
Novel Harmicines with Improved Potency against Plasmodium

 ,
,  , ,
, ,  , and
, and
Abstract
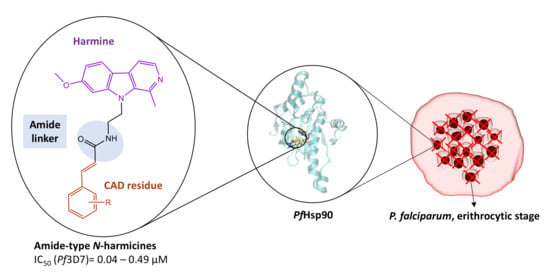
1. Introduction
2. Results and Discussion
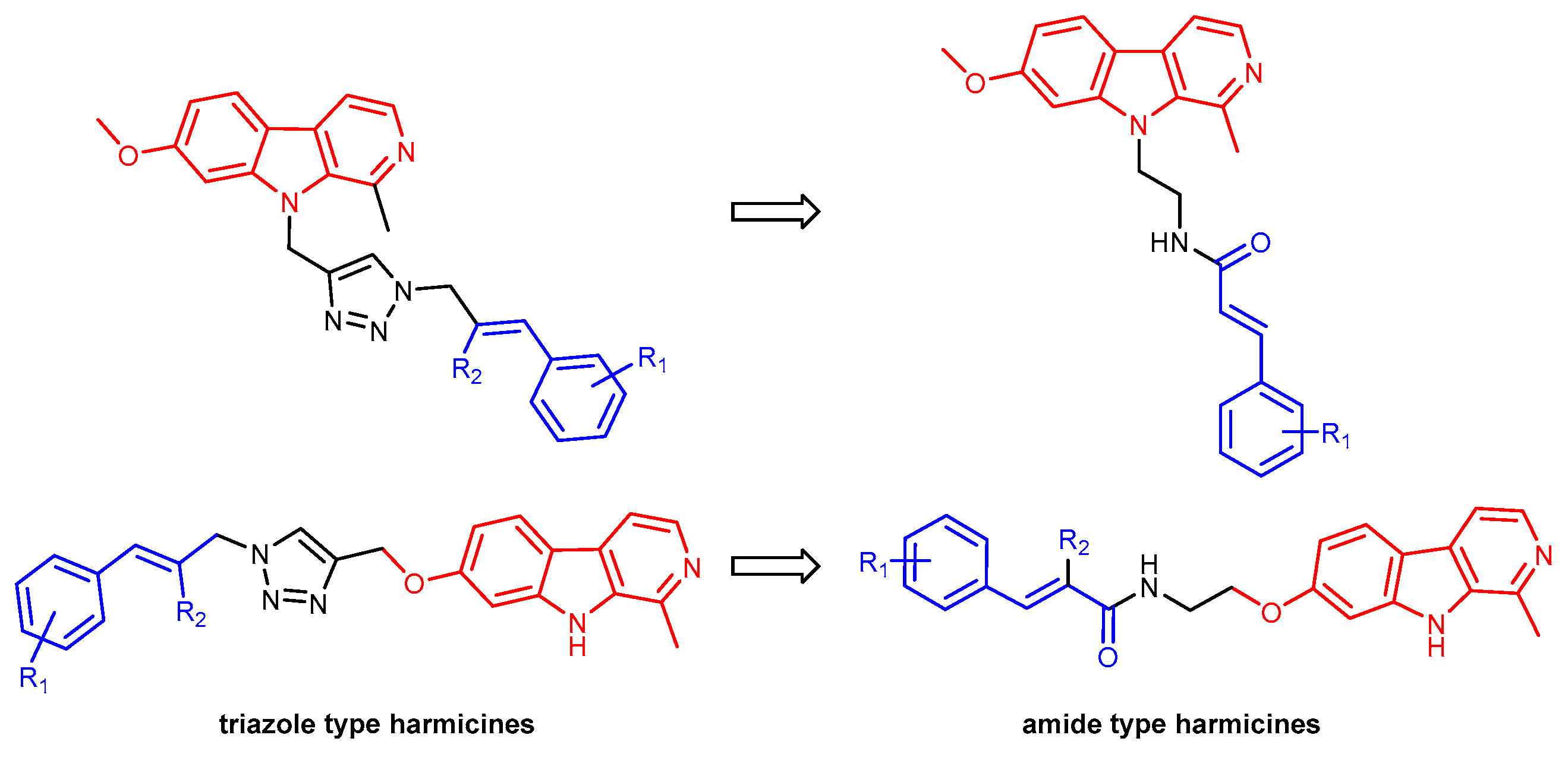
2.1. Chemistry
2.2. Biological Evaluations
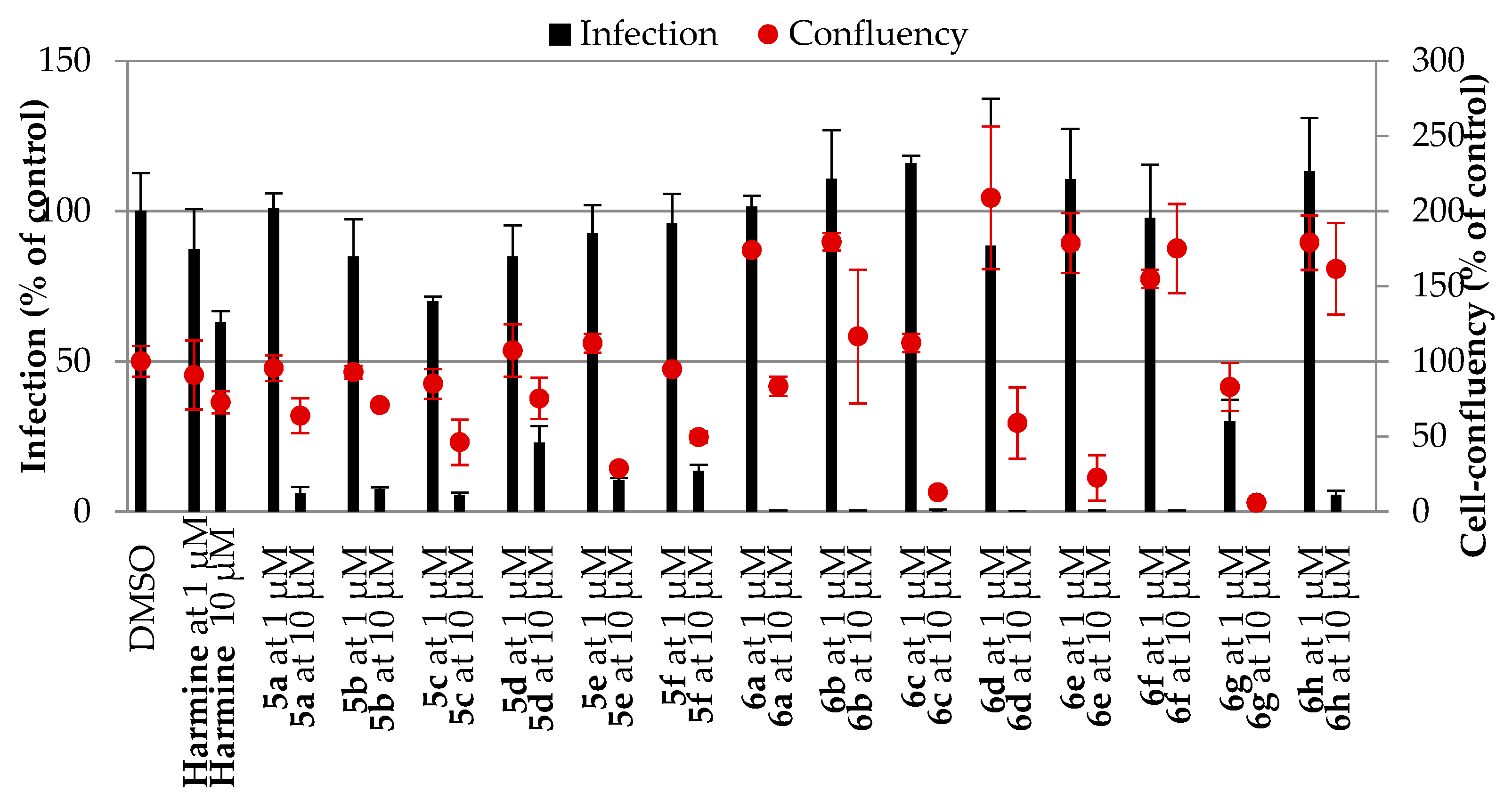
2.2.1. Antiplasmodial Activity
In Vitro Activity against P. falciparum Erythrocytic Stage
In Vitro Activity against P. berghei Hepatic Stages
2.2.2. Cytotoxicity Assay
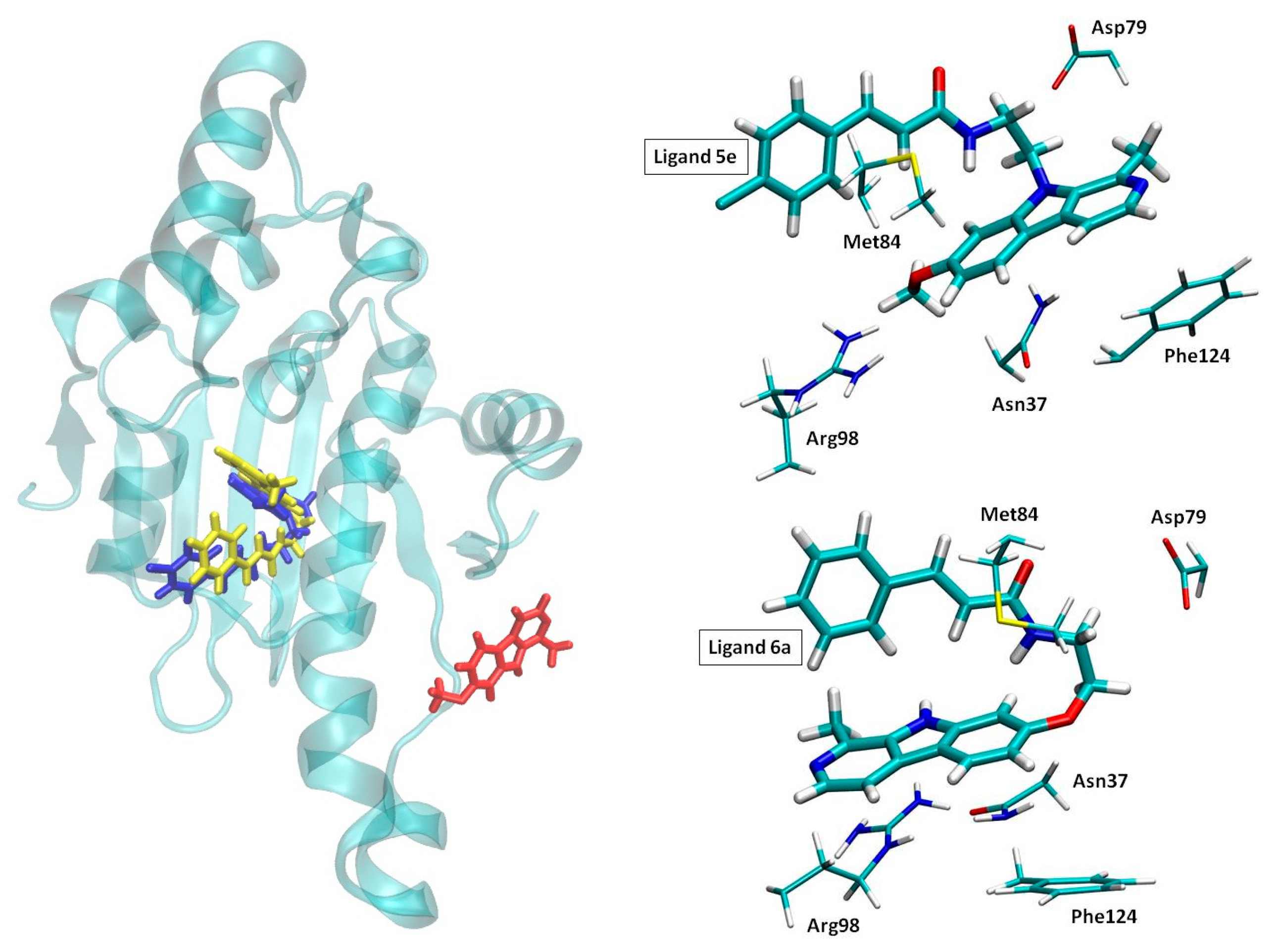
2.3. Molecular Dynamics Simulations
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Synthesis of tert-butyl (2-(7-methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)ethyl)carbamate (1)
3.1.3. Synthesis of tert-butyl (2-((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethyl)carbamate (2)
3.1.4. General Procedure for the Synthesis of Amines 3 and 4
2-(7-Methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)ethan-1-amine (3)
2-((1-Methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethan-1-amine (4)
3.1.5. General Procedure for the Synthesis of Harmicines 5a–f
N-(2-(7-methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)ethyl)cinnamamide (5a)
(E)-3-(3-fluorophenyl)-N-(2-(7-methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)ethyl)acrylamide (5b)
