
Nucleophilic Synthesis of 6-l-[18F]FDOPA. Is Copper-Mediated Radiofluorination the Answer?


Abstract
1. Introduction
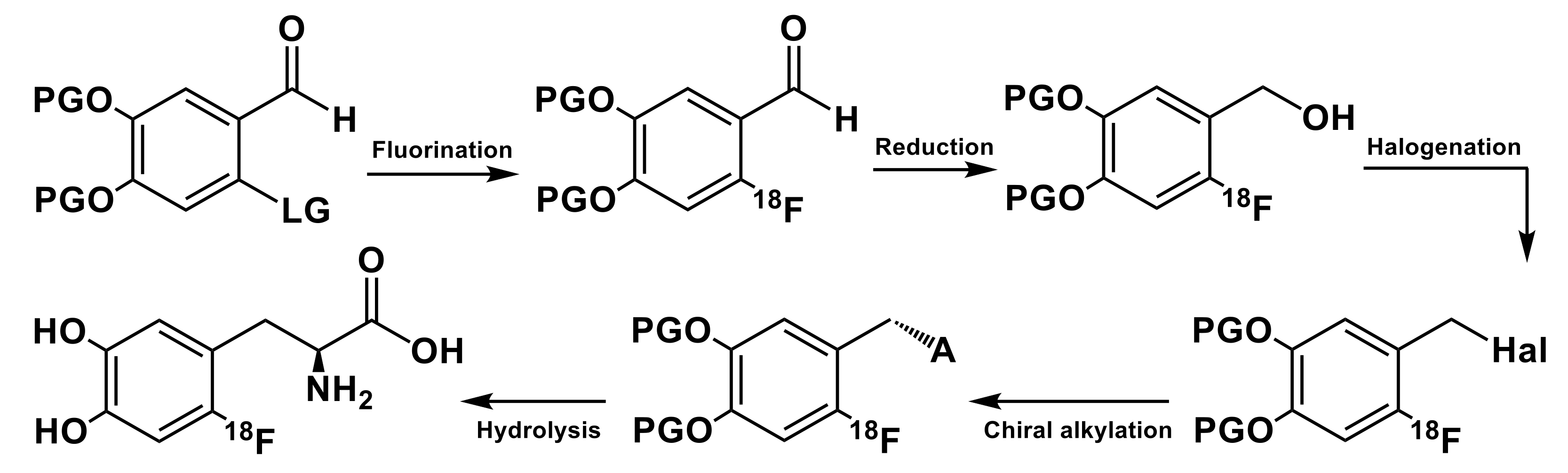
2. General Concepts for Nucleophilic Synthesis of 6-l-[18F]FDOPA
3. Late Stage Fluorinations
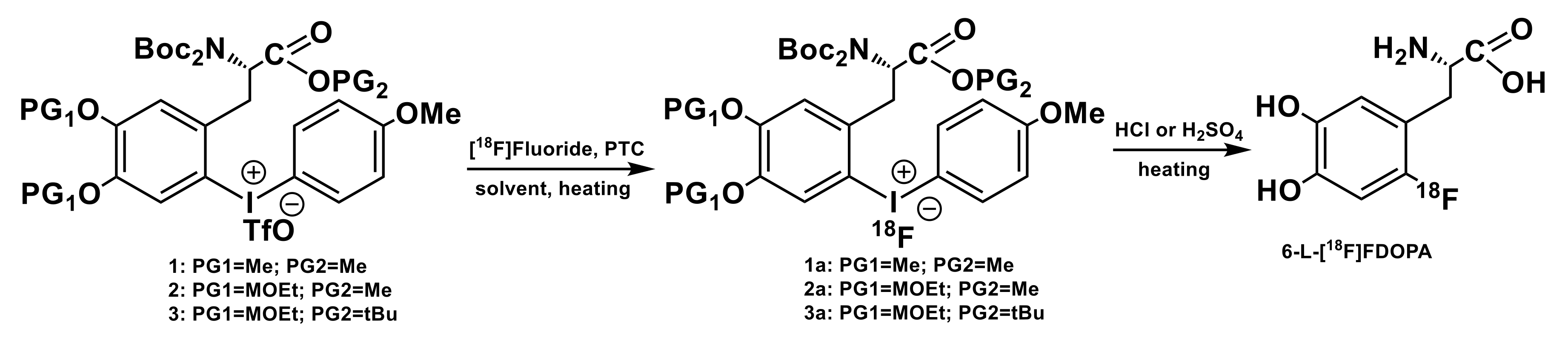
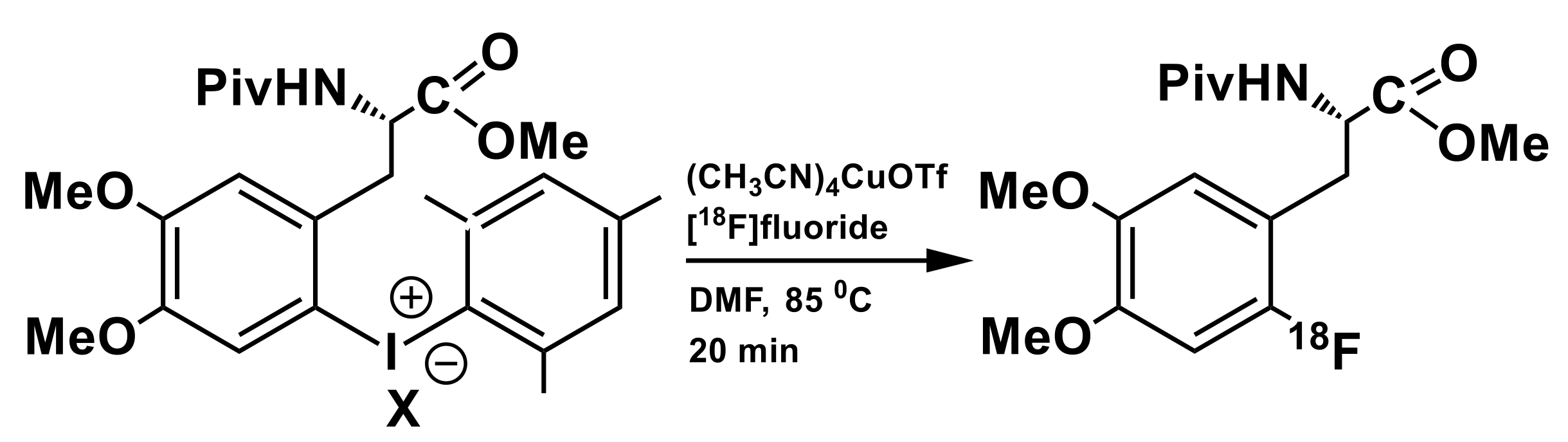
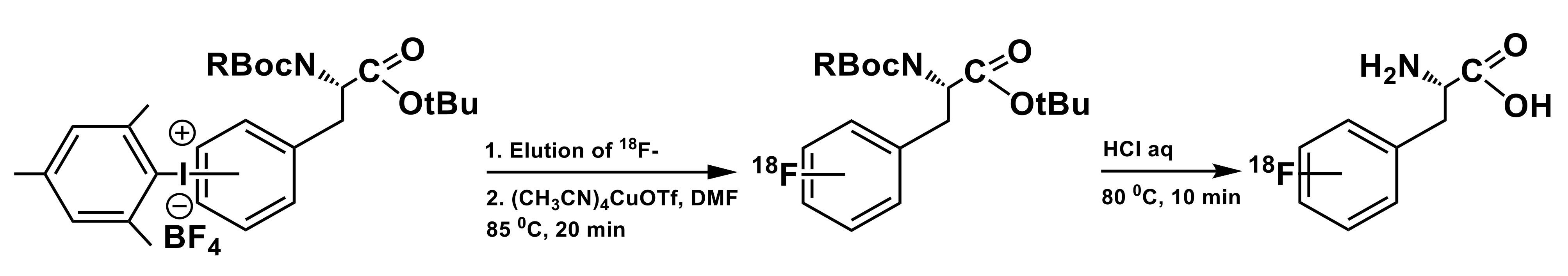
3.1. Iodonium Salts
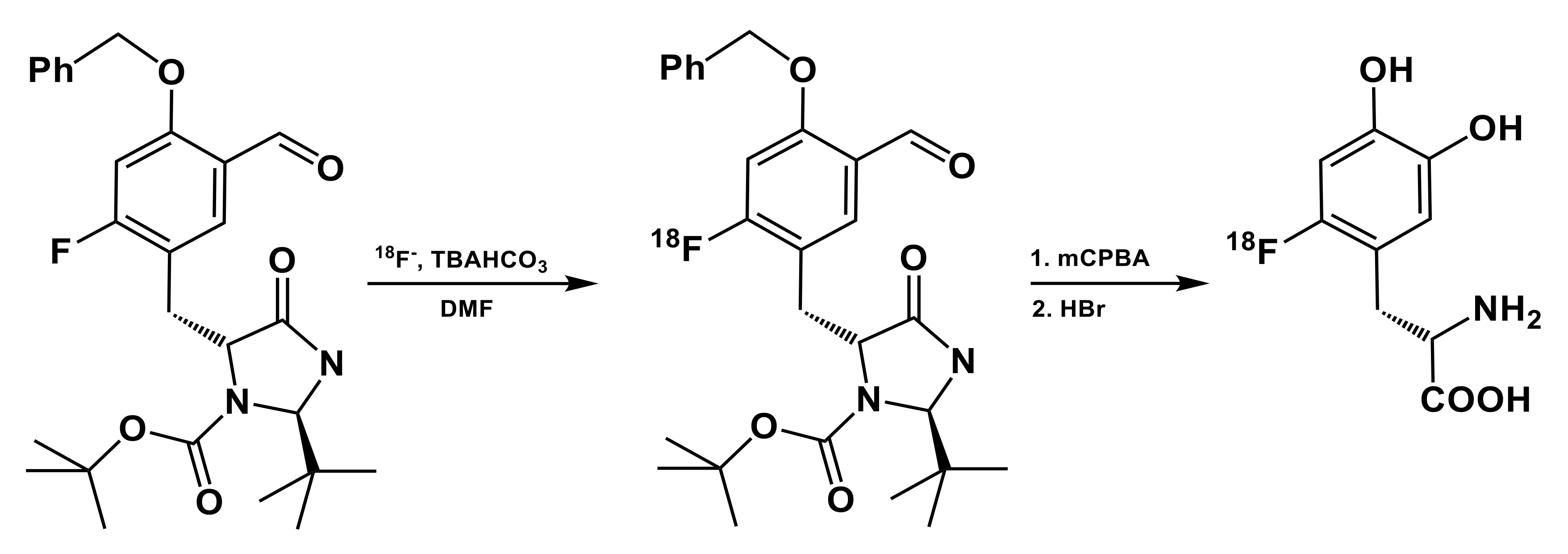
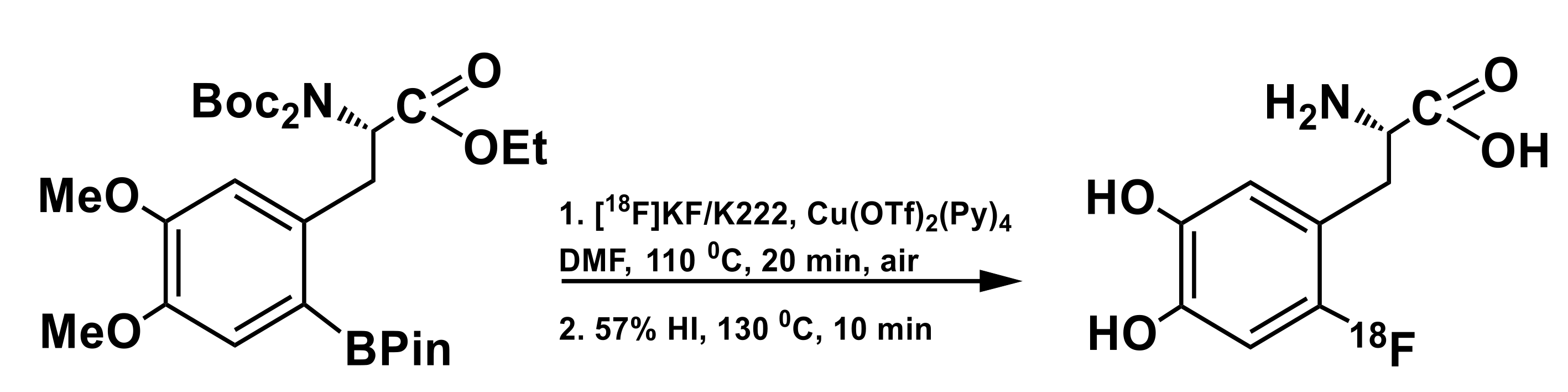
3.2. Arylboronic Acid Pinacol Esters (ArylBPin)
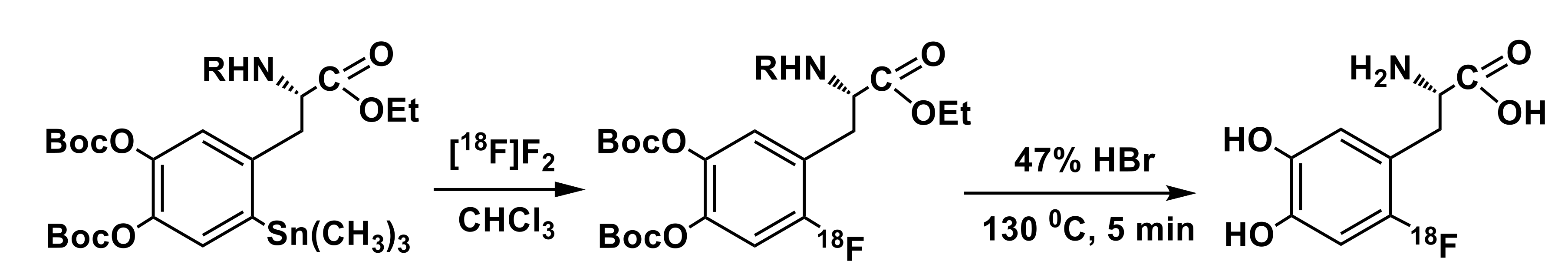
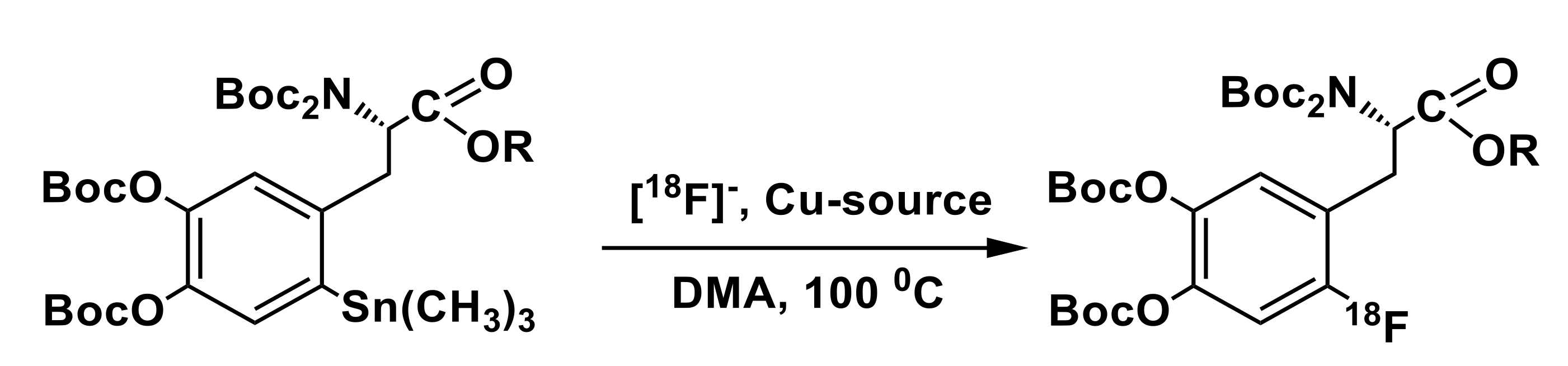
3.3. Organostannanes
4. Summary and Conclusions
Funding
Conflicts of Interest
References
- Laverman, P.; Boerman, O.C.; Corstens, F.H.; Oyen, W.J. Fluorinated Amino Acids for Tumour Imaging with Positron Emission Tomography. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 681–690. [Google Scholar] [CrossRef]
- Galldiks, N.; Langen, K.-J. Amino Acid PET—An Imaging Option to Identify Treatment Response, Posttherapeutic Effects, and Tumor Recurrence? Front. Neurol. 2016, 7. [Google Scholar] [CrossRef]
- Ermert, J.; Coenen, H.H. Methods for 11C- and 18F-Labelling of Amino Acids and Derivatives for Positron Emission Tomography Imaging. J. Label. Compd. Radiopharm. 2013, 56, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Minn, H.; Kauhanen, S.; Seppanen, M.; Nuutila, P. 18F-FDOPA: A Multiple-Target Molecule. J. Nucl. Med. 2009, 50, 1915–1918. [Google Scholar] [CrossRef] [PubMed]
- Taïeb, D.; Imperiale, A.; Pacak, K. 18F-DOPA: The Versatile Radiopharmaceutical. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1187–1189. [Google Scholar] [CrossRef]
- Garnett, E.S.; Firnau, G.; Nahmias, C. Dopamine Visualized in the Basal Ganglia of Living Man. Nature 1983, 305, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Sioka, C.; Fotopoulos, A.; Kyritsis, A.P. Recent Advances in PET Imaging for Evaluation of Parkinson’s Disease. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1594–1603. [Google Scholar] [CrossRef]
- Politis, M. Neuroimaging in Parkinson Disease: From Research Setting to Clinical Practice. Nat. Rev. Neurol. 2014, 10, 708–722. [Google Scholar] [CrossRef]
- Seibyl, J.P.; Chen, W.; Silverman, D.H. 3,4-Dihydroxy-6-[18F]-Fluoro-L-Phenylalanine Positron Emission Tomography in Patients with Central Motor Disorders and in Evaluation of Brain and Other Tumors. Semin. Nucl. Med. 2007, 37, 440–450. [Google Scholar] [CrossRef]
