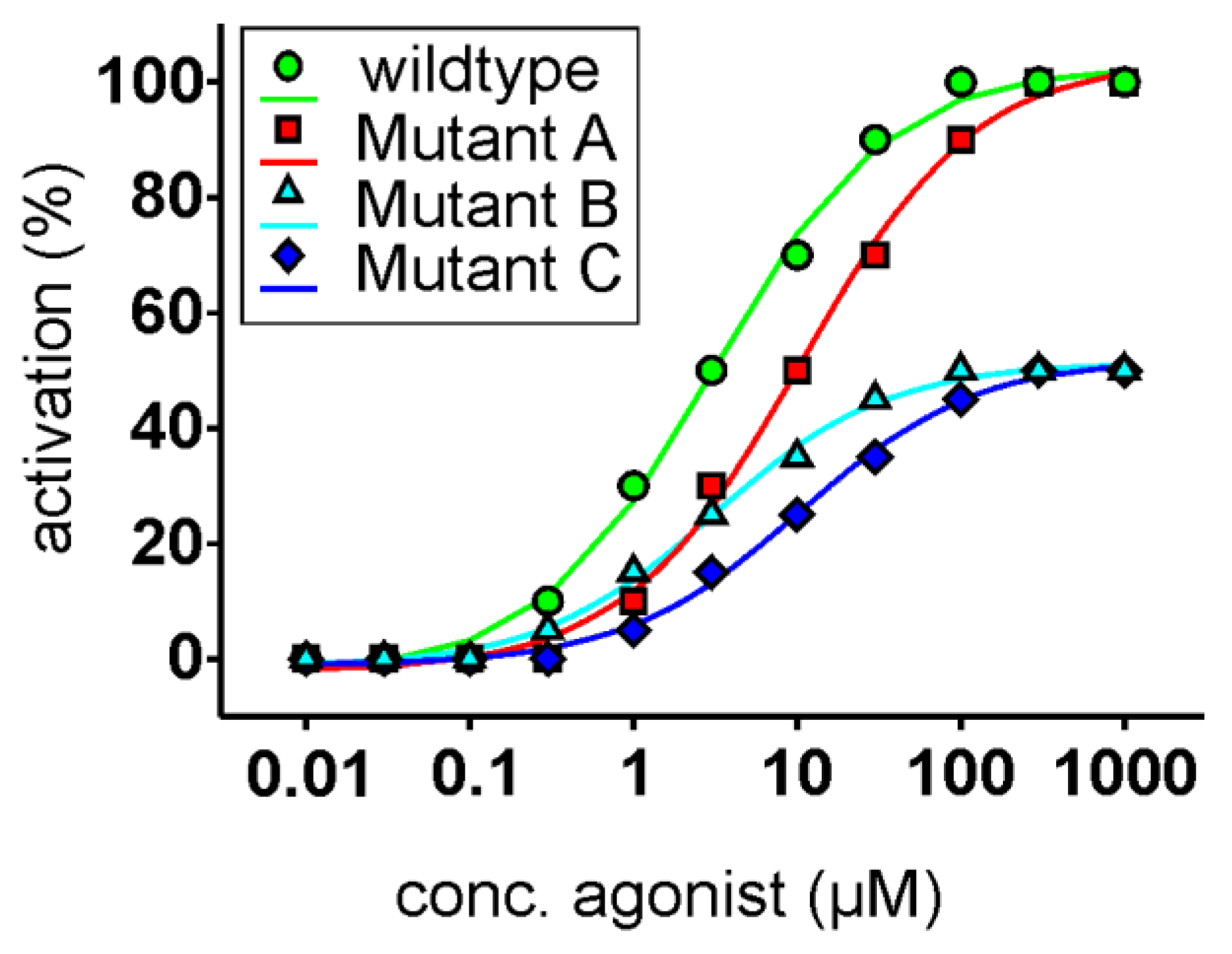
3.3. Functional Heterologous Expression in Combination with In Vitro Mutagenesis
4. Localization of TAS2R Binding Pockets
5. Receptor Activation
6. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Lindemann, B. Taste reception. Physiol. Rev. 1996, 76, 718–766. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.; Roper, S.D. The cell biology of taste. J. Cell Biol. 2010, 190, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, B.; Wilson, C.E.; Tu, Y.H.; Joshi, N.R.; Kinnamon, S.C.; Liman, E.R. Cellular and Neural Responses to Sour Stimuli Require the Proton Channel Otop1. Curr. Biol. 2019, 29, 3647–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Y.H.; Cooper, A.J.; Teng, B.; Chang, R.B.; Artiga, D.J.; Turner, H.N.; Mulhall, E.M.; Ye, W.; Smith, A.D.; Liman, E.R. An evolutionarily conserved gene family encodes proton-selective ion channels. Science 2018, 359, 1047–1050. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jin, H.; Zhang, W.; Ding, C.; O’Keeffe, S.; Ye, M.; Zuker, C.S. Sour Sensing from the Tongue to the Brain. Cell 2019, 179, 392–402. [Google Scholar] [CrossRef]
- Chandrashekar, J.; Kuhn, C.; Oka, Y.; Yarmolinsky, D.A.; Hummler, E.; Ryba, N.J.; Zuker, C.S. The cells and peripheral representation of sodium taste in mice. Nature 2010, 464, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Behrens, M.; Meyerhof, W. Chapter 13 - G Protein–Coupled Taste Receptors. In Chemosensory Transduction; Zufall, F., Munger, S.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 227–244. [Google Scholar]
- Max, M.; Shanker, Y.G.; Huang, L.; Rong, M.; Liu, Z.; Campagne, F.; Weinstein, H.; Damak, S.; Margolskee, R.F. Tas1r3, encoding a new candidate taste receptor, is allelic to the sweet responsiveness locus Sac. Nat. Genet. 2001, 28, 58–63. [Google Scholar] [CrossRef]
- Montmayeur, J.P.; Liberles, S.D.; Matsunami, H.; Buck, L.B. A candidate taste receptor gene near a sweet taste locus. Nat. Neurosci. 2001, 4, 492–498. [Google Scholar] [CrossRef]
- Nelson, G.; Hoon, M.A.; Chandrashekar, J.; Zhang, Y.; Ryba, N.J.; Zuker, C.S. Mammalian sweet taste receptors. Cell 2001, 106, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Staszewski, L.; Xu, H.; Durick, K.; Zoller, M.; Adler, E. Human receptors for sweet and umami taste. Proc. Natl. Acad. Sci. USA 2002, 99, 4692–4696. [Google Scholar] [CrossRef] [Green Version]
- Nelson, G.; Chandrashekar, J.; Hoon, M.A.; Feng, L.; Zhao, G.; Ryba, N.J.; Zuker, C.S. An amino-acid taste receptor. Nature 2002, 416, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Adler, E.; Hoon, M.A.; Mueller, K.L.; Chandrashekar, J.; Ryba, N.J.; Zuker, C.S. A novel family of mammalian taste receptors. Cell 2000, 100, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, J.; Mueller, K.L.; Hoon, M.A.; Adler, E.; Feng, L.; Guo, W.; Zuker, C.S.; Ryba, N.J. T2Rs function as bitter taste receptors. Cell 2000, 100, 703–711. [Google Scholar] [CrossRef] [Green Version]
- Matsunami, H.; Montmayeur, J.P.; Buck, L.B. A family of candidate taste receptors in human and mouse. Nature 2000, 404, 601–604. [Google Scholar] [CrossRef]
- Meyerhof, W.; Batram, C.; Kuhn, C.; Brockhoff, A.; Chudoba, E.; Bufe, B.; Appendino, G.; Behrens, M. The molecular receptive ranges of human TAS2R bitter taste receptors. Chem. Senses 2010, 35, 157–170. [Google Scholar] [CrossRef]
- Behrens, M.; Meyerhof, W. Vertebrate Bitter Taste Receptors: Keys for Survival in Changing Environments. J. Agric. Food Chem. 2018, 66, 2204–2213. [Google Scholar] [CrossRef]
- Behrens, M.; Korsching, S.I.; Meyerhof, W. Tuning properties of avian and frog bitter taste receptors dynamically fit gene repertoire sizes. Mol. Biol. Evol. 2014, 31, 3216–3227. [Google Scholar] [CrossRef] [Green Version]
