
Expanding the Scope of Orthogonal Translation with Pyrrolysyl-tRNA Synthetases Dedicated to Aromatic Amino Acids


 ,
,  , and
, and 
Abstract
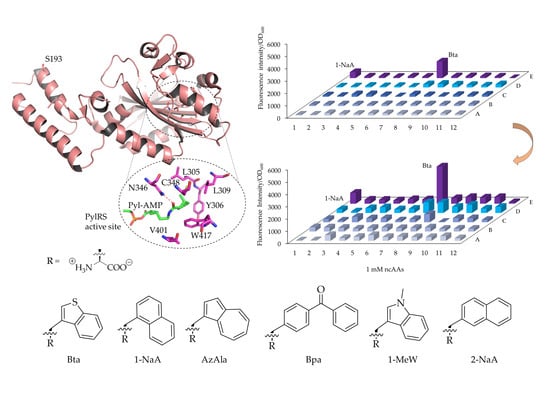
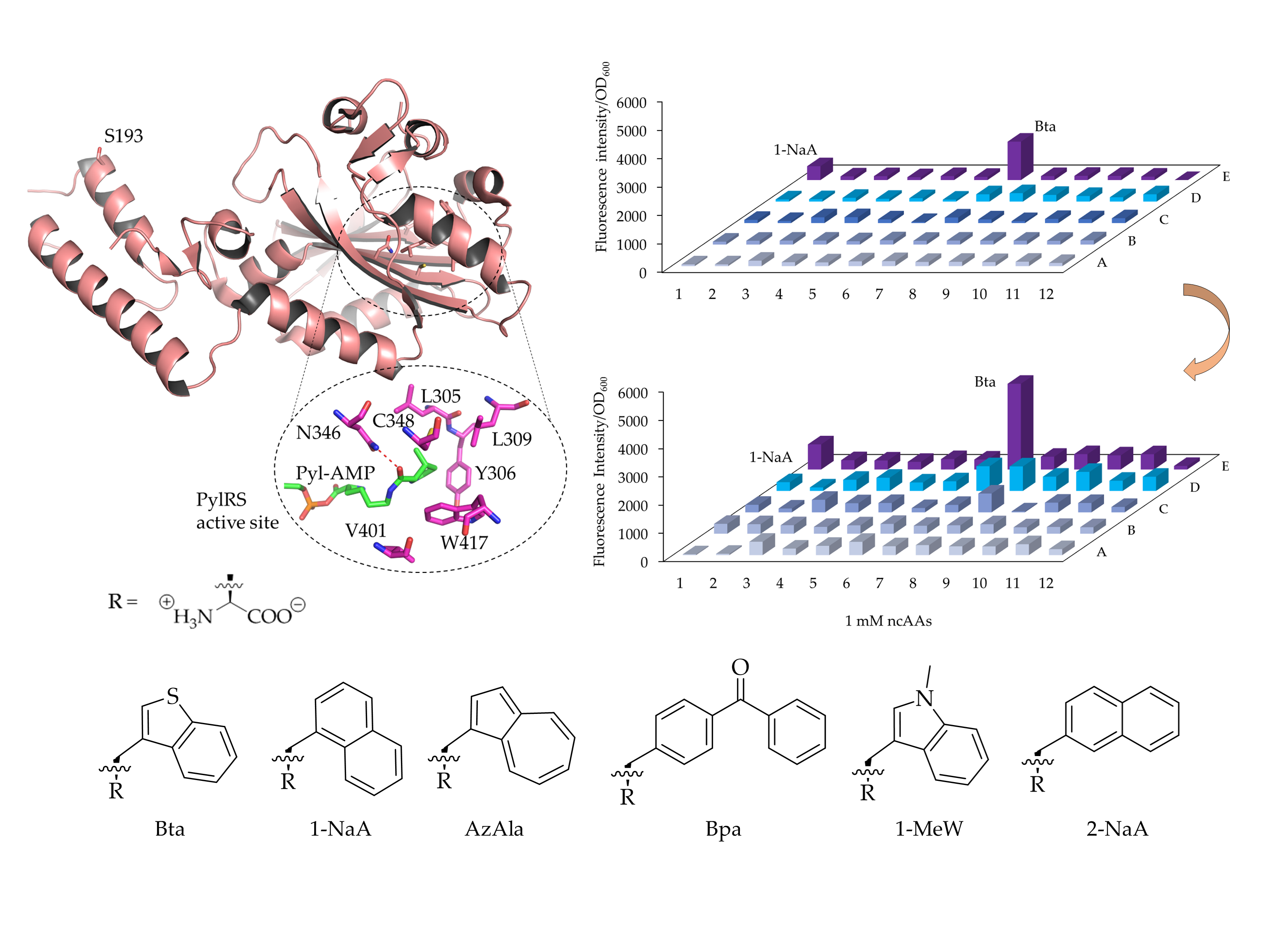{kind=link}
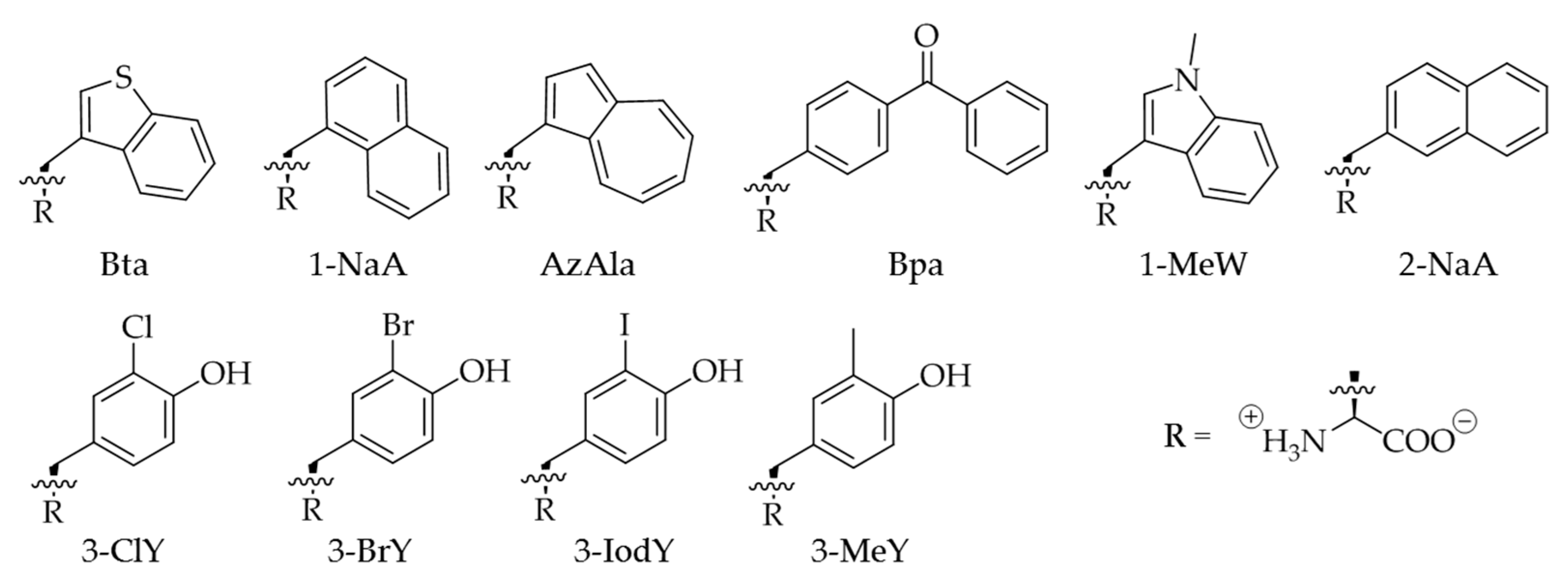{kind=link}
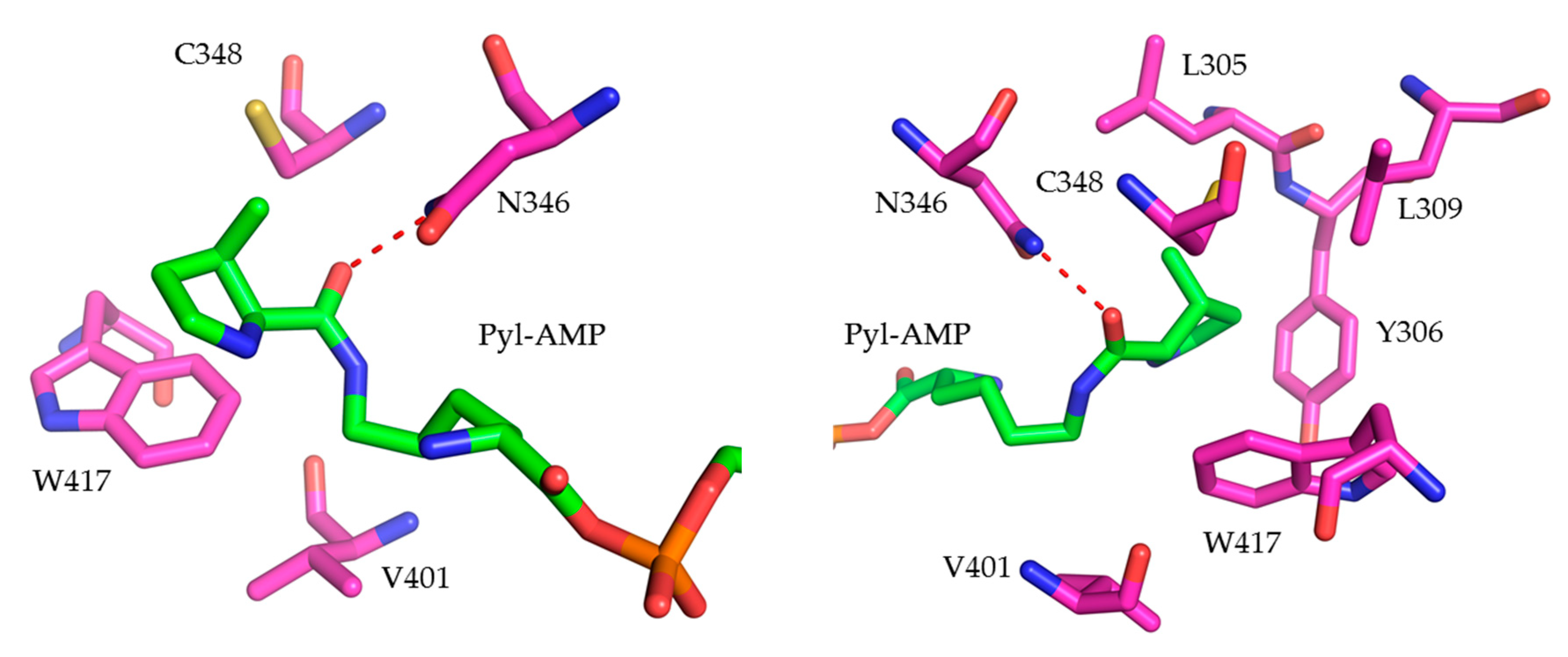{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Engineering MmPylRS Variants for Aromatic Non-Canonical Amino Acids
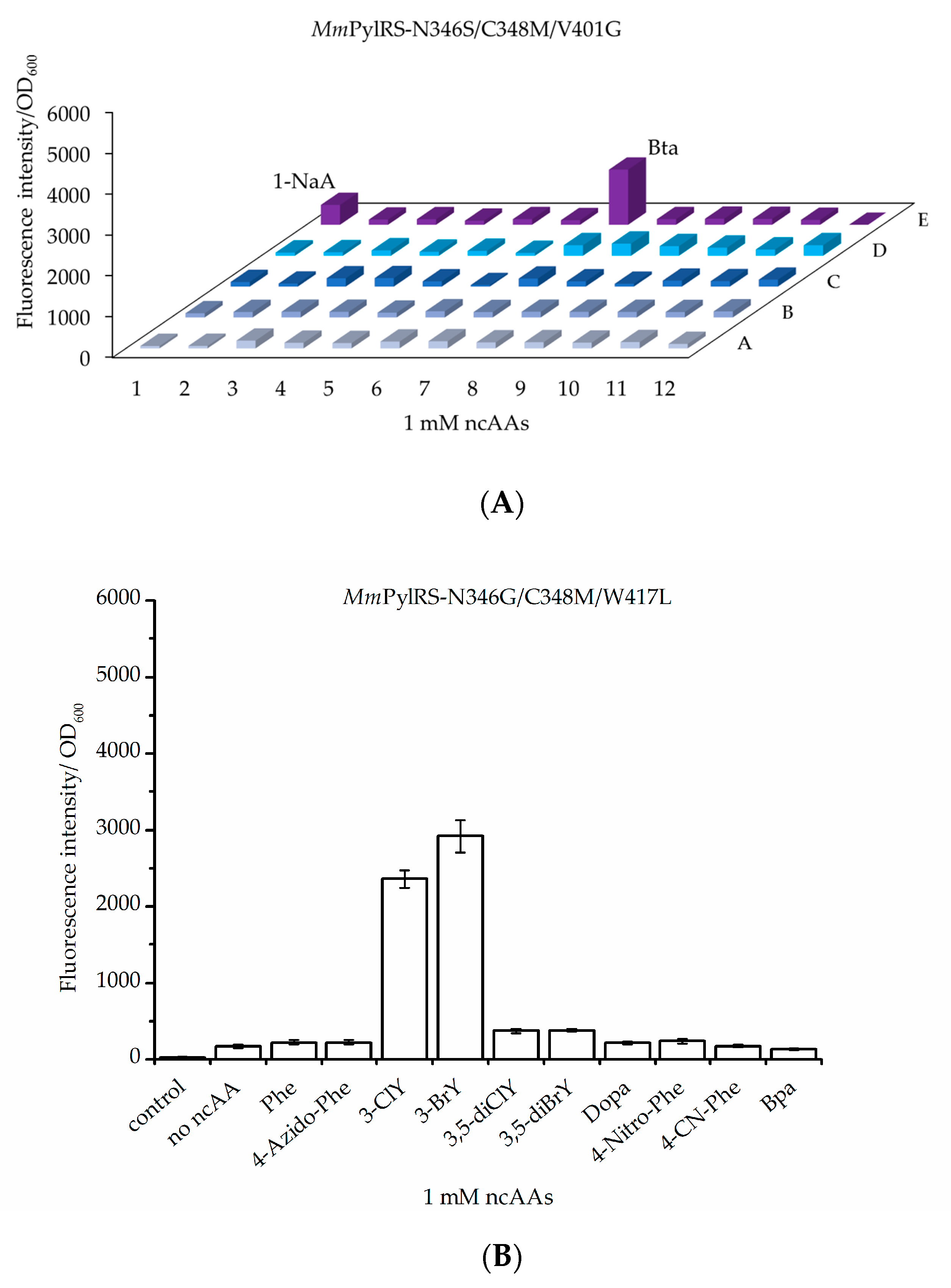
2.1.1. MmPylRS Mutagenesis Strategy and Screening towards Tryptophan and Tyrosine Analogs
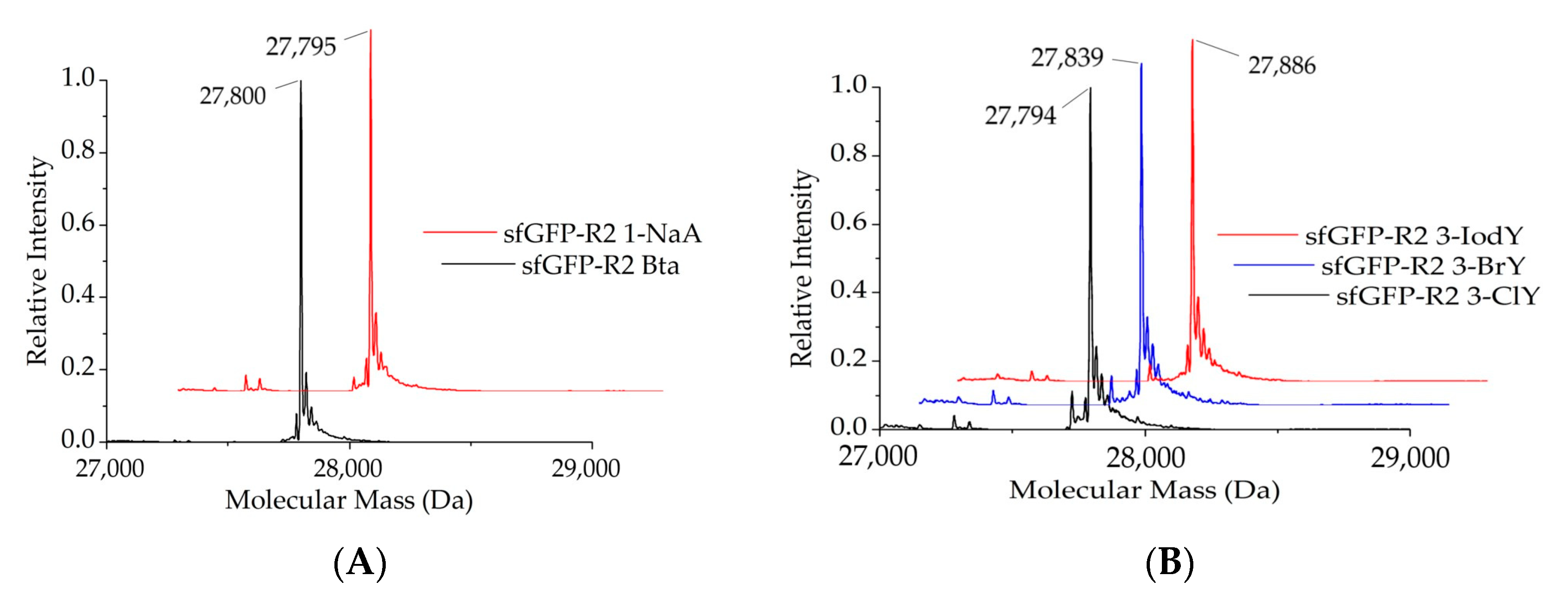
2.1.2. Intact Protein Mass Analysis of Aromatic ncAA Incorporation
2.2. Improvement of Amber Suppression Efficiency
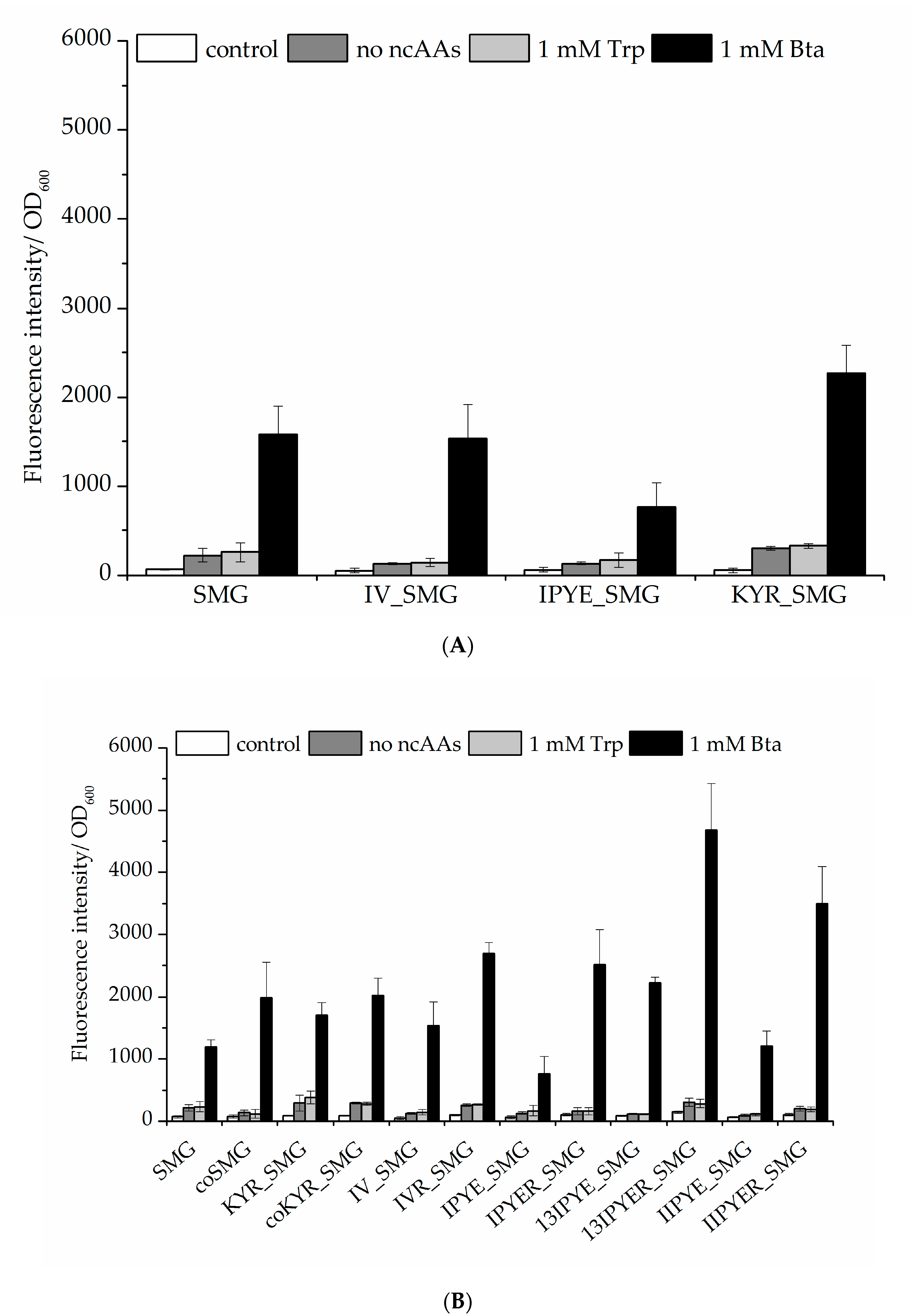
2.2.1. Bta Incorporation Efficiency Compared to Other MmPylRS Variants
2.2.2. Enhancing the Efficiency of Ribosomal Bta Incorporation and Target Protein Yields
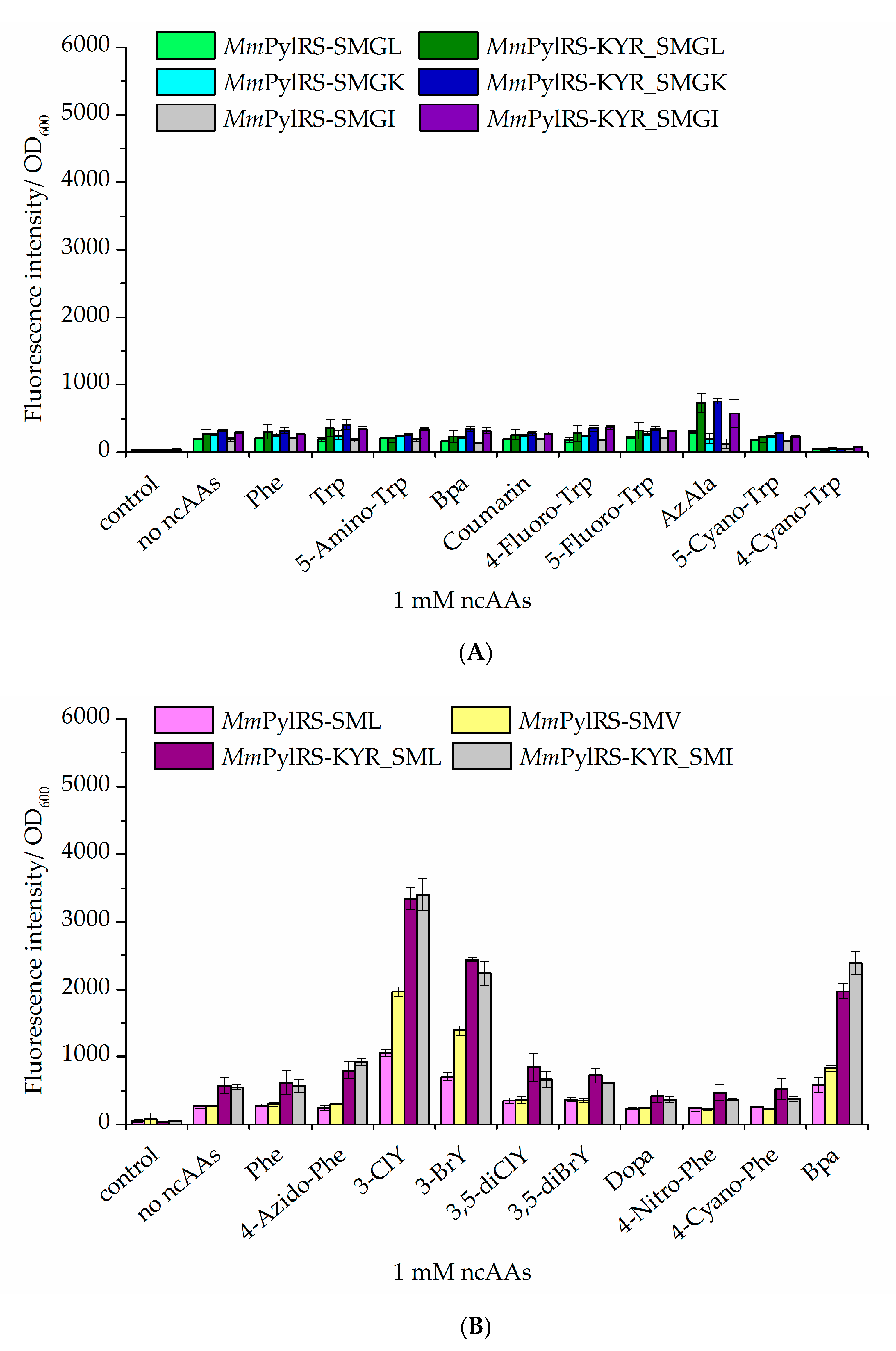
2.3. Improvement of Enzyme Efficiency Toward Other ncAAs
2.4. Evaluation of Amber Suppression Efficiency and Other Growth Media
3. Discussion
4. Materials and Methods
4.1. Canonical and Non-Canonical Amino Acids
4.2. Site-Directed Mutagenesis, Strains, and Screening Library Construction
4.3. Screening aaRS Variants for Activity
4.4. Protein Production and Purification
4.5. Mass Spectrometry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A

References
- Hammerling, M.J.; Krüger, A.; Jewett, M.C. Strategies for in vitro engineering of the translation machinery. Nucleic Acids Res. 2019, 48, 1068–1083. [Google Scholar] [CrossRef] [PubMed]
- Ros, E.; Torres, A.G.; Ribas de Pouplana, L. Learning from Nature to Expand the Genetic Code. Trends Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.M. Post-Translational Modifications of Protein Backbones: Unique Functions, Mechanisms, and Challenges. Biochemistry 2018, 57, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Renner, C.; Alefelder, S.; Bae, J.H.; Budisa, N.; Huber, R.; Moroder, L. Fluoroprolines as Tools for Protein Design and Engineering. Angew. Chem. Int. Ed. 2001, 40, 923–925. [Google Scholar] [CrossRef]