(E)-3-(3-bromophenyl)-N-(2-(7-methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)ethyl)acrylamide (5c)
(E)-3-(4-fluorophenyl)-N-(2-(7-methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)ethyl)acrylamide (5d)
(E)-3-(4-chlorophenyl)-N-(2-(7-methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)ethyl)acrylamide (5e)
(E)-N-(2-(7-methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)ethyl)-3-(4-methoxyphenyl)acrylamide (5f)
3.1.6. General Procedure for the Synthesis of Harmicines 6a–h
N-(2-((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethyl)cinnamamide (6a)
(E)-3-(3-fluorophenyl)-N-(2-((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethyl)acrylamide (6b)
(E)-3-(3-bromophenyl)-N-(2-((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethyl)acrylamide (6c)
(E)-3-(4-fluorophenyl)-N-(2-((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethyl)acrylamide (6d)
(E)-3-(4-chlorophenyl)-N-(2-((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethyl)acrylamide (6e)
(E)-3-(4-methoxyphenyl)-N-(2-((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethyl)acrylamide (6f)
(E)-3-(2-fluorophenyl)-N-(2-((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethyl)acrylamide (6g)
(E)-2-methyl-N-(2-((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethyl)-3-phenylacrylamide (6h)
3.2. In Vitro Drug Sensitivity Assay against Erythrocytic Stages of P. falciparum
3.3. In Vitro Activity against P. berghei Hepatic Stages
3.4. In Vitro Cytotoxicity Assay
3.5. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | adenosine diphosphate |
| ATR | attenuated total reflectance |
| Boc | tert-butyloxycarbonyl |
| CAD | cinnamic acid derivative |
| DCM | dichloromethane |
| DIEA | N,N-diisopropylethylamine |
| DMF | N,N-dimethylformamide |
| ESI | electrospray ionization |
| GAFF | generalized Amber force field |
| HATU | 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxidhexafluorophosphate |
| HEPES | 2-(4-(2-hydroxyethyl)piperazin-1-yl)ethanesulfonic acid |
| HRP2 | histidine-rich protein 2 |
| HepG2 | human liver hepatocellular carcinoma cell line |
| IC50 | the concentration of the tested compound necessary for 50% growth inhibition |
| MD | chloroquine-sensitive strain of P. falciparum |
| MM-GBSA | molecular mechanics/generalized born surface area |
| MW | microwave |
| PfDd2 | chloroquine-resistant strain of P. falciparum |
| PfHsp90 | P. falciparum heat shock protein 90 |
| QSAR | quantitative structure–activity relationship |