- Lossow, K.; Hubner, S.; Roudnitzky, N.; Slack, J.P.; Pollastro, F.; Behrens, M.; Meyerhof, W. Comprehensive Analysis of Mouse Bitter Taste Receptors Reveals Different Molecular Receptive Ranges for Orthologous Receptors in Mice and Humans. J. Biol. Chem. 2016, 291, 15358–15377. [Google Scholar] [CrossRef] [Green Version]
- Lei, W.; Ravoninjohary, A.; Li, X.; Margolskee, R.F.; Reed, D.R.; Beauchamp, G.K.; Jiang, P. Functional Analyses of Bitter Taste Receptors in Domestic Cats (Felis catus). PLoS ONE 2015, 10, 0139670. [Google Scholar] [CrossRef] [Green Version]
- Sandau, M.M.; Goodman, J.R.; Thomas, A.; Rucker, J.B.; Rawson, N.E. A functional comparison of the domestic cat bitter receptors Tas2r38 and Tas2r43 with their human orthologs. BMC Neurosci. 2015, 16, 33. [Google Scholar] [CrossRef] [Green Version]
- Bufe, B.; Hofmann, T.; Krautwurst, D.; Raguse, J.D.; Meyerhof, W. The human TAS2R16 receptor mediates bitter taste in response to beta-glucopyranosides. Nat. Genet. 2002, 32, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.R.; Blank, K.; See Hoe, L.E.; Behrens, M.; Meyerhof, W.; Peart, J.N.; Thomas, W.G. Bitter taste receptor agonists elicit G-protein-dependent negative inotropy in the murine heart. Faseb J. 2014, 28, 4497–4508. [Google Scholar] [CrossRef] [PubMed]
- Oike, H.; Nagai, T.; Furuyama, A.; Okada, S.; Aihara, Y.; Ishimaru, Y.; Marui, T.; Matsumoto, I.; Misaka, T.; Abe, K. Characterization of ligands for fish taste receptors. J. Neurosci. 2007, 27, 5584–5592. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Wang, Y.; Zhang, L.; Jiang, P.; Zhao, H. Lineage-specific duplication and adaptive evolution of bitter taste receptor genes in bats. Mol. Ecol. 2018, 27, 4475–4488. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Suzuki, N.; Ishimaru, Y.; Sakurai, T.; Yin, L.; Pan, W.; Abe, K.; Misaka, T.; Hirai, H. Functional diversity of bitter taste receptor TAS2R16 in primates. Biol. Lett. 2012, 8, 652–656. [Google Scholar] [CrossRef]
- Itoigawa, A.; Hayakawa, T.; Suzuki-Hashido, N.; Imai, H. A natural point mutation in the bitter taste receptor TAS2R16 causes inverse agonism of arbutin in lemur gustation. Proc. Biol. Sci. 2019, 286, 20190884. [Google Scholar] [CrossRef] [Green Version]
- Purba, L.H.; Widayati, K.A.; Tsutsui, K.; Suzuki-Hashido, N.; Hayakawa, T.; Nila, S.; Suryobroto, B.; Imai, H. Functional characterization of the TAS2R38 bitter taste receptor for phenylthiocarbamide in colobine monkeys. Biol. Lett. 2017, 13. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Sugawara, T.; Matsui, A.; Go, Y.; Hirai, H.; Imai, H. Identification of non-taster Japanese macaques for a specific bitter taste. Primates 2010, 51, 285–289. [Google Scholar] [CrossRef]
- Tsutsui, K.; Otoh, M.; Sakurai, K.; Suzuki-Hashido, N.; Hayakawa, T.; Misaka, T.; Ishimaru, Y.; Aureli, F.; Melin, A.D.; Kawamura, S.; et al. Variation in ligand responses of the bitter taste receptors TAS2R1 and TAS2R4 among New World monkeys. Bmc. Evol. Biol. 2016, 16, 208. [Google Scholar] [CrossRef] [Green Version]
- Wooding, S.; Bufe, B.; Grassi, C.; Howard, M.T.; Stone, A.C.; Vazquez, M.; Dunn, D.M.; Meyerhof, W.; Weiss, R.B.; Bamshad, M.J. Independent evolution of bitter-taste sensitivity in humans and chimpanzees. Nature 2006, 440, 930–934. [Google Scholar] [CrossRef]
- Di Pizio, A.; Levit, A.; Slutzki, M.; Behrens, M.; Karaman, R.; Niv, M.Y. Comparing Class A GPCRs to bitter taste receptors: Structural motifs, ligand interactions and agonist-to-antagonist ratios. Methods Cell Biol. 2016, 132, 401–427. [Google Scholar] [PubMed]
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharm. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronin, A.N.; Tang, H.; Connor, J.; Keung, W. Identification of ligands for two human bitter T2R receptors. Chem. Senses 2004, 29, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floriano, W.B.; Hall, S.; Vaidehi, N.; Kim, U.; Drayna, D.; Goddard, W.A., 3rd. Modeling the human PTC bitter-taste receptor interactions with bitter tastants. J. Mol. Model 2006, 12, 931–941. [Google Scholar] [CrossRef]