- Larregola, M.; Moore, S.; Budisa, N. Congeneric bio-adhesive mussel foot proteins designed by modified prolines revealed a chiral bias in unnatural translation. Biochem. Biophys. Res. Commun. 2012, 421, 646–650. [Google Scholar] [CrossRef]
- Doerfel, L.K.; Wohlgemuth, I.; Kubyshkin, V.; Starosta, A.L.; Wilson, D.N.; Budisa, N.; Rodnina, M.V. Entropic Contribution of Elongation Factor P to Proline Positioning at the Catalytic Center of the Ribosome. J. Am. Chem. Soc. 2015, 137, 12997–13006. [Google Scholar] [CrossRef]
- Kobayashi, T.; Yanagisawa, T.; Sakamoto, K.; Yokoyama, S. Recognition of Non-α-amino Substrates by Pyrrolysyl-tRNA Synthetase. J. Mol. Biol. 2009, 385, 1352–1360. [Google Scholar] [CrossRef]
- Srinivasan, G.; James, C.M.; Krzycki, J.A. Pyrrolysine Encoded by UAG in Archaea: Charging of a UAG-Decoding Specialized tRNA. Science 2002, 296, 1459. [Google Scholar] [CrossRef]
- Hao, B.; Zhao, G.; Kang, P.T.; Soares, J.A.; Ferguson, T.K.; Gallucci, J.; Krzycki, J.A.; Chan, M.K. Reactivity and chemical synthesis of L-pyrrolysine- the 22(nd) genetically encoded amino acid. Chem. Biol. 2004, 11, 1317–1324. [Google Scholar] [CrossRef]
- Brabham, R.; Fascione, M.A. Pyrrolysine Amber Stop-Codon Suppression: Development and Applications. Chembiochem 2017, 18, 1973–1983. [Google Scholar] [CrossRef]
- Polycarpo, C.R.; Herring, S.; Bérubé, A.; Wood, J.L.; Söll, D.; Ambrogelly, A. Pyrrolysine analogues as substrates for pyrrolysyl-tRNA synthetase. FEBS Lett. 2006, 580, 6695–6700. [Google Scholar] [CrossRef] [PubMed]
- Hoesl, M.G.; Budisa, N. In Vivo Incorporation of Multiple Noncanonical Amino Acids into Proteins. Angew. Chem. Int. Ed. 2011, 50, 2896–2902. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.-T.; Wang, Y.-S.; Nakamura, A.; Eiler, D.; Kavran, J.M.; Wong, M.; Kiessling, L.L.; Steitz, T.A.; O’Donoghue, P.; Söll, D. Polyspecific pyrrolysyl-tRNA synthetases from directed evolution. Proc. Natl. Acad. Sci. USA 2014, 111, 16724–16729. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Schultz, P.G. Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef]