- Calabria, F.; Calabria, E.; Gangemi, V.; Cascini, L.G. Current Status and Future Challenges of Brain Imaging with (18)F-DOPA PET for Movement Disorders. Hell. J. Nucl Med. 2016, 19, 33–41. [Google Scholar]
- Hoegerle, S.; Altehoefer, C.; Ghanem, N.; Brink, I.; Moser, E.; Nitzsche, E. 18F-DOPA Positron Emission Tomography for Tumour Detection in Patients with Medullary Thyroid Carcinoma and Elevated Calcitonin Levels. Eur. J. Nucl. Med. Mol. Imaging 2001, 28, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Jager, P.L.; Chirakal, R.; Marriott, C.J.; Brouwers, A.H.; Gulenchyn, K.Y.; Koopmans, K.P. 6-L-18F-Fluorodihydroxyphenylalanine PET in Neuroendocrine Tumors: Basic Aspects and Emerging Clinical Applications*. J. Nucl. Med. 2008, 49, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Santhanam, P.; Taïeb, D. Role of 18F-FDOPA PET/CT Imaging in Endocrinology. Clin. Endocrinol. 2014, 81, 789–798. [Google Scholar] [CrossRef]
- Bozkurt, M.F.; Virgolini, I.; Balogova, S.; Beheshti, M.; Rubello, M.; Decristoforo, C.; Ambrosini, V.; Kjaer, A.; Delgado-Bolton, R.; Kunikowska, J.; et al. Guideline for PET/CT Imaging of Neuroendocrine Neoplasms with 68Ga-DOTA-Conjugated Somatostatin Receptor Targeting Peptides and 18F-DOPA. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1588–1601. [Google Scholar] [CrossRef] [PubMed]
- Fueger, B.J.; Czernin, J.; Cloughesy, T.; Silverman, D.H.; Geist, C.L.; Walter, M.A.; Schiepers, C.; Nghiemphu, P.; Lai, A.; Phelps, M.E.; et al. Correlation of 6-18F-Fluoro-L-Dopa PET Uptake with Proliferation and Tumor Grade in Newly Diagnosed and Recurrent Gliomas. J. Nucl. Med. 2010, 51, 1532–1538. [Google Scholar] [CrossRef]
- Bell, C.; Dowson, N.; Puttick, S.; Gal, Y.; Thomas, P.; Fay, M.; Smith, J.; Rose, S. Increasing Feasibility and Utility of 18F-FDOPA PET for the Management of Glioma. Nucl. Med. Biol. 2015, 42, 788–795. [Google Scholar] [CrossRef]
- Morana, G.; Piccardo, A.; Garrè, M.L.; Nozza, P.; Consales, A.; Rossi, A. Multimodal Magnetic Resonance Imaging and 18F-L-Dihydroxyphenylalanine Positron Emission Tomography in Early Characterization of Pseudoresponse and Nonenhancing Tumor Progression in a Pediatric Patient with Malignant Transformation of Ganglioglioma Treated with Bevacizumab. J. Clin. Oncol. 2013, 31, e1–e5. [Google Scholar]
- Albert, N.L.; Weller, M.; Suchorska, B.; Galldiks, N.; Soffietti, R.; Kim, M.M.; La Fougère, C.; Pope, W.; Law, I.; Arbizu, J.; et al. Response Assessment in Neuro-Oncology Working Group and European Association for Neuro-Oncology Recommendations for the Clinical Use of PET Imaging in Gliomas. Neuro-Oncology 2016, 18, 1199–1208. [Google Scholar] [CrossRef]