- Miguet, L.; Zhang, Z.; Grigorov, M.G. Computational studies of ligand-receptor interactions in bitter taste receptors. J. Recept. Signal Transduct. Res. 2006, 26, 611–630. [Google Scholar] [CrossRef]
- Brockhoff, A.; Behrens, M.; Niv, M.Y.; Meyerhof, W. Structural requirements of bitter taste receptor activation. Proc. Natl. Acad Sci. USA 2010, 107, 11110–11115. [Google Scholar] [CrossRef] [Green Version]
- Di Pizio, A.; Kruetzfeldt, L.M.; Cheled-Shoval, S.; Meyerhof, W.; Behrens, M.; Niv, M.Y. Ligand binding modes from low resolution GPCR models and mutagenesis: Chicken bitter taste receptor as a test-case. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Behrens, M.; Briand, L.; de March, C.A.; Matsunami, H.; Yamashita, A.; Meyerhof, W.; Weyand, S. Structure-Function Relationships of Olfactory and Taste Receptors. Chem. Senses 2018, 43, 81–87. [Google Scholar] [CrossRef]
- Loconto, J.; Papes, F.; Chang, E.; Stowers, L.; Jones, E.P.; Takada, T.; Kumanovics, A.; Fischer Lindahl, K.; Dulac, C. Functional expression of murine V2R pheromone receptors involves selective association with the M10 and M1 families of MHC class Ib molecules. Cell 2003, 112, 607–618. [Google Scholar] [CrossRef] [Green Version]
- McClintock, T.S.; Landers, T.M.; Gimelbrant, A.A.; Fuller, L.Z.; Jackson, B.A.; Jayawickreme, C.K.; Lerner, M.R. Functional expression of olfactory-adrenergic receptor chimeras and intracellular retention of heterologously expressed olfactory receptors. Brain Res. Mol. Brain Res. 1997, 48, 270–278. [Google Scholar] [CrossRef]
- Reichling, C.; Meyerhof, W.; Behrens, M. Functions of human bitter taste receptors depend on N-glycosylation. J. Neurochem. 2008, 106, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Krautwurst, D.; Yau, K.W.; Reed, R.R. Identification of ligands for olfactory receptors by functional expression of a receptor library. Cell 1998, 95, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Ammon, C.; Schafer, J.; Kreuzer, O.J.; Meyerhof, W. Presence of a plasma membrane targeting sequence in the amino-terminal region of the rat somatostatin receptor 3. Arch. Physiol. Biochem. 2002, 110, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Kubota, M.; Roberts, R.W.; Chi, Q.; Matsunami, H. RTP family members induce functional expression of mammalian odorant receptors. Cell 2004, 119, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Behrens, M.; Bartelt, J.; Reichling, C.; Winnig, M.; Kuhn, C.; Meyerhof, W. Members of RTP and REEP gene families influence functional bitter taste receptor expression. J. Biol. Chem. 2006, 281, 20650–20659. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, J.D.; Chakraborty, R.; Shaik, F.A.; Jaggupilli, A.; Bhullar, R.P.; Chelikani, P. The Pharmacochaperone Activity of Quinine on Bitter Taste Receptors. PLoS ONE 2016, 11, 0156347. [Google Scholar] [CrossRef]
- Milligan, G. G protein-coupled receptor dimerization: Function and ligand pharmacology. Mol. Pharm. 2004, 66, 1–7. [Google Scholar] [CrossRef]
- Kuhn, C.; Bufe, B.; Batram, C.; Meyerhof, W. Oligomerization of TAS2R bitter taste receptors. Chem. Senses 2010, 35, 395–406. [Google Scholar] [CrossRef]
- Kuhn, C.; Meyerhof, W. Oligomerization of sweet and bitter taste receptors. Methods Cell Biol. 2013, 117, 229–242. [Google Scholar]
- Behrens, M.; Brockhoff, A.; Kuhn, C.; Bufe, B.; Winnig, M.; Meyerhof, W. The human taste receptor hTAS2R14 responds to a variety of different bitter compounds. Biochem. Biophys. Res. Commun. 2004, 319, 479–485. [Google Scholar] [CrossRef]
- Brockhoff, A.; Behrens, M.; Massarotti, A.; Appendino, G.; Meyerhof, W. Broad tuning of the human bitter taste receptor hTAS2R46 to various sesquiterpene lactones, clerodane and labdane diterpenoids, strychnine, and denatonium. J. Agric Food Chem. 2007, 55, 6236–6243. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, S.; Behrens, M.; Meyerhof, W. Major haplotypes of the human bitter taste receptor TAS2R41 encode functional receptors for chloramphenicol. Biochem. Biophys. Res. Commun. 2013, 435, 267–273. [Google Scholar] [CrossRef]