| RESP | restrained electrostatic potential |
| SI | selectivity index |
| TIP3P | transferable intermolecular potential with 3 points |
| TMS | tetramethylsilane |
| UATR | universal attenuated total reflectance |
References
- WHO. World Malaria Report 2019. World Health Organisation, 2019. Available online: https://apps.who.int/iris/bitstream/handle/10665/330011/9789241565721-eng.pdf?sequence=1&isAllowed=y (accessed on 15 July 2020).
- Mishra, M.; Mishra, V.K.; Kashaw, V.; Iyer, A.K.; Kashaw, S.K. Comprehensive review on various strategies for antimalarial drug discovery. Eur. J. Med. Chem. 2017, 125, 1300–1320. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Álvaro, E.; Hong, W.D.; Nixon, G.L.; O’Neill, P.M.; Calderón, F. Antimalarial Chemotherapy: Natural Product Inspired Development of Preclinical and Clinical Candidates with Diverse Mechanisms of Action. J. Med. Chem. 2016, 59, 5587–5603. [Google Scholar] [CrossRef] [PubMed]
- Grimberg, B.T.; Mehlotra, R.K. Expanding the antimalarial drug arsenal-now, but how? Pharmaceuticals 2011, 4, 681–712. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ema.europa.eu/en/news/first-malaria-vaccine-receives-positive-scientific-opinion-ema (accessed on 15 July 2020).