- Uttamkumar, S.; Debnath, P.; Pinak, C. Environment of tryptophan side chains in proteins. Proteins 2000, 38, 288–300. [Google Scholar]
- O’Donoghue, P.; Ling, J.; Wang, Y.-S.; Söll, D. Upgrading protein synthesis for synthetic biology. Nat. Chem. Biol. 2013, 9, 594–598. [Google Scholar] [CrossRef]
- Agostini, F.; Völler, J.-S.; Koksch, B.; Acevedo-Rocha, C.G.; Kubyshkin, V.; Budisa, N. Biocatalysis with Unnatural Amino Acids: Enzymology Meets Xenobiology. Angew. Chem. Int. Ed. 2017, 56, 9680–9703. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Kobayashi, T.; Sakamoto, K.; Yokoyama, S. Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N(epsilon)-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem. Biol. 2008, 15, 1187–1197. [Google Scholar] [CrossRef]
- Schmidt, M.J.; Weber, A.; Pott, M.; Welte, W.; Summerer, D. Structural basis of furan-amino acid recognition by a polyspecific aminoacyl-tRNA-synthetase and its genetic encoding in human cells. Chembiochem 2014, 15, 1755–1760. [Google Scholar] [CrossRef]
- Ko, J.-h.; Wang, Y.-S.; Nakamura, A.; Guo, L.-T.; Söll, D.; Umehara, T. Pyrrolysyl-tRNA synthetase variants reveal ancestral aminoacylation function. FEBS Lett. 2013, 587, 3243–3248. [Google Scholar] [CrossRef]
- Lacey, V.K.; Louie, G.V.; Noel, J.P.; Wang, L. Expanding the library and substrate diversity of the pyrrolysyl-tRNA synthetase to incorporate unnatural amino acids containing conjugated rings. Chembiochem 2013, 14, 2100–2105. [Google Scholar] [CrossRef] [PubMed]
- Baumann, T.; Nickling, J.H.; Bartholomae, M.; Buivydas, A.; Kuipers, O.P.; Budisa, N. Prospects of In vivo Incorporation of Non-canonical Amino Acids for the Chemical Diversification of Antimicrobial Peptides. Front. Microbiol. 2017, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Baumann, T.; Hauf, M.; Richter, F.; Albers, S.; Möglich, A.; Ignatova, Z.; Budisa, N. Computational Aminoacyl-tRNA Synthetase Library Design for Photocaged Tyrosine. Int. J. Mol. Sci. 2019, 20, 2343. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, T.; Kuratani, M.; Seki, E.; Hino, N.; Sakamoto, K.; Yokoyama, S. Structural Basis for Genetic-Code Expansion with Bulky Lysine Derivatives by an Engineered Pyrrolysyl-tRNA Synthetase. Cell Chem. Biol. 2019, 26, 936–949. [Google Scholar] [CrossRef]