- Behrens, M. Bitter Taste. Reference Module in Neuroscience and Biobehavioral Psychology, 2020. [Google Scholar] [CrossRef]
- Behrens, M.; Meyerhof, W. A role for taste receptors in (neuro)endocrinology? J. Neuroendocrinol. 2019, 31, 12691. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.; Somoza, V. Gastrointestinal taste receptors: Could tastants become drugs? Curr. Opin. Endocrinol Diabetes Obes. 2020, 27, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Zhang, C.H.; Lifshitz, L.M.; ZhuGe, R. Extraoral bitter taste receptors in health and disease. J. Gen. Physiol. 2017, 149, 181–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.N.; Workman, A.D.; Cohen, N.A. Role of Taste Receptors as Sentinels of Innate Immunity in the Upper Airway. J. Pathog. 2018, 2018, 9541987. [Google Scholar] [CrossRef] [Green Version]
- Shaik, F.A.; Singh, N.; Arakawa, M.; Duan, K.; Bhullar, R.P.; Chelikani, P. Bitter taste receptors: Extraoral roles in pathophysiology. Int. J. Biochem. Cell Biol. 2016, 77, 197–204. [Google Scholar]
- Steensels, S.; Depoortere, I. Chemoreceptors in the Gut. Annu. Rev. Physiol. 2018, 80, 117–141. [Google Scholar]
- Behrens, M.; Meyerhof, W. Bitter taste receptors and human bitter taste perception. Cell Mol. Life Sci. 2006, 63, 1501–1509. [Google Scholar]
- Kinnamon, S.; Finger, T. Recent advances in taste transduction and signaling [version 1; peer review: 2 approved]. F1000Research 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Roper, S.D. Signal transduction and information processing in mammalian taste buds. Pflug. Arch. 2007, 454, 759–776. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.K.; McKinnon, P.J.; Margolskee, R.F. Gustducin is a taste-cell-specific G protein closely related to the transducins. Nature 1992, 357, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Rossler, P.; Boekhoff, I.; Tareilus, E.; Beck, S.; Breer, H.; Freitag, J. G protein betagamma complexes in circumvallate taste cells involved in bitter transduction. Chem. Senses 2000, 25, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Shanker, Y.G.; Dubauskaite, J.; Zheng, J.Z.; Yan, W.; Rosenzweig, S.; Spielman, A.I.; Max, M.; Margolskee, R.F. Ggamma13 colocalizes with gustducin in taste receptor cells and mediates IP3 responses to bitter denatonium. Nat. Neurosci. 1999, 2, 1055–1062. [Google Scholar] [PubMed]
- Rossler, P.; Kroner, C.; Freitag, J.; Noe, J.; Breer, H. Identification of a phospholipase C beta subtype in rat taste cells. Eur. J. Cell Biol. 1998, 77, 253–261. [Google Scholar] [PubMed]
- Clapp, T.R.; Stone, L.M.; Margolskee, R.F.; Kinnamon, S.C. Immunocytochemical evidence for co-expression of Type III IP3 receptor with signaling components of bitter taste transduction. BMC Neurosci. 2001, 2, 6. [Google Scholar]
- Ogura, T.; Mackay-Sim, A.; Kinnamon, S.C. Bitter taste transduction of denatonium in the mudpuppy Necturus maculosus. J. Neurosci. 1997, 17, 3580–3587. [Google Scholar]
- Perez, C.A.; Huang, L.; Rong, M.; Kozak, J.A.; Preuss, A.K.; Zhang, H.; Max, M.; Margolskee, R.F. A transient receptor potential channel expressed in taste receptor cells. Nat. Neurosci. 2002, 5, 1169–1176. [Google Scholar] [CrossRef]
- Gao, N.; Lu, M.; Echeverri, F.; Laita, B.; Kalabat, D.; Williams, M.E.; Hevezi, P.; Zlotnik, A.; Moyer, B.D. Voltage-gated sodium channels in taste bud cells. BMC Neurosci. 2009, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Finger, T.E.; Danilova, V.; Barrows, J.; Bartel, D.L.; Vigers, A.J.; Stone, L.; Hellekant, G.; Kinnamon, S.C. ATP signaling is crucial for communication from taste buds to gustatory nerves. Science 2005, 310, 1495–1499. [Google Scholar] [CrossRef]