- Wilby, K.J.; Lau, T.T.; Gilchrist, S.E.; Ensom, M.H. Mosquirix (RTS,S): A Novel Vaccine for the Prevention of Plasmodium falciparum Malaria. Ann. Pharmacother. 2012, 46, 384–393. [Google Scholar] [CrossRef]
- Malaria Vaccine Pilot Launched in Malawi. Available online: https://www.who.int/news-room/detail/23-04-2019-malaria-vaccine-pilot-launched-in-malawi (accessed on 14 July 2020).
- Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar] [CrossRef]
- Meunier, B. Hybrid molecules with a dual mode of action: Dream or reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Pérez, B.C.; Teixeira, C.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Gomes, J.R.B.; Prudeîncio, M.; Gomes, P. N-Cinnamoylated chloroquine analogues as dual-stage antimalarial leads. J. Med. Chem. 2013, 56, 556–567. [Google Scholar] [CrossRef]
- Pérez, B.; Teixeira, C.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Prudêncio, M.; Gomes, P. Primacins, N-cinnamoyl-primaquine conjugates, with improved liver-stage antimalarial activity. Medchemcomm 2012, 3, 1170–1172. [Google Scholar] [CrossRef]
- Pérez, B.; Teixeira, C.; Gut, J.; Rosenthal, P.J.; Gomes, J.R.B.; Gomes, P. Cinnamic Acid/Chloroquinoline Conjugates as Potent Agents against Chloroquine-Resistant Plasmodium falciparum. ChemMedChem 2012, 7, 1537–1540. [Google Scholar] [CrossRef]
- Pérez, B.; Teixeira, C.; Gomes, A.S.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Prudêncio, M.; Gomes, P. In Vitro efficiency of 9-(N-cinnamoylbutyl)aminoacridines against blood- and liver-stage malaria parasites. Bioorg. Med. Chem. Lett. 2013, 23, 610–613. [Google Scholar] [CrossRef]
- Pérez, B.C.; Teixeira, C.; Figueiras, M.; Gut, J.; Rosenthal, P.J.; Gomes, J.R.B.; Gomes, P. Novel cinnamic acid/4-aminoquinoline conjugates bearing non-proteinogenic amino acids: Towards the development of potential dual action antimalarials. Eur. J. Med. Chem. 2012, 54, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Perković, I.; Raić-Malić, S.; Fontinha, D.; Prudêncio, M.; Pessanha de Carvalho, L.; Held, J.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Harmicines—Harmine and cinnamic acid hybrids as novel antiplasmodial hits. Eur. J. Med. Chem. 2020, 187. [Google Scholar] [CrossRef] [PubMed]
- Shahinas, D.; MacMullin, G.; Benedict, C.; Crandall, I.; Pillaid, D.R. Harmine is a potent antimalarial targeting Hsp90 and synergizes with chloroquine and artemisinin. Antimicrob. Agents Chemother. 2012, 56, 4207–4213. [Google Scholar] [CrossRef] [PubMed]
- Shahinas, D.; Liang, M.; Datti, A.; Pillai, D.R. A repurposing strategy identifies novel synergistic inhibitors of plasmodium falciparum heat shock protein 90. J. Med. Chem. 2010, 53, 3552–3557. [Google Scholar] [CrossRef]
- Bayih, A.G.; Folefoc, A.; Mohon, A.N.; Eagon, S.; Anderson, M.; Pillai, D.R. In vitro and in vivo anti-malarial activity of novel harmine-analog heat shock protein 90 inhibitors: A possible partner for artemisinin. Malar. J. 2016, 15, 1–11. [Google Scholar] [CrossRef]
- Shahinas, D.; Folefoc, A.; Pillai, D.R. Targeting plasmodium falciparum Hsp90: Towards reversing antimalarial resistance. Pathogens 2013, 2, 33–54. [Google Scholar] [CrossRef]
- Wang, T.; Mäser, P.; Picard, D. Inhibition of Plasmodium falciparum Hsp90 Contributes to the Antimalarial Activities of Aminoalcohol-carbazoles. J. Med. Chem. 2016, 59, 6344–6352. [Google Scholar] [CrossRef]
- Chemicalize. Available online: https://chemicalize.com/ (accessed on 20 July 2020).
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.r-project.org/ (accessed on 20 July 2020).