- Jennifer, M.; Kavran, S.G.; Patrick, O.; Markus, E.; Dieter, S. Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc. Natl. Acad. Sci. USA 2007, 104, 11268–11273. [Google Scholar]
- Englert, M.; Nakamura, A.; Wang, Y.-S.; Eiler, D.; Söll, D.; Guo, L.-T. Probing the active site tryptophan of Staphylococcus aureus thioredoxin with an analog. Nucleic Acids Res. 2015, 43, 11061–11067. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Russell, W.K.; Wang, Z.; Wan, W.; Dodd, L.E.; Pai, P.-J.; Russell, D.H.; Liu, W.R. The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of l-phenylalanine and its derivatives. Mol. Syst. Biol. 2011, 7, 714–717. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Fang, X.; Wallace, A.L.; Wu, B.; Liu, W.R. A rationally designed pyrrolysyl-tRNA synthetase mutant with a broad substrate spectrum. J. Am. Chem. Soc. 2012, 134, 2950–2953. [Google Scholar] [CrossRef]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the genetic code of Escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [CrossRef]
- Fan, C.; Ho, J.M.L.; Chirathivat, N.; Söll, D.; Wang, Y.-S. Exploring the substrate range of wild-type aminoacyl-tRNA synthetases. Chembiochem 2014, 15, 1805–1809. [Google Scholar] [CrossRef]
- Jiang, H.-K.; Wang, Y.-H.; Weng, J.-H.; Kurkute, P.; Li, C.-L.; Lee, M.-N.; Chen, P.-J.; Tseng, H.-W.; Tsai, M.-D.; Wang, Y.-S. Probing the Active Site of Deubiquitinase USP30 with Noncanonical Tryptophan Analogues. Biochemistry 2020. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Gu, X.; Cooley, R.B.; Porter, J.J.; Henson, R.L.; Willi, T.; DiDonato, J.A.; Hazen, S.L.; Mehl, R.A. Efficient Site-Specific Prokaryotic and Eukaryotic Incorporation of Halotyrosine Amino Acids into Proteins. ACS Chem. Biol. 2020, 15, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Smolskaya, S.; Andreev, Y.A. Site-Specific Incorporation of Unnatural Amino Acids into Escherichia coli Recombinant Protein: Methodology Development and Recent Achievement. Biomolecules 2019, 9, 255. [Google Scholar] [CrossRef] [PubMed]