- Taruno, A.; Vingtdeux, V.; Ohmoto, M.; Ma, Z.; Dvoryanchikov, G.; Li, A.; Adrien, L.; Zhao, H.; Leung, S.; Abernethy, M.; et al. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature 2013, 495, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Nayak, A.P.; Shah, S.D.; Michael, J.V.; Deshpande, D.A. Bitter Taste Receptors for Asthma Therapeutics. Front. Physiol. 2019, 10, 884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kok, B.P.; Galmozzi, A.; Littlejohn, N.K.; Albert, V.; Godio, C.; Kim, W.; Kim, S.M.; Bland, J.S.; Grayson, N.; Fang, M.; et al. Intestinal bitter taste receptor activation alters hormone secretion and imparts metabolic benefits. Mol. Metab. 2018, 16, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Shaik, F.A.; Myal, Y.; Chelikani, P. Chemosensory bitter taste receptors T2R4 and T2R14 activation attenuates proliferation and migration of breast cancer cells. Mol. Cell Biochem. 2020, 465, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Brisson, A.; Unwin, P.N. Quaternary structure of the acetylcholine receptor. Nature 1985, 315, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Dennis, J.C.; Aono, S.; Vodyanoy, V.J.; Morrison, E.E. Development, Morphology, and Functional Anatomy of the Olfactory Epithelium. In Handbook of Olfaction and Gustation, 3rd ed.; Doty, R.L., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 93–107. [Google Scholar]
- Stowers, L.; Spehr, M. The Vomeronasal Organ. In Handbook of Olfaction and Gustation; Doty, R.L., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 1113–1132. [Google Scholar]
- Witt, M.; Reutter, K. Anatomy of the Tongue and Taste Buds. In Handbook of Olfaction and Gustation; Doty, R.L., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 637–663. [Google Scholar]
- Son, M.; Kim, D.; Ko, H.J.; Hong, S.; Park, T.H. A portable and multiplexed bioelectronic sensor using human olfactory and taste receptors. Biosens. Bioelectron. 2017, 87, 901–907. [Google Scholar] [CrossRef]
- Maîtrepierre, E.; Sigoillot, M.; Le Pessot, L.; Briand, L. Recombinant expression, in vitro refolding, and biophysical characterization of the N-terminal domain of T1R3 taste receptor. Protein Expr. Purif. 2012, 83, 75–83. [Google Scholar] [CrossRef]
- Nie, Y.; Vigues, S.; Hobbs, J.R.; Conn, G.L.; Munger, S.D. Distinct contributions of T1R2 and T1R3 taste receptor subunits to the detection of sweet stimuli. Curr. Biol. 2005, 15, 1948–1952. [Google Scholar] [CrossRef]
- Levit, A.; Barak, D.; Behrens, M.; Meyerhof, W.; Niv, M.Y. Homology model-assisted elucidation of binding sites in GPCRs. Methods Mol. Biol. 2012, 914, 179–205. [Google Scholar]
- Goddard, W.A., 3rd; Abrol, R. 3-Dimensional structures of G protein-coupled receptors and binding sites of agonists and antagonists. J. Nutr. 2007, 137, 1528–1538. [Google Scholar] [CrossRef]
- Behrens, M.; Meyerhof, W. Bitter taste receptor research comes of age: From characterization to modulation of TAS2Rs. Semin Cell Dev. Biol. 2013, 24, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Dagan-Wiener, A.; Di Pizio, A.; Nissim, I.; Bahia, M.S.; Dubovski, N.; Margulis, E.; Niv, M.Y. BitterDB: Taste ligands and receptors database in 2019. Nucleic Acids Res. 2019, 47, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Di Pizio, A.; Niv, M.Y. Promiscuity and selectivity of bitter molecules and their receptors. Bioorg. Med. Chem. 2015, 23, 4082–4091. [Google Scholar] [CrossRef]
- Sandal, M.; Behrens, M.; Brockhoff, A.; Musiani, F.; Giorgetti, A.; Carloni, P.; Meyerhof, W. Evidence for a Transient Additional Ligand Binding Site in the TAS2R46 Bitter Taste Receptor. J. Chem. Theory. Comput. 2015, 11, 4439–4449. [Google Scholar] [CrossRef] [PubMed]
- Born, S.; Levit, A.; Niv, M.Y.; Meyerhof, W.; Behrens, M. The human bitter taste receptor TAS2R10 is tailored to accommodate numerous diverse ligands. J. Neurosci. 2013, 33, 201–213. [Google Scholar] [CrossRef]