- Noedl, H.; Bronnert, J.; Yingyuen, K.; Attlmayr, B.; Kollaritsch, H.; Fukuda, M. Simple histidine-rich protein 2 double-site sandwich enzyme-linked immunosorbent assay for use in malaria drug sensitivity testing. Antimicrob. Agents Chemother. 2005, 49, 3575–3577. [Google Scholar] [CrossRef]
- Held, J.; Gebru, T.; Kalesse, M.; Jansen, R.; Gerth, K.; Müller, R.; Mordmüller, B. Antimalarial activity of the myxobacterial macrolide chlorotonil A. Antimicrob. Agents Chemother. 2014, 58, 6378–6384. [Google Scholar] [CrossRef]
- Ploemen, I.H.J.; Prudêncio, M.; Douradinha, B.G.; Ramesar, J.; Fonager, J.; van Gemert, G.-J.; Luty, A.J.F.; Hermsen, C.C.; Sauerwein, R.W.; Baptista, F.G.; et al. Visualisation and Quantitative Analysis of the Rodent Malaria Liver Stage by Real Time Imaging. PLoS ONE 2009, 4, e7881. [Google Scholar] [CrossRef]
- Machado, M.; Sanches-Vaz, M.; Cruz, J.P.; Mendes, A.M.; Prudêncio, M. Inhibition of Plasmodium hepatic infection by antiretroviral compounds. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Borenfreund, E.; Puerner, J.A. A simple quantitative procedure using monolayer cultures for cytotoxicity assays (HTD/NR-90). J. Tissue Cult. Methods 1985, 9, 7–9. [Google Scholar] [CrossRef]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Berger, J.M. Structure of the ATP-binding domain of Plasmodium falciparum Hsp90. Proteins Struct. Funct. Bioinform. 2010, 78, 2738–2744. [Google Scholar] [CrossRef]
- Frédérick, R.; Bruyére, C.; Vancraeynest, C.; Reniers, J.; Meinguet, C.; Pochet, L.; Backlund, A.; Masereel, B.; Kiss, R.; Wouters, J. Novel trisubstituted harmine derivatives with original in vitro anticancer activity. J. Med. Chem. 2012, 55, 6489–6501. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; Izadi, S.; et al. Amber 2016; University of California, San Francisco: San Francisco, CA, USA, 2016. [Google Scholar]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Tandarić, T.; Vianello, R. Computational Insight into the Mechanism of the Irreversible Inhibition of Monoamine Oxidase Enzymes by the Antiparkinsonian Propargylamine Inhibitors Rasagiline and Selegiline. ACS Chem. Neurosci. 2019, 10, 3532–3542. [Google Scholar] [CrossRef]
- Matić, J.; Šupljika, F.; Tandarić, T.; Dukši, M.; Piotrowski, P.; Vianello, R.; Brozovic, A.; Piantanida, I.; Schmuck, C.; Stojković, M.R. DNA/RNA recognition controlled by the glycine linker and the guanidine moiety of phenanthridine peptides. Int. J. Biol. Macromol. 2019, 134, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | IC50 1 (µM) | SI 2 | ||
|---|---|---|---|---|
| Pf3D7 | PfDd2 | HepG2 | ||
| 5a | 0.49 ± 0.25 3 | 1.11 ± 0.15 3 | 350.31 ± 13.02 3 | 715 |
| 5b | 0.07 ± 0.03 | 0.78 ± 0.32 | 54.11 ± 13.36 | 773 |
| 5c | 0.07 ± 0.03 | 0.41 ± 0.01 | 2.88 ± 0.42 | 41 |
| 5d | 0.09 ± 0.06 | 0.33 ± 0.11 | 20.99 ± 0.88 | 233 |
| 5e | 0.04 ± 0.02 | 0.17 ± 0.01 | 2.91 ± 1.75 | 73 |
| 5f | 0.26 ± 0.001 | 0.49 ± 0.24 | 74.69 ± 7.61 | 287 |
| 6a | 0.98 ± 0.12 | 4.7 ± 2.65 | 12.74 ± 0.66 | 13 |