- Langen, K.-J.; Galldiks, N. Update on Amino Acid PET of Brain Tumours. Curr. Opin. Neurol. 2018, 31, 354–361. [Google Scholar] [CrossRef]
- Firnau, G.; Chirakal, R.; Garnett, E.S. Aromatic Radiofluorination with [18F]fluorine Gas: 6-[18F]fluoro-L-Dopa. J. Nucl. Med. 1984, 25, 1228–1233. [Google Scholar]
- Namavari, M.; Bishop, A.; Satyamurthy, N.; Bida, G.; Barrio, J.R. Regioselective Radiofluorodestannylation with [18F]F2 and [18F]CH3COOF: A High Yield Synthesis of 6-[18F]fluoro-L-Dopa. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1992, 43, 989–996. [Google Scholar] [CrossRef]
- De Vries, E.F.J.; Luurtsema, G.; Brüssermann, M.; Elsinga, P.H.; Vaalburg, W. Fully Automated Synthesis Module for the High Yield One-Pot Preparation of 6-[18F]fluoro-L-DOPA. Appl. Radiat. Isot. 1999, 51, 389–394. [Google Scholar] [CrossRef]
- Antuganov, D.O.; Zykov, M.P.; Ryzhkova, D.V.; Zykova, T.A.; Vinal’Ev, A.A.; Antuganova, Y.O.; Samburov, O.P. Synthesis of [18F]-L-DOPA Radiopharmaceutical on a Modified GE TracerLAB Fx F-E Platform. Radiochemistry 2016, 58, 649–653. [Google Scholar] [CrossRef]
- Luurtsema, G.; Boersma, H.H.; Schepers, M.; De Vries, A.M.T.; Maas, B.; Zijlma, R.; De Vries, E.F.J.; Elsinga, P.H. Improved GMP-Compliant Multi-Dose Production and Quality Control of 6-[18F]fluoro-L-DOPA. EJNMMI Radiopharm. Chem. 2016, 1, 677. [Google Scholar] [CrossRef][Green Version]
- Wagner, M.; Wuest, F. The Radiopharmaceutical Chemistry of Fluorine-18: Electrophilic Fluorinations. In Analytical and Chromatographic Techniques in Radiopharmaceutical Chemistry; Springer Science and Business Media LLC: Berlin, Germany, 2019; pp. 285–295. [Google Scholar] [CrossRef]
- Akamatsu, G.; Ohnishi, A.; Aita, K.; Nishida, H.; Ikari, Y.; Sasaki, M.; Kohara, N.; Senda, M. A Revisit to Quantitative PET with 18F-FDOPA of High Specific Activity Using a High-Resolution Condition in View of Application to Regenerative Therapy. Ann. Nucl. Med. 2016, 31, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, K.P.; Brouwers, A.H.; De Hooge, M.N.; Van Der Horst-Schrivers, A.N.; Kema, I.P.; Wolffenbuttel, B.H.; De Vries, E.G.; Jager, P.L. Carcinoid Crisis After Injection of 6-18F-Fluorodihydroxyphenylalanine in a Patient with Metastatic Carcinoid. J. Nucl. Med. 2005, 46, 1240–1243. [Google Scholar]