- Nowak, S.; Di Pizio, A.; Levit, A.; Niv, M.Y.; Meyerhof, W.; Behrens, M. Reengineering the ligand sensitivity of the broadly tuned human bitter taste receptor TAS2R14. J. Biochim. Biophys. Acta. Gen. Sub. 2018, 1862, 2162–2173. [Google Scholar] [CrossRef]
- Slack, J.P.; Brockhoff, A.; Batram, C.; Menzel, S.; Sonnabend, C.; Born, S.; Galindo, M.M.; Kohl, S.; Thalmann, S.; Ostopovici-Halip, L.; et al. Modulation of bitter taste perception by a small molecule hTAS2R antagonist. Curr. Biol. 2010, 20, 1104–1109. [Google Scholar] [CrossRef] [Green Version]
- Dunkel, A.; Hofmann, T.; Di Pizio, A. In Silico Investigation of Bitter Hop-Derived Compounds and Their Cognate Bitter Taste Receptors. J. Agric. Food Chem. 2020. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Singh, N.; De Jesus, V.C.; Gounni, M.S.; Dhanaraj, P.; Chelikani, P. Chemosensory bitter taste receptors (T2Rs) are activated by multiple antibiotics. Faseb J. 2019, 33, 501–517. [Google Scholar] [CrossRef]
- Xue, A.Y.; Di Pizio, A.; Levit, A.; Yarnitzky, T.; Penn, O.; Pupko, T.; Niv, M.Y. Independent Evolution of Strychnine Recognition by Bitter Taste Receptor Subtypes. Front. Mol. Biosci. 2018, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaman, R.; Nowak, S.; Di Pizio, A.; Kitaneh, H.; Abu-Jaish, A.; Meyerhof, W.; Niv, M.Y.; Behrens, M. Probing the Binding Pocket of the Broadly Tuned Human Bitter Taste Receptor TAS2R14 by Chemical Modification of Cognate Agonists. Chem. Biol. Drug Des. 2016, 88, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Ley, J.P.; Dessoy, M.; Paetz, S.; Blings, M.; Hoffmann-Lücke, P.; Reichelt, K.V.; Krammer, G.E.; Pienkny, S.; Brandt, W.; Wessjohann, L. Identification of enterodiol as a masker for caffeine bitterness by using a pharmacophore model based on structural analogues of homoeriodictyol. J. Agric. Food Chem. 2012, 60, 6303–6311. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.; Redel, U.; Blank, K.; Meyerhof, W. The human bitter taste receptor TAS2R7 facilitates the detection of bitter salts. Biochem. Biophys. Res. Commun. 2019, 512, 877–881. [Google Scholar] [CrossRef]
- Wang, Y.; Zajac, A.L.; Lei, W.; Christensen, C.M.; Margolskee, R.F.; Bouysset, C.; Golebiowski, J.; Zhao, H.; Fiorucci, S.; Jiang, P. Metal Ions Activate the Human Taste Receptor TAS2R7. Chem. Senses 2019, 44, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Jaggupilli, A.; Premnath, D.; Chelikani, P. Plasticity of the ligand binding pocket in the bitter taste receptor T2R7. Biochim. Biophys. Acta Biomembr. 2018, 1860, 991–999. [Google Scholar] [CrossRef]
- Chen, Z.; Dong, S.; Meng, F.; Liang, Y.; Zhang, S.; Sun, J. Insights into the binding of agonist and antagonist to TAS2R16 receptor: A molecular simulation study. Mol. Simul. 2018, 44, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, T.; Misaka, T.; Ishiguro, M.; Masuda, K.; Sugawara, T.; Ito, K.; Kobayashi, T.; Matsuo, S.; Ishimaru, Y.; Asakura, T.; et al. Characterization of the beta-D-glucopyranoside binding site of the human bitter taste receptor hTAS2R16. J. Biol. Chem. 2010, 285, 28373–28378. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, T.; Misaka, T.; Ueno, Y.; Ishiguro, M.; Matsuo, S.; Ishimaru, Y.; Asakura, T.; Abe, K. The human bitter taste receptor, hTAS2R16, discriminates slight differences in the configuration of disaccharides. Biochem. Biophys. Res. Commun. 2010, 402, 595–601. [Google Scholar] [CrossRef]
- Thomas, A.; Sulli, C.; Davidson, E.; Berdougo, E.; Phillips, M.; Puffer, B.A.; Paes, C.; Doranz, B.J.; Rucker, J.B. The Bitter Taste Receptor TAS2R16 Achieves High Specificity and Accommodates Diverse Glycoside Ligands by using a Two-faced Binding Pocket. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Fierro, F.; Giorgetti, A.; Carloni, P.; Meyerhof, W.; Alfonso-Prieto, M. Dual binding mode of “bitter sugars” to their human bitter taste receptor target. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biarnes, X.; Marchiori, A.; Giorgetti, A.; Lanzara, C.; Gasparini, P.; Carloni, P.; Born, S.; Brockhoff, A.; Behrens, M.; Meyerhof, W. Insights into the binding of Phenyltiocarbamide (PTC) agonist to its target human TAS2R38 bitter receptor. PLoS ONE 2010, 5, 12394. [Google Scholar] [CrossRef] [PubMed]