- Bryson, D.I.; Fan, C.; Guo, L.-T.; Miller, C.; Söll, D.; Liu, D.R. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat. Chem. Biol. 2017, 13, 1253–1260. [Google Scholar] [CrossRef]
- Chatterjee, A.; Sun, S.B.; Furman, J.L.; Xiao, H.; Schultz, P.G. A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 2013, 52, 1828–1837. [Google Scholar] [CrossRef]
- Mukai, T.; Yamaguchi, A.; Ohtake, K.; Takahashi, M.; Hayashi, A.; Iraha, F.; Kira, S.; Yanagisawa, T.; Yokoyama, S.; Hoshi, H.; et al. Reassignment of a rare sense codon to a non-canonical amino acid in Escherichia coli. Nucleic Acids Res. 2015, 43, 8111–8122. [Google Scholar] [CrossRef]
- Owens, A.E.; Grasso, K.T.; Ziegler, C.A.; Fasan, R. Two-Tier Screening Platform for Directed Evolution of Aminoacyl-tRNA Synthetases with Enhanced Stop Codon Suppression Efficiency. Chembiochem 2017, 18, 1109–1116. [Google Scholar] [CrossRef]
- Sharma, V.; Zeng, Y.; Wang, W.W.; Qiao, Y.; Kurra, Y.; Liu, W.R. Evolving the N-Terminal Domain of Pyrrolysyl-tRNA Synthetase for Improved Incorporation of Noncanonical Amino Acids. Chembiochem 2018, 19, 26–30. [Google Scholar] [CrossRef]
- Suzuki, T.; Miller, C.; Guo, L.-T.; Ho, J.M.L.; Bryson, D.I.; Wang, Y.-S.; Liu, D.R.; Söll, D. Crystal structures reveal an elusive functional domain of pyrrolysyl-tRNA synthetase. Nat. Chem. Biol. 2017, 13, 1261–1266. [Google Scholar] [CrossRef]
- Jiang, H.-K.; Lee, M.-N.; Tsou, J.-C.; Chang, K.-W.; Tseng, H.-W.; Chen, K.-P.; Li, Y.-K.; Wang, Y.-S. Linker and N-Terminal Domain Engineering of Pyrrolysyl-tRNA Synthetase for Substrate Range Shifting and Activity Enhancement. Front. Bioeng. Biotechnol. 2020, 8, 235. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Fang, X.; Chen, H.-Y.; Wu, B.; Wang, Z.U.; Hilty, C.; Liu, W.R. Genetic Incorporation of Twelve meta-Substituted Phenylalanine Derivatives Using a Single Pyrrolysyl-tRNA Synthetase Mutant. ACS Chem. Biol. 2013, 8, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Muzika, M.; Muskat, N.H.; Sarid, S.; Ben-David, O.; Mehl, R.A.; Arbely, E. Chemically-defined lactose-based autoinduction medium for site-specific incorporation of non-canonical amino acids into proteins. RSC Adv. 2018, 8, 25558–25567. [Google Scholar] [CrossRef] [PubMed]