- Bergman, J.; Solin, O. Fluorine-18-Labeled Fluorine Gas for Synthesis of Tracer Molecules. Nucl. Med. Biol. 1997, 24, 677–683. [Google Scholar] [CrossRef]
- Forsback, S.; Eskola, O.; Haaparanta, M.; Bergmann, J.; Solin, O. Electrophilic Synthesis of 6-[18F]fluoro-L-DOPA Using Post-Target Produced [18F]F2. Radiochim. Acta 2008, 96, 845–848. [Google Scholar] [CrossRef]
- Krzyczmonik, A.; Keller, T.; Kirjavainen, A.K.; Forsback, S.; Solin, O. Vacuum Ultraviolet photon–mediated Production of [18F]F2. J. Label. Compd. Radiopharm. 2017, 60, 186–193. [Google Scholar] [CrossRef]
- Stenhagen, I.S.R.; Kirjavainen, A.K.; Forsback, S.; Jørgensen, C.G.; Robins, E.G.; Luthra, S.K.; Solin, O.; Gouverneur, V. [18F]Fluorination of an Arylboronic Ester Using [18F]selectfluor bis(triflate): Application to 6-[18F]fluoro-L-DOPA. Chem. Commun. 2013, 49, 1386. [Google Scholar] [CrossRef] [PubMed]
- Council of Europe. Fluorodopa (18F) (prepared by electrophilic substitution) injection. European Pharmacopoeia, 9th ed.; Council of Europe: Strasbourg, France; Volume 3, pp. 1141–1143.
- Pretze, M.; Wängler, C.; Wängler, B. 6-[18F]Fluoro-L-DOPA: A Well-Established Neurotracer with Expanding Application Spectrum and Strongly Improved Radiosyntheses. Bio. Med. Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Edwards, R.; Wirth, T. [18F]-6-Fluoro-3,4-Dihydroxy-L-Phenylalanine - Recent Modern Syntheses for an Elusive Radiotracer. J. Label. Compd. Radiopharm. 2015, 58, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Orlovskaya, V.V.; Fedorova, O.S.; Krasikova, R.N. Methods for the Synthesis of Fluorine-18-Labeled Aromatic Amino Acids, Radiotracers for Positron Emission Tomography (PET). Russ. Chem. Bull. 2015, 64, 1518–1535. [Google Scholar] [CrossRef]
- Pretze, M.; Franck, D.; Kunkel, F.; Foßhag, E.; Wängler, C.; Wängler, B. Evaluation of Two Nucleophilic Syntheses Routes for the Automated Synthesis of 6-[18F]fluoro-L-DOPA. Nucl. Med. Biol. 2017, 45, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.S.; Kaur, T.; Preshlock, S.; Tanzey, S.S.; Winton, W.P.; Sharninghausen, L.S.; Wiesner, N.; Brooks, A.F.; Sanford, M.S.; Scott, P. Copper-Mediated Late-Stage Radiofluorination: Five Years of Impact on Preclinical and Clinical PET Imaging. Clin. Transl. Imaging 2020, 8, 1–40. [Google Scholar] [CrossRef]
- Cai, L.; Lu, S.-Y.; Pike, V.W. Chemistry with [18F]Fluoride Ion. Eur. J. Org. Chem. 2008, 2008, 2853–2873. [Google Scholar] [CrossRef]