| 6b | 2.75 ± 1.6 | 5.01 ± 0.83 | 12.86 ± 3.31 | 5 |
| 6c | 0.37 ± 0.22 | 0.48 ± 0.28 | 6.11 ± 2.07 | 16 |
| 6d | 0.15 ± 0.06 | 0.69 ± 0.18 | 7.63 ± 2.47 | 51 |
| 6e | 0.32 ± 0.03 | 0.4 ± 0.24 | 7.72 ± 3.04 | 24 |
| 6f | 0.21 ± 0.14 | 1.09 ± 0.49 | 10.53 ± 0.43 | 50 |
| 6g | 0.93 ± 0.28 | 3.92 ± 1.35 | 16.17 ± 1.86 | 17 |
| 6h | 6.78 ± 0.72 | 19.53 ± 11.83 | 61.28 ± 2.75 | 9 |
| CQ 4 | 0.003 ± 0.002 | 0.20 ± 0.10 | n.d. | n.d. |
| Harmine | 8.25 ± 2.83 | >27.7 | > 250 | 30 |
| Residue | 5a | 5d | 5e | 6a | 6d | 6e | Harmine |
|---|---|---|---|---|---|---|---|
| Asn37 | −1.82 | −1.33 | −2.01 | −2.09 | −1.69 | −1.81 | 0.00 |
| Asp79 | 1.47 | 1.36 | 1.58 | 1.11 | 1.30 | 0.74 | 0.00 |
| Arg98 | −0.05 | −0.93 | −1.39 | −2.00 | −2.45 | −2.24 | 0.00 |
| Phe124 | −1.44 | −1.68 | −0.64 | −0.58 | −0.35 | −0.37 | 0.00 |
| Met84 | −2.50 | −2.63 | −2.70 | −2.05 | −2.25 | −2.02 | 0.00 |
| Gly83 | −0.79 | −1.28 | −1.63 | −1.00 | −1.69 | −1.56 | 0.00 |
| Ile82 | −0.88 | −1.50 | −1.51 | −1.28 | −1.49 | −1.43 | 0.00 |
| Thr171 | −1.60 | −1.92 | −1.51 | −0.74 | −0.51 | −0.53 | 0.00 |
| Ala41 | −1.39 | −1.37 | −1.44 | −1.32 | −1.40 | −1.27 | 0.00 |
| Ile173 | −1.05 | −1.13 | −1.18 | −0.46 | −0.42 | −0.32 | −0.01 |
| Asn92 | −0.96 | −0.94 | −1.06 | −1.64 | −1.43 | −1.41 | 0.00 |
| Ala38 | −0.90 | −0.91 | −0.89 | −0.56 | −0.53 | −0.50 | 0.00 |
| Leu93 | −0.63 | −0.60 | −0.80 | −0.46 | −0.52 | −0.59 | 0.00 |
| Leu34 | −0.68 | −0.82 | −0.75 | −0.16 | −0.14 | −0.12 | 0.00 |
| Val136 | −0.67 | −0.72 | −0.64 | −0.11 | −0.14 | −0.12 | −0.02 |
| Gly81 | −0.33 | −0.49 | −0.54 | −0.37 | −0.42 | −0.35 | 0.00 |
| Thr95 | 0.03 | 0.03 | 0.03 | 0.03 | 0.04 | 0.04 | 0.00 |
| Asp142 | 0.04 | 0.03 | 0.04 | 0.03 | 0.03 | 0.03 | 0.00 |
| Glu48 | 0.03 | 0.05 | 0.04 | 0.05 | 0.06 | 0.06 | 0.00 |
| Total ΔGBIND | −33.9 | −38.1 | −40.9 | −34.6 | −35.7 | −37.5 | −7.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marinović, M.; Perković, I.; Fontinha, D.; Prudêncio, M.; Held, J.; Pessanha de Carvalho, L.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Novel Harmicines with Improved Potency against Plasmodium. Molecules 2020, 25, 4376. https://doi.org/10.3390/molecules25194376
Marinović M, Perković I, Fontinha D, Prudêncio M, Held J, Pessanha de Carvalho L, Tandarić T, Vianello R, Zorc B, Rajić Z. Novel Harmicines with Improved Potency against Plasmodium. Molecules. 2020; 25(19):4376. https://doi.org/10.3390/molecules25194376
Chicago/Turabian StyleMarinović, Marina, Ivana Perković, Diana Fontinha, Miguel Prudêncio, Jana Held, Lais Pessanha de Carvalho, Tana Tandarić, Robert Vianello, Branka Zorc, and Zrinka Rajić. 2020. "Novel Harmicines with Improved Potency against Plasmodium" Molecules 25, no. 19: 4376. https://doi.org/10.3390/molecules25194376
APA StyleMarinović, M., Perković, I., Fontinha, D., Prudêncio, M., Held, J., Pessanha de Carvalho, L., Tandarić, T., Vianello, R., Zorc, B., & Rajić, Z. (2020). Novel Harmicines with Improved Potency against Plasmodium. Molecules, 25(19), 4376. https://doi.org/10.3390/molecules25194376