- Budisa, N. Expanded genetic code for the engineering of ribosomally synthetized and post-translationally modified peptide natural products (RiPPs). Curr. Opin. Biotechnol. 2013, 24, 591–598. [Google Scholar] [CrossRef]
- Hoesl, M.G.; Budisa, N. Recent advances in genetic code engineering in Escherichia coli. Curr. Opin. Biotechnol. 2012, 23, 751–757. [Google Scholar] [CrossRef]
- Exner, M.P.; Kuenzl, T.; To, T.M.T.; Ouyang, Z.; Schwagerus, S.; Hoesl, M.G.; Hackenberger, C.P.R.; Lensen, M.C.; Panke, S.; Budisa, N. Design of S-Allylcysteine in Situ Production and Incorporation Based on a Novel Pyrrolysyl-tRNA Synthetase Variant. Chembiochem 2017, 18, 85–90. [Google Scholar] [CrossRef]
- Acuner Ozbabacan, S.E.; Engin, H.B.; Gursoy, A.; Keskin, O. Transient protein–protein interactions. Protein Eng. Des. Sel. 2011, 24, 635–648. [Google Scholar] [CrossRef]
- Ma, B.; Elkayam, T.; Wolfson, H.; Nussinov, R. Protein–protein interactions: Structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Nat. Acad. Sci. USA 2003, 100, 5772. [Google Scholar] [CrossRef]
- Budisa, N.; Pal, P.P. Designing novel spectral classes of proteins with a tryptophan-expanded genetic code. Biol. Chem. 2004, 385, 893–904. [Google Scholar] [CrossRef]
- Hyun Bae, J.; Rubini, M.; Jung, G.; Wiegand, G.; Seifert, M.H.J.; Azim, M.K.; Kim, J.-S.; Zumbusch, A.; Holak, T.A.; Moroder, L.; et al. Expansion of the Genetic Code Enables Design of a Novel “Gold” Class of Green Fluorescent Proteins. J. Mol. Biol. 2003, 328, 1071–1081. [Google Scholar] [CrossRef]
- Minks, C.; Huber, R.; Moroder, L.; Budisa, N. Atomic mutations at the single tryptophan residue of human recombinant annexin V: Effects on structure, stability, and activity. Biochemistry 1999, 38, 10649–10659. [Google Scholar] [CrossRef]
- Moroder, L.; Budisa, N. Synthetic Biology of Protein Folding. ChemPhysChem 2010, 11, 1181–1187. [Google Scholar] [CrossRef]
- Moroz, Y.S.; Binder, W.; Nygren, P.; Caputo, G.A.; Korendovych, I.V. Painting proteins blue: β-(1-azulenyl)-l-alanine as a probe for studying protein–protein interactions. Chem. Commun. 2013, 49, 490–492. [Google Scholar] [CrossRef]