- Marchiori, A.; Capece, L.; Giorgetti, A.; Gasparini, P.; Behrens, M.; Carloni, P.; Meyerhof, W. Coarse-grained/molecular mechanics of the TAS2R38 bitter taste receptor: Experimentally-validated detailed structural prediction of agonist binding. PLoS ONE 2013, 8, 64675. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Abrol, R.; Trzaskowski, B.; Goddard, W.A., 3rd. 3D structure prediction of TAS2R38 bitter receptors bound to agonists phenylthiocarbamide (PTC) and 6-n-propylthiouracil (PROP). J. Chem. Inf. Model. 2012, 52, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, J.; Pydi, S.P.; Singh, N.; Aluko, R.E.; Chelikani, P. Bitter taste receptor T2R1 is activated by dipeptides and tripeptides. Biochem. Biophys. Res. Commun. 2010, 398, 331–335. [Google Scholar] [CrossRef]
- Stoeger, V.; Holik, A.K.; Hölz, K.; Dingjan, T.; Hans, J.; Ley, J.P.; Krammer, G.E.; Niv, M.Y.; Somoza, M.M.; Somoza, V. Bitter-Tasting Amino Acids l-Arginine and l-Isoleucine Differentially Regulate Proton Secretion via T2R1 Signaling in Human Parietal Cells in Culture. J. Agric. Food. Chem. 2020, 68, 3434–3444. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; You, Z.; Zhou, H.; Zhang, J.; Hu, Y. Structure-function relationships of the human bitter taste receptor hTAS2R1: Insights from molecular modeling studies. J. Recept. Signal. Transduct. Res. 2011, 31, 229–240. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Singh, N.; Jesus, V.C.; Duan, K.; Chelikani, P. Characterization of the Binding Sites for Bacterial Acyl Homoserine Lactones (AHLs) on Human Bitter Taste Receptors (T2Rs). Acs. Infect. Dis. 2018, 4, 1146–1156. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Li, X.; Peng, S.; Wang, S.; Huang, C.Z.; Huang, C.Z.; Zhang, Q.; Li, D.; Jiang, J.; et al. Identification of a specific agonist of human TAS2R14 from Radix Bupleuri through virtual screening, functional evaluation and binding studies. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Singh, N.; Pydi, P.; Upadhyaya, J.; Chelikani, P. Structural basis of activation of bitter taste receptor T2R1 and comparison with class A GPCRs. J. Biol. Chem. 2011, 286, 36032–36041. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Hall, S.E.; Vaidehi, N. Agonist-induced conformational changes in bovine rhodopsin: Insight into activation of G-protein-coupled receptors. J. Mol. Biol. 2008, 382, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Pydi, S.P.; Bhullar, R.P.; Chelikani, P. Constitutively active mutant gives novel insights into the mechanism of bitter taste receptor activation. J. Neurochem. 2012, 122, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Pydi, S.P.; Bhullar, R.P.; Chelikani, P. Constitutive activity of bitter taste receptors (T2Rs). Adv. Pharm. 2014, 70, 303–326. [Google Scholar]
- Scheerer, P.; Park, J.H.; Hildebrand, P.W.; Kim, Y.J.; Krauss, N.; Choe, H.W.; Hofmann, K.P.; Ernst, O.P. Crystal structure of opsin in its G-protein-interacting conformation. Nature 2008, 455, 497–502. [Google Scholar] [CrossRef]
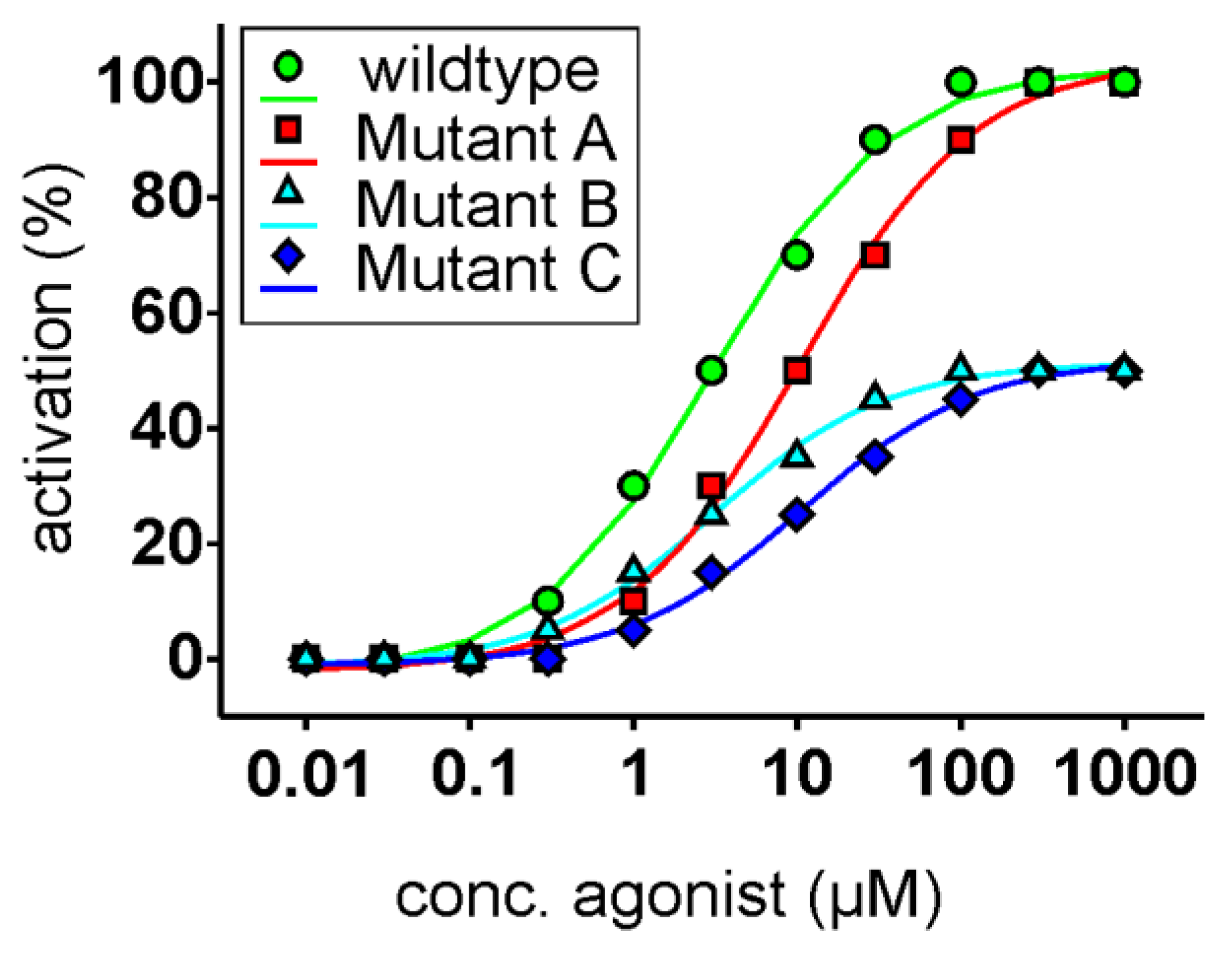
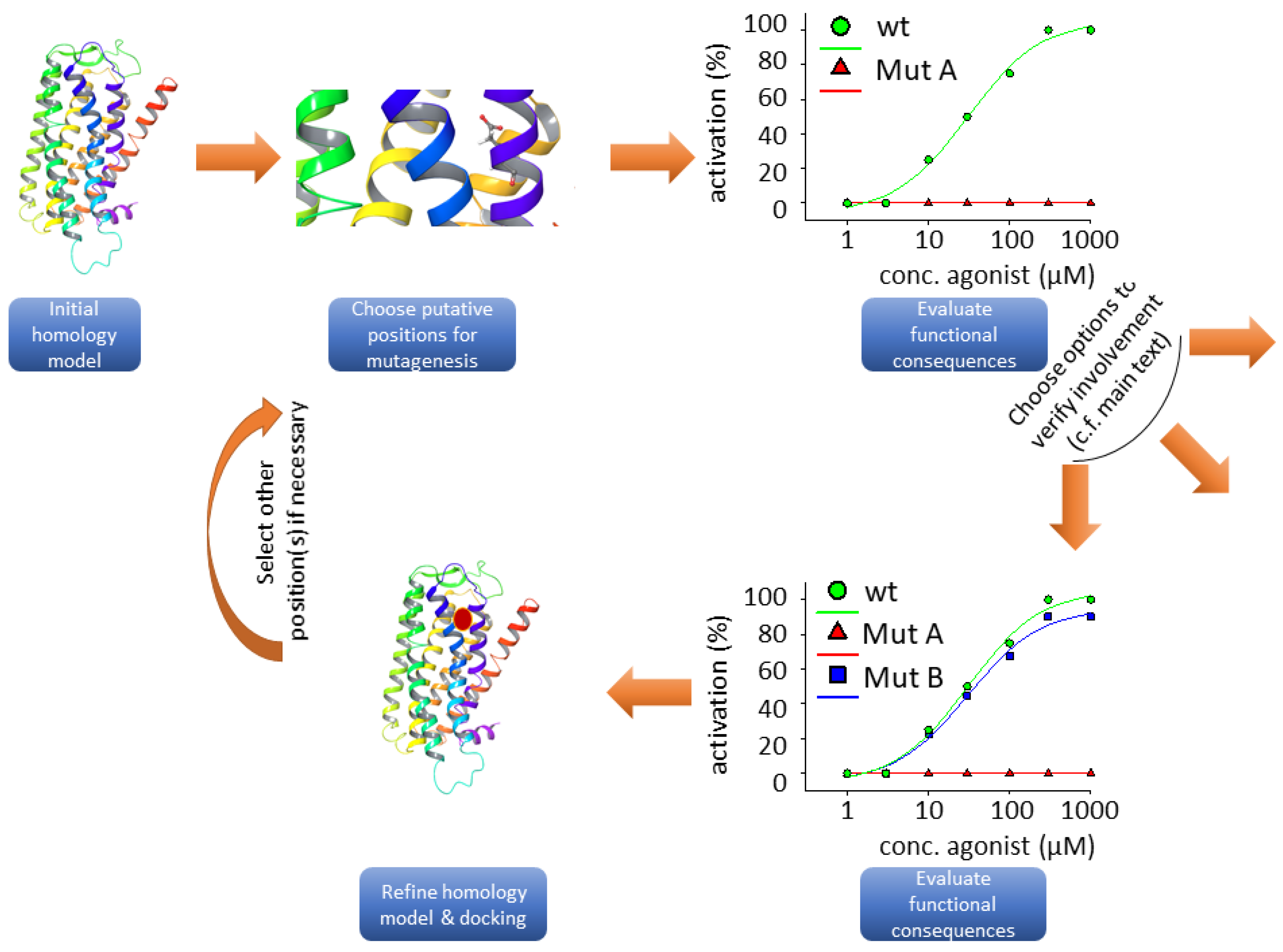

{kind=link}
{kind=link}
{kind=link}
| BW Pos. | TAS2R | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 4 | 7 | 10 | 14 | 16 | 20 | 38 | 40 | 46 | |
| 2.53 | L59 [105 #, 106 *] | |||||||||
| 2.57 | S63 [106 *] | |||||||||
| 2.60 | D65 [101 #] | H65 [95 #] | N65 [90] | |||||||
| 2.61 | N66 [110 *, 115] | W66 [92,94 *] | N67 [106 *] | W66 [37, 90, 96 *] | ||||||
| 2.65 | E70 [37, 90, 96 *] | |||||||||
| 2.66 | L71 [37, 90, 96 *] a | |||||||||
| ECL1 | E74 [110 *, 115] b | T74 [95] | ||||||||
| 3.24 | V77 [105 #] | |||||||||
| 3.25 | S81 [95] | I82 [37, 90, 96 *] c | ||||||||