- Coenen, H.H.; Ermert, J. 18F-Labelling Innovations and Their Potential for Clinical Application. Clin. Transl. Imaging 2018, 6, 169–193. [Google Scholar] [CrossRef]
- Deng, X.; Rong, J.; Wang, L.; Vasdev, N.; Zhang, L.; Josephson, L.; Liang, S.H. Chemistry for Positron Emission Tomography: Recent Advances in 11C-, 18F-, 13N-, and 15O-Labeling Reactions. Angew. Chem. Int. Ed. 2019, 58, 2580–2605. [Google Scholar] [CrossRef]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjugate Chem. 2014, 26, 1–18. [Google Scholar] [CrossRef]
- Eberl, S.; Eriksson, T.; Svedberg, O.; Norling, J.; Henderson, D.; Lam, P.; Fulham, M.J. High Beam Current Operation of a PETtraceTM Cyclotron for 18F− Production. Appl. Radiat. Isot. 2012, 70, 922–930. [Google Scholar] [CrossRef]
- Lemaire, C.; Damhaut, P.; Plenevaux, A.; Comar, D. Enantioselective Synthesis of 6-[fluorine-18]-Fluoro-L-Dopa from No-Carrier-Added Fluorine-18-Fluoride. J. Nucl. Med. 1994, 35, 1996–2002. [Google Scholar]
- Krasikova, R.N.; Fedorova, O.S.; Mosevich, I.K.; Kuznetsova, O.F.; Korsakov, M.V.; Ametamey, S.M.; Schubiger, P.A. Asymmetric Synthesis of 6-[18F]Fluoro-L-DOPA Using a Chiral Nickel Complex of the Schiff Base of (S)-O-[(N-Benzylprolyl)-amino]benzophenone and Glycine. J. Labelled Comp. Radiopharm. 1999, 42, S102–S104. [Google Scholar]
- Lemaire, C.F.; Gillet, S.; Guillouet, S.; Plenevaux, A.; Aerts, J.; Luxen, A. Highly Enantioselective Synthesis of No-Carrier-Added 6-[18F]Fluoro-L-Dopa by Chiral Phase-Transfer Alkylation. Eur. J. Org. Chem. 2004, 2004, 2899–2904. [Google Scholar] [CrossRef]
- Krasikova, R.; Zaitsev, V.; Ametamey, S.; Kuznetsova, O.; Fedorova, O.; Mosevich, I.; Belokon, Y.; Vyskočil, Š.; Shatik, S.; Nader, M.; et al. Catalytic Enantioselective Synthesis of 18F-Fluorinated α-Amino Acids under Phase-Transfer Conditions Using (S)-NOBIN. Nucl. Med. Biol. 2004, 31, 597–603. [Google Scholar] [CrossRef]
- Libert, L.C.; Franci, X.; Plenevaux, A.R.; Ooi, T.; Maruoka, K.; Luxen, A.J.; Lemaire, C. Production at the Curie Level of No-Carrier-Added 6-18F-Fluoro-L-Dopa. J. Nucl. Med. 2013, 54, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, C.F.; Libert, L.; Franci, X.; Genon, J.-L.; Kuçi, S.; Giacomelli, F.; Luxen, A. Automated Production at the Curie Level of No-Carrier-Added 6-[18F]fluoro-L-Dopa and 2-[18F]fluoro-L-Tyrosine on a FASTlab Synthesizer. J. Label. Compd. Radiopharm. 2015, 58, 281–290. [Google Scholar] [CrossRef]
- [18F]FDOPA Nucleophilic Process. Available online: http://www.trasis.com/Tracers/18ffdopa (accessed on 18 September 2020).
