Pyrroloquinoline Quinone Inhibits Rotenone-Induced Microglia Inflammation by Enhancing Autophagy


Abstract
1. Introduction
2. Results
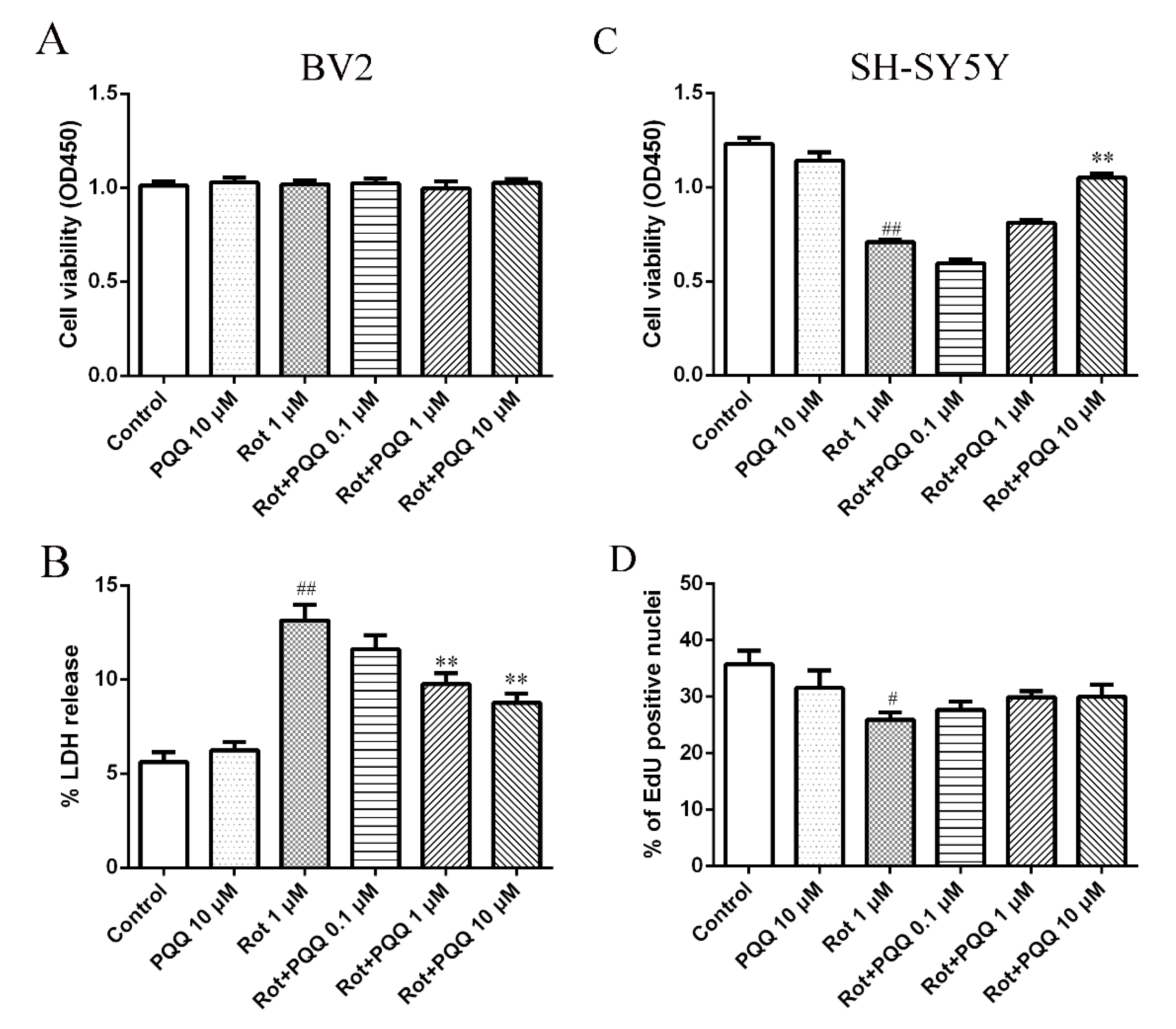