| 3.28 | L85 [92] | L81 [105 #] | ||||||||
| 3.29 | C82 [111 *] | D86 [101 #] | S85 [91, 96 *] | T86 [92, 95 #] | T82 [105 #] | K98 [94 *] | Y85 [90] | |||
| 3.30 | N87 [92] | N86 [90] | ||||||||
| 3.32 | L85 [94 *] | W89 [101 #] | W88 [91] | W89 [92, 114 *] | W85 [106 *] | W88 [95 #] | T101 [94 *] | W88 [37, 90, 96 *] | ||
| 3.33 | L86 [94 *, 112 *] | V89 [91, 96 *] | T90 [92] | E86 [103, 106 *] | A89 [90] | |||||
| 3.35 | F88 [106 *] | |||||||||
| 3.36 | N89 [94 *, 110 *, 115 *d] | N92 [91, 96 *] | N93 [92, 94 *] | N89 [106 *] | N103 [107, 108] | N105 [94 *] | N92 [37, 90, 96 *] | |||
| 3.37 | E90 [110 *, 112 *] | H94 [100] | Q93 [96 *] | H94 [92] | I90 [105 #] | H93 [90] | ||||
| 3.39 | T92 [106 *] | |||||||||
| 3.40 | F93 [103 #, 106 *] | N96 [90] | ||||||||
| 3.41 | W94 [103 #] | W108 [109 *#] | ||||||||
| 3.42 | L109 [109 *#] | |||||||||
| 3.45 | C112 [109 *#] | |||||||||
| 4.60 | I140 [94 *] | F156 [94 *] | I147 [90] | |||||||
| 4.62 | S144 [105 #] | |||||||||
| 4.64 | H144 [94 *] | I148 [94 *, 95 #] | ||||||||
| 4.65 | N150 [37, 96 *] | |||||||||
| ECL2 | N148 [105] | |||||||||
| ECL2 | Q152 [95] | |||||||||
| ECL2 | S154 [95] | |||||||||
| ECL2 | R160 [95] | |||||||||
| ECL2 | R161 [95] | N161 [96 *] | ||||||||
| ECL2 | N163 [110 *] | R163 [95] | K163 [95] | |||||||
| ECL2 | A164 [111 *] | W164 [95] | ||||||||
| ECL2 | N165 [95] | |||||||||
| ECL2 | T166 [95] | I166 [95] | ||||||||
| ECL2 | N167 [101] | |||||||||
| ECL2 | K168 [112 *] | |||||||||
| ECL2 | T169 [101] | |||||||||
| ECL2 | W170 [101] | |||||||||
| 5.38 | S181 [101 #] | L178 [94 *] | S175 [90] | |||||||
| 5.39 | Q175 [94 *] | K174 [96 *] | I179 [95 #] | Q177 [103 #] | L194 [94 *] | N176 [37, 90, 96 *] | ||||
| 5.40 | Q175 [91, 96 *] | |||||||||
| 5.42 | S178 [94 *] | L177 [96 *] | T182 [92, 95 #] | F197 [108] | V179 [96 *] | |||||
| 5.43 | L178 [91, 96 *] | S183 [92] | H181 [103 #] | T180 [90] | ||||||
| 5.44 | Y199 [109 *#] | |||||||||
| 5.46 | E182 [94 *, 112 *] | F186 [92] | A184 [105 #] | W201 [108] | ||||||
| 5.47 | I187 [92] | N184 [90] | ||||||||
| 5.48 | V203 [109 *#] | |||||||||
| 5.49 | P204 [109 *#] | |||||||||
| 6.48 | Y237 [112 *] | Y240 [92] | F236 [106 *] | |||||||
| 6.49 | A241 [92] | |||||||||
| 6.51 | Y239 [91] | F243 [92, 94 *] | Y239 [106 *] | Y241 [37, 90, 96 *] | ||||||
| 6.52 | F240 [103 #] | S260 [108] | F242 [90] | |||||||
| 6.54 | I243 [94 *] | A262 [109 *#] | S244 [90] | |||||||
| 6.55 | K244 [94 *, 112 *] | F247 [92, 94 *] | I243 [103#] | A263 [109 *#] | L263 [44 *] | I245 [90] | ||||
| 6.56 | F264 [108] | |||||||||
| 6.57 | I165 [109 *#] | |||||||||
| 6.58 | Q249 [95] | S266 [109 *#] | S248 [90] | |||||||
| 6.59 | T255 [101 #] | V251 [95 #] | F249 [95#] | V249 [90] | ||||||
| 6.60 | S248 [111 *] | |||||||||
| 6.63 | E253 [37, 96 *] | |||||||||
| 7.32 | E264 [100] | |||||||||
| 7.35 | I258 [94 *] | K262 [95] | I262 [94 *] | L258 [105 #] | F261 [90] | |||||
| 7.36 | I263 [92] | |||||||||
| 7.38 | F261 [94 *] | W261 [105 #] | ||||||||
| 7.39 | F262 [94 *] | E271 [101 #] | M263 [96 *] | Q266 [92] | E262 [105 #, 106 *] | Q265 [95 #] | K282 [94 *] | E265 [37, 90, 96 *] | ||
| 7.42 | T266 [96 *] | G269 [92] | V265 [106 *] | A268 [37, 90, 96 *] | ||||||
| 7.43 | Y266 [106 *] | F269 [37, 90, 96 *] | ||||||||
| 7.45 | F268 [106 *] | |||||||||
| 7.46 | I269 [106 *] | |||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behrens, M.; Ziegler, F. Structure-Function Analyses of Human Bitter Taste Receptors—Where Do We Stand? Molecules 2020, 25, 4423. https://doi.org/10.3390/molecules25194423
Behrens M, Ziegler F. Structure-Function Analyses of Human Bitter Taste Receptors—Where Do We Stand? Molecules. 2020; 25(19):4423. https://doi.org/10.3390/molecules25194423
Chicago/Turabian StyleBehrens, Maik, and Florian Ziegler. 2020. "Structure-Function Analyses of Human Bitter Taste Receptors—Where Do We Stand?" Molecules 25, no. 19: 4423. https://doi.org/10.3390/molecules25194423