- Baumann, T.; Hauf, M.; Schildhauer, F.; Eberl, K.B.; Durkin, P.M.; Deniz, E.; Löffler, J.G.; Acevedo-Rocha, C.G.; Jaric, J.; Martins, B.M.; et al. Site-Resolved Observation of Vibrational Energy Transfer Using a Genetically Encoded Ultrafast Heater. Angew. Chem. Int. Ed. Engl. 2019, 58, 2899–2903. [Google Scholar] [CrossRef]
- Shao, J.; Korendovych, I.V.; Broos, J. Biosynthetic incorporation of the azulene moiety in proteins with high efficiency. Amino Acids 2015, 47, 213–216. [Google Scholar] [CrossRef]
- Völler, J.-S.; Biava, H.; Hildebrandt, P.; Budisa, N. An expanded genetic code for probing the role of electrostatics in enzyme catalysis by vibrational Stark spectroscopy. BBA-Gen. Subj. 2017, 1861, 3053–3059. [Google Scholar] [CrossRef]
- Kasahara, Y.; Sakurai, T.; Matsuda, R.; Narukawa, M.; Yasuoka, A.; Mori, N.; Watanabe, H.; Okabe, T.; Kojima, H.; Abe, K.; et al. Novel indole and benzothiophene ring derivatives showing differential modulatory activity against human epithelial sodium channel subunits, ENaC β and γ. Biosci. Biotechnol. Biochem. 2019, 83, 243–250. [Google Scholar] [CrossRef]
- Stempel, E.; Kaml, R.F.-X.; Budisa, N.; Kalesse, M. Painting argyrins blue: Negishi cross-coupling for synthesis of deep-blue tryptophan analogue β-(1-azulenyl)-l alanine and its incorporation into argyrin C. Bioorg. Med. Chem. 2018, 26, 5259–5269. [Google Scholar] [CrossRef]
- Zhou, L.; Shao, J.; Li, Q.; van Heel, A.J.; de Vries, M.P.; Broos, J.; Kuipers, O.P. Incorporation of tryptophan analogues into the lantibiotic nisin. Amino Acids 2016, 48, 1309–1318. [Google Scholar] [CrossRef]
- Lepthien, S.; Wiltschi, B.; Bolic, B.; Budisa, N. In vivo engineering of proteins with nitrogen-containing tryptophan analogs. Appl. Microbiol. Biotechnol. 2006, 73, 740–754. [Google Scholar] [CrossRef]
- Hauf, M.; Richter, F.; Schneider, T.; Faidt, T.; Martins, B.M.; Baumann, T.; Durkin, P.; Dobbek, H.; Jacobs, K.; Möglich, A.; et al. Photoactivatable Mussel-Based Underwater Adhesive Proteins by an Expanded Genetic Code. Chembiochem 2017, 18, 1819–1823. [Google Scholar] [CrossRef]