- Wagner, F.M.; Ermert, J.; Coenen, H.H. Three-Step, “One-Pot” Radiosynthesis of 6-Fluoro-3,4-Dihydroxy-L-Phenylalanine by Isotopic Exchange. J. Nucl. Med. 2009, 50, 1724–1729. [Google Scholar] [CrossRef]
- Hoepping, A.; Müller, M.; Smits, R.; Mollitor, J.; Baumgart, D.; Clausnitzer, A. Precursors and process for the production of 18F-labelled amino acids. EP2746250A1, 25 June 2014. [Google Scholar]
- Lee, E.; Kamlet, A.S.; Powers, D.C.; Neumann, C.N.; Boursalian, G.B.; Furuya, T.; Choi, D.C.; Hooker, J.M.; Ritter, T. A Fluoride-Derived Electrophilic Late-Stage Fluorination Reagent for PET Imaging. Science 2011, 334, 639–642. [Google Scholar] [CrossRef]
- Brooks, A.F.; Topczewski, J.J.; Ichiishi, N.; Sanford, M.S.; Scott, P. Late-Stage [18F]fluorination: New Solutions to Old Problems. Chem. Sci. 2014, 5, 4545–4553. [Google Scholar] [CrossRef]
- Taylor, N.J.; Emer, E.; Preshlock, S.; Schedler, M.; Tredwell, M.; Verhoog, S.; Mercier, J.; Genicot, C.; Gouverneur, V. Derisking the Cu-Mediated 18F-Fluorination of Heterocyclic Positron Emission Tomography Radioligands. J. Am. Chem. Soc. 2017, 139, 8267–8276. [Google Scholar] [CrossRef]
- Cole, E.L.; Stewart, M.N.; Littich, R.; Hoareau, R.; Scott, P. Radiosyntheses Using Fluorine-18: The Art and Science of Late Stage Fluorination. Curr. Top. Med. Chem. 2014, 14, 875–900. [Google Scholar] [CrossRef] [PubMed]
- Preshlock, S.; Tredwell, M.; Gouverneur, V. 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef] [PubMed]
- Zarganes-Tzitzikas, T.; Clemente, G.S.; Elsinga, P.H.; Dömling, A. MCR Scaffolds Get Hotter with 18F-Labeling. Molecules 2019, 24, 1327. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W.; Aigbirhio, F.I. Reactions of Cyclotron-Produced [18F]fluoride with Diaryliodonium salts?A Novel Single-Step Route to No-Carrier-Added [18F]fluoroarenes. J. Chem. Soc. Chem. Commun. 1995, 21, 2215. [Google Scholar] [CrossRef]
- Pike, V.W. Hypervalent Aryliodine Compounds As Precursors for Radiofluorination. J. Label. Compd. Radiopharm. 2018, 61, 196–227. [Google Scholar] [CrossRef] [PubMed]
- DiMagno, S.G. Fluorination of Aromatic Ring Systems. US20110313170A1, 22 December 2011. [Google Scholar]
- Neumann, K.D.; Qin, L.; Vavere, A.L.; Snyder, S.E.; DiMagno, S.G. New Rapid Fluorination Process for the Production of Carrier-Free-F-18 6-[18F]FDA and 6-[18F]-l-DOPA. J. Nucl. Med. 2012, 53, 71. [Google Scholar]
- Ground Fluor Pharmaceuticals, Inc., Lincoln, Nebraska, USA, 2014. Technical Specification Sheet (PDF). Available online: http://www.gfpharma.com/pubs/FDOPA_CutSheet.Pdf (accessed on 4 December 2014).