2.1. PQQ Exhibited Protective Effect against Microglia-Mediated SH-SY5Y Cell Death
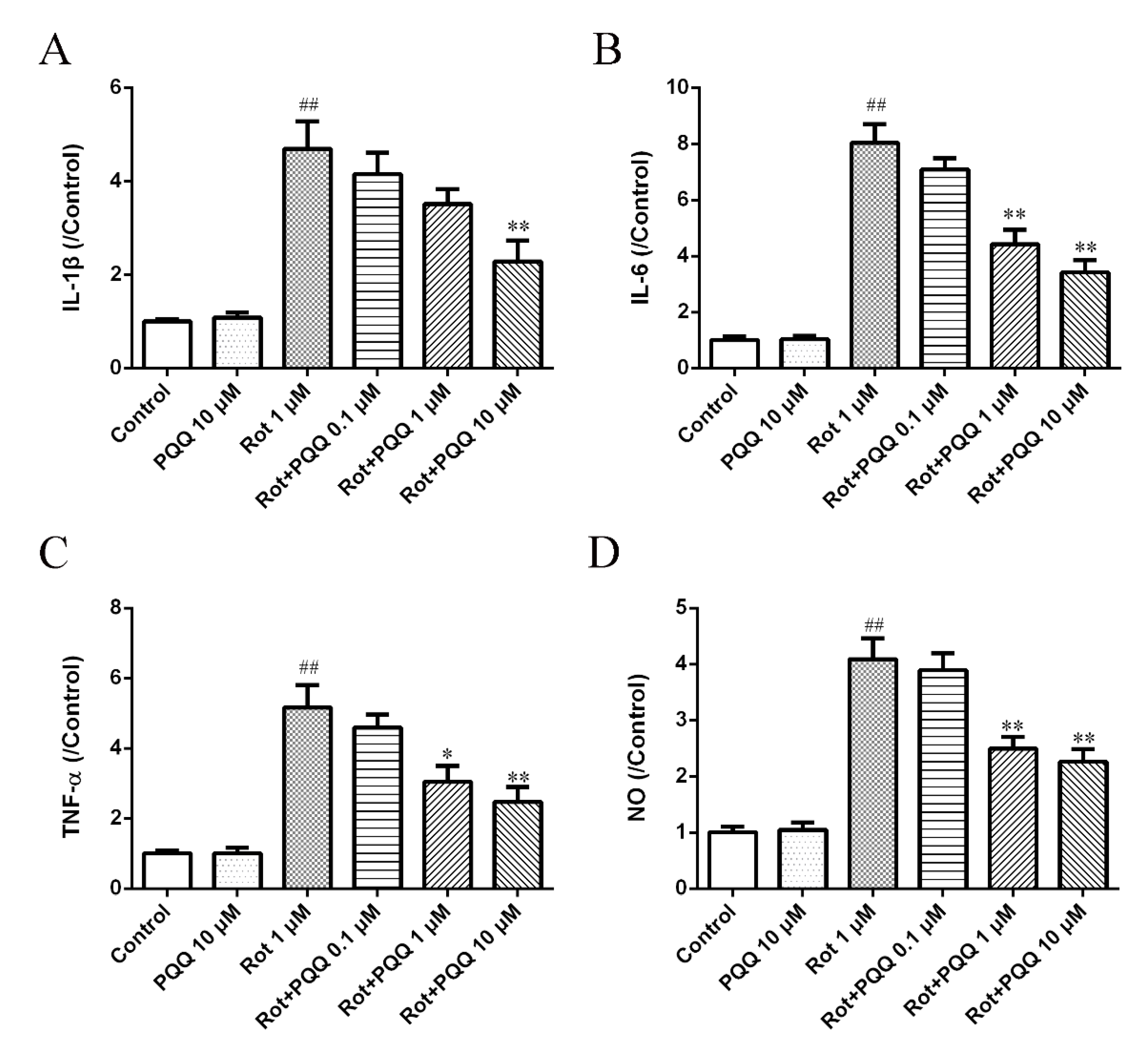
2.2. PQQ Suppressed Rotenone-Induced Inflammation in BV2 Microglia