- Wan, W.; Tharp, J.M.; Liu, W.R. Pyrrolysyl-tRNA synthetase: An ordinary enzyme but an outstanding genetic code expansion tool. BBA-Proteins Proteom. 2014, 1844, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, J.K.; Dellas, N.; Noel, J.P.; Wang, L. Stereochemical Basis for Engineered Pyrrolysyl-tRNA Synthetase and the Efficient in Vivo Incorporation of Structurally Divergent Non-native Amino Acids. ACS Chem. Biol. 2011, 6, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Beyer, J.N.; Hosseinzadeh, P.; Gottfried-Lee, I.; van Fossen, E.M.; Zhu, P.; Bednar, R.M.; Karplus, P.A.; Mehl, R.A.; Cooley, R.B. Overcoming Near-Cognate Suppression in a Release Factor 1-Deficient Host with an Improved Nitro-Tyrosine tRNA Synthetase. J. Mol. Biol. 2020, 432, 4690–4704. [Google Scholar] [CrossRef]
- Komarov, A.G.; Linn, K.M.; Devereaux, J.J.; Valiyaveetil, F.I. Modular strategy for the semisynthesis of a K+ channel: Investigating interactions of the pore helix. ACS Chem. Biol. 2009, 4, 1029–1038. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, H.-W.; Baumann, T.; Sun, H.; Wang, Y.-S.; Ignatova, Z.; Budisa, N. Expanding the Scope of Orthogonal Translation with Pyrrolysyl-tRNA Synthetases Dedicated to Aromatic Amino Acids. Molecules 2020, 25, 4418. https://doi.org/10.3390/molecules25194418
Tseng H-W, Baumann T, Sun H, Wang Y-S, Ignatova Z, Budisa N. Expanding the Scope of Orthogonal Translation with Pyrrolysyl-tRNA Synthetases Dedicated to Aromatic Amino Acids. Molecules. 2020; 25(19):4418. https://doi.org/10.3390/molecules25194418
Chicago/Turabian StyleTseng, Hsueh-Wei, Tobias Baumann, Huan Sun, Yane-Shih Wang, Zoya Ignatova, and Nediljko Budisa. 2020. "Expanding the Scope of Orthogonal Translation with Pyrrolysyl-tRNA Synthetases Dedicated to Aromatic Amino Acids" Molecules 25, no. 19: 4418. https://doi.org/10.3390/molecules25194418
APA StyleTseng, H.-W., Baumann, T., Sun, H., Wang, Y.-S., Ignatova, Z., & Budisa, N. (2020). Expanding the Scope of Orthogonal Translation with Pyrrolysyl-tRNA Synthetases Dedicated to Aromatic Amino Acids. Molecules, 25(19), 4418. https://doi.org/10.3390/molecules25194418