- Kuik, W.-J.; Kema, I.P.; Brouwers, A.H.; Zijlma, R.; Neumann, K.D.; Dierckx, R.A.J.O.; DiMagno, S.G.; Elsinga, P.H. In Vivo Biodistribution of No-Carrier-Added 6-18F-Fluoro-3,4-Dihydroxy-L-Phenylalanine (18F-DOPA), Produced by a New Nucleophilic Substitution Approach, Compared with Carrier-Added 18F-DOPA, Prepared by Conventional Electrophilic Substitution. J. Nucl. Med. 2014, 56, 106–112. [Google Scholar] [CrossRef]
- Collins, J.; Waldmann, C.M.; Drake, C.; Slavik, R.; Ha, N.S.; Sergeev, M.; Lazari, M.; Shen, B.; Chin, F.T.; Moore, M.; et al. Production of Diverse PET Probes with Limited Resources: 24 18F-Labeled Compounds Prepared with a Single Radiosynthesizer. Proc. Natl. Acad. Sci. USA 2017, 114, 11309–11314. [Google Scholar] [CrossRef]
- Wang, J.; Holloway, T.; Lisova, K.; Van Dam, R.M. Green and Efficient Synthesis of the Radiopharmaceutical [18F]FDOPA Using a Microdroplet Reactor. React. Chem. Eng. 2020, 5, 320–329. [Google Scholar] [CrossRef]
- Edwards, R.; Westwell, A.D.; Daniels, S.; Wirth, T. Convenient Synthesis of Diaryliodonium Salts for the Production of [18F]F-DOPA. Eur. J. Org. Chem. 2014, 2015, 625–630. [Google Scholar] [CrossRef]
- Maisonial, A.; Serre, A.; Ouadi, A.; Schmitt, S.; Canitrot, D.; Léal, F.; Miot-Noirault, E.; Brasse, D.; Marchand, P.; Chezal, J.M. Base/Cryptand/Metal-Free Automated Nucleophilic Radiofluorination of [18 F]FDOPA from Iodonium Salts: Importance of Hydrogen Carbonate Counterion. Eur. J. Org. Chem. 2018, 2018, 7058–7065. [Google Scholar] [CrossRef]
- Zischler, J.; Krapf, P.; Richarz, R.; Zlatopolskiy, B.D.; Neumaier, B. Automated Synthesis of 4-[18 F]fluoroanisole, [18F]DAA1106 and 4-[18F]FPhe Using Cu-Mediated Radiofluorination under “minimalist” Conditions. Appl. Radiat. Isot. 2016, 115, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Ichiishi, N.; Canty, A.J.; Yates, B.F.; Sanford, M.S. Cu-Catalyzed Fluorination of Diaryliodonium Salts with KF. Org. Lett. 2013, 15, 5134–5137. [Google Scholar] [CrossRef] [PubMed]
- Ichiishi, N.; Brooks, A.F.; Topczewski, J.J.; Rodnick, M.E.; Sanford, M.S.; Scott, P. Copper-Catalyzed [18F]Fluorination of (Mesityl)(aryl)iodonium Salts. Org. Lett. 2014, 16, 3224–3227. [Google Scholar] [CrossRef]
- Orlovskaya, V.V.; Modemann, D.J.; Kuznetsova, O.F.; Fedorova, O.S.; Urusova, E.A.; Kolks, N.; Neumaier, B.; Krasikova, R.N.; Zlatopolskiy, B.D. Alcohol-Supported Cu-Mediated 18F-Fluorination of Iodonium Salts under “Minimalist” Conditions. Molecules. 2019, 24, 3197. [Google Scholar] [CrossRef]
- Tredwell, M.; Preshlock, S.M.; Taylor, N.J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Genicot, C.; Gouverneur, V. A General Copper-Mediated Nucleophilic18F Fluorination of Arenes. Angew. Chem. Int. Ed. 2014, 53, 7751–7755. [Google Scholar] [CrossRef]
- Ye, Y.; Schimler, S.D.; Hanley, P.S.; Sanford, M.S. Cu(OTf)2-Mediated Fluorination of Aryltrifluoroborates with Potassium Fluoride. J. Am. Chem. Soc. 2013, 135, 16292–16295. [Google Scholar] [CrossRef]
- Qiao, J.X.; Lam, P.Y.S. Copper-Promoted Carbon-Heteroatom Bond Cross-Coupling with Boronic Acids and Derivatives. Synthesis 2010, 2011, 829–856. [Google Scholar] [CrossRef]
- Mossine, A.V.; Brooks, A.F.; Makaravage, K.J.; Miller, J.M.; Ichiishi, N.; Sanford, M.S.; Scott, P. Synthesis of [18F]Arenes via the Copper-Mediated [18F]Fluorination of Boronic Acids. Org. Lett. 2015, 17, 5780–5783. [Google Scholar] [CrossRef]
- Makaravage, K.J.; Brooks, A.F.; Mossine, A.V.; Sanford, M.S.; Scott, P. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef]
- Orlovskaya, V.V.; Craig, A.S.; Krasikova, R.N.; Fedorova, O.S.; Kuznetsova, O.F.; Neumaier, B.; Zlatopolskiy, B.D. Facile Synthesis of 6-L-[18F]fluoro-m-Tyrosine via Alcohol-Enhanced Cu-Mediated Radiofluorination of Bpin-Substituted Chiral Ni-BPB-AA Complex. Presented at the 19th European Symposium on Radiopharmacy and Radiopharmaceuticals (ESRR’18), Groningen, The Netherlands, 5–8 April 2018. [Google Scholar]
- Craig, A.; Kolks, N.; Urusova, E.A.; Zischler, J.; Brugger, M.; Endepols, H.; Neumaier, B.; Zlatopolskiy, B.D. Preparation of Labeled Aromatic Amino Acids via Late-Stage 18F-Fluorination of Chiral Nickel and Copper Complexes. Chem. Commun. 2020, 56, 9505–9508. [Google Scholar] [CrossRef] [PubMed]
- Preshlock, S.; Calderwood, S.; Verhoog, S.; Hienzsch, A.; Cailly, T.; Schedler, M.; Mollitor, J.; Hoepping, A.; Genicot, C.; Tredwell, M.; et al. Enhanced Copper-Mediated 18F-Fluorination of Aryl Boronic Esters Provides Eight Radiotracers for PET Applications. Chem. Commun. 2016, 52, 8361–8364. [Google Scholar] [CrossRef] [PubMed]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Kordys, E.; Endepols, H.; Neumaier, B. Copper-Mediated Aromatic Radiofluorination Revisited: Efficient Production of PET Tracers on a Preparative Scale. Chem. A Eur. J. 2015, 21, 5972–5979. [Google Scholar] [CrossRef] [PubMed]
- Zischler, J.; Kolks, N.; Modemann, D.; Neumaier, B.; Zlatopolskiy, B.D. Alcohol-Enhanced Cu-Mediated Radiofluorination. Chem. A Eur. J. 2017, 23, 3251–3256. [Google Scholar] [CrossRef]
- Krasikova, R.; Fedorova, O.; Kuznetsova, O.; Orlovskaya, V. Nucleophilic Synthesis of 6-[18F]fluoro-L-DOPA via Copper Mediated Radiofluorination. Presented at the International Symposium on Trends in Radiopharmaceuticals (ISTR-2019), IAEA Headquarters, Vienna, Austria, 28 October–1 November 2019. [Google Scholar]
- Antuganov, D.O.; Zykov, M.; Timofeev, V.; Timofeeva, K.; Antuganova, Y.; Orlovskaya, V.; Fedorova, O.S.; Krasikova, R.N. Copper-Mediated Radiofluorination of Aryl Pinacolboronate Esters: A Straightforward Protocol by Using Pyridinium Sulfonates. Eur. J. Org. Chem. 2018, 2019, 918–922. [Google Scholar] [CrossRef]
- Mossine, A.V.; Tanzey, S.S.; Brooks, A.F.; Makaravage, K.J.; Ichiishi, N.; Miller, J.M.; Henderson, B.D.; Skaddan, M.B.; Sanford, M.S.; Scott, P. One-Pot Synthesis of High Molar Activity 6-[18F]fluoro-L-DOPA by Cu-Mediated Fluorination of a BPin Precursor. Org. Biomol. Chem. 2019, 17, 8701–8705. [Google Scholar] [CrossRef]
- Mossine, A.V.; Tanzey, S.S.; Brooks, A.F.; Makaravage, K.J.; Ichiishi, N.; Miller, J.M.; Henderson, B.D.; Erhard, T.; Bruetting, C.; Skaddan, M.B.; et al. Synthesis of High-Molar-Activity [18F]-6-Fluoro-L-DOPA Suitable for Human Use via Cu-Mediated Fluorination of a BPin Precursor. Nat. Protoc. 2020, 15, 1742–1759. [Google Scholar] [CrossRef]
- Zarrad, F.; Zlatopolskiy, B.D.; Krapf, P.; Zischler, J.; Neumaier, B. A Practical Method for the Preparation of 18F-Labeled Aromatic Amino Acids from Nucleophilic [18F]Fluoride and Stannyl Precursors for Electrophilic Radiohalogenation. Molecules 2017, 22, 2231. [Google Scholar] [CrossRef]
- ICH Guideline of Elemental Impurities Q3D. Available online: https://www.ema.europa.eu/en/ich-q3d-elemental-impurities (accessed on 22 September 2020).
- Antuganov, D.O.; Antuganova, Y.; Zykova, T.; Krasikova, R.N. Use of Capillary Electrophoresis for the Determination of Impurities in Preparations of Fluorine-18 Labelled PET Radiopharmaceuticals. J. Pharm. Biomed. Anal. 2019, 173, 68–74. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 18F source | Precursor, mg/µmol | Trapping/ Reaction Solvent | RCY, % (EOS); Synthesis Time, min | Molar Activity (Am) | Reference |
|---|---|---|---|---|---|
| 20Ne(d,α)18F 0.35% F2 | 60/97 | CFCl3 | 25 ± 3; 45 min | 10.6 ± 2.3 GBq/mmol | [22] |
| 20Ne(d,α)18F 0.25% F2 | 55–65/ 89–105 | CHCl3 | 15 ± 5 | 8.5 ± 3.3 GBq/mmol | [24] |
| 18O(p,n)18F 0.5% F2 | 30/48.5 | CHCl3 | 23 ± 4 | 121 ± 27 GBq/mmol | [24] |
| 20Ne(d,α)18F 0.5% F2 | 45/73 | CDCl3 | 17.7 ± 2.3; 45 min | 14.5 ± 3.5 GBq/mmol | [23] |
| 18O(p,n)18F 18O-water target | 6.2/10 | acetone-d6 | 12.1 ± 3.7 | 3.4± 0.1 GBq/µmol | [31] * |
| Precursor Structure | [18F]Fluoride Elution | Precursor/ Cu-Complex, µmol | Reaction Solvent | Hydrolysis | RCY,% Synthesis Time | Ref. |
|---|---|---|---|---|---|---|
 | K222/K2C2O4/ K2CO3 /CH3CN/ H2O | 20/20 | DMF | 57% HI, 150 °C, 10 min | 8.7 146 min | [72] |
 | Et4NHCO3/ n-BuOH | 60/53 | DMA/ n-BuOH | 12 M HCl,130 °C, 5 min | 40 ± 4 a - | [81] |
 | Bu4NOTf/ Cs2CO3/H2O | 4/20 b | DMF | 12 M HCl, 0.25M ascorbic acid 130 °C, 5 min | 6 ± 1 110 min | [84] |
| Bu4NOTf/ i-PrOH | 5/8 | DMA/ i-PrOH | 6 M HCl 120 °C, 10 min | 12 80 min | [82] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasikova, R.N. Nucleophilic Synthesis of 6-l-[18F]FDOPA. Is Copper-Mediated Radiofluorination the Answer? Molecules 2020, 25, 4365. https://doi.org/10.3390/molecules25194365
Krasikova RN. Nucleophilic Synthesis of 6-l-[18F]FDOPA. Is Copper-Mediated Radiofluorination the Answer? Molecules. 2020; 25(19):4365. https://doi.org/10.3390/molecules25194365
Chicago/Turabian StyleKrasikova, Raisa N. 2020. "Nucleophilic Synthesis of 6-l-[18F]FDOPA. Is Copper-Mediated Radiofluorination the Answer?" Molecules 25, no. 19: 4365. https://doi.org/10.3390/molecules25194365
APA StyleKrasikova, R. N. (2020). Nucleophilic Synthesis of 6-l-[18F]FDOPA. Is Copper-Mediated Radiofluorination the Answer? Molecules, 25(19), 4365. https://doi.org/10.3390/molecules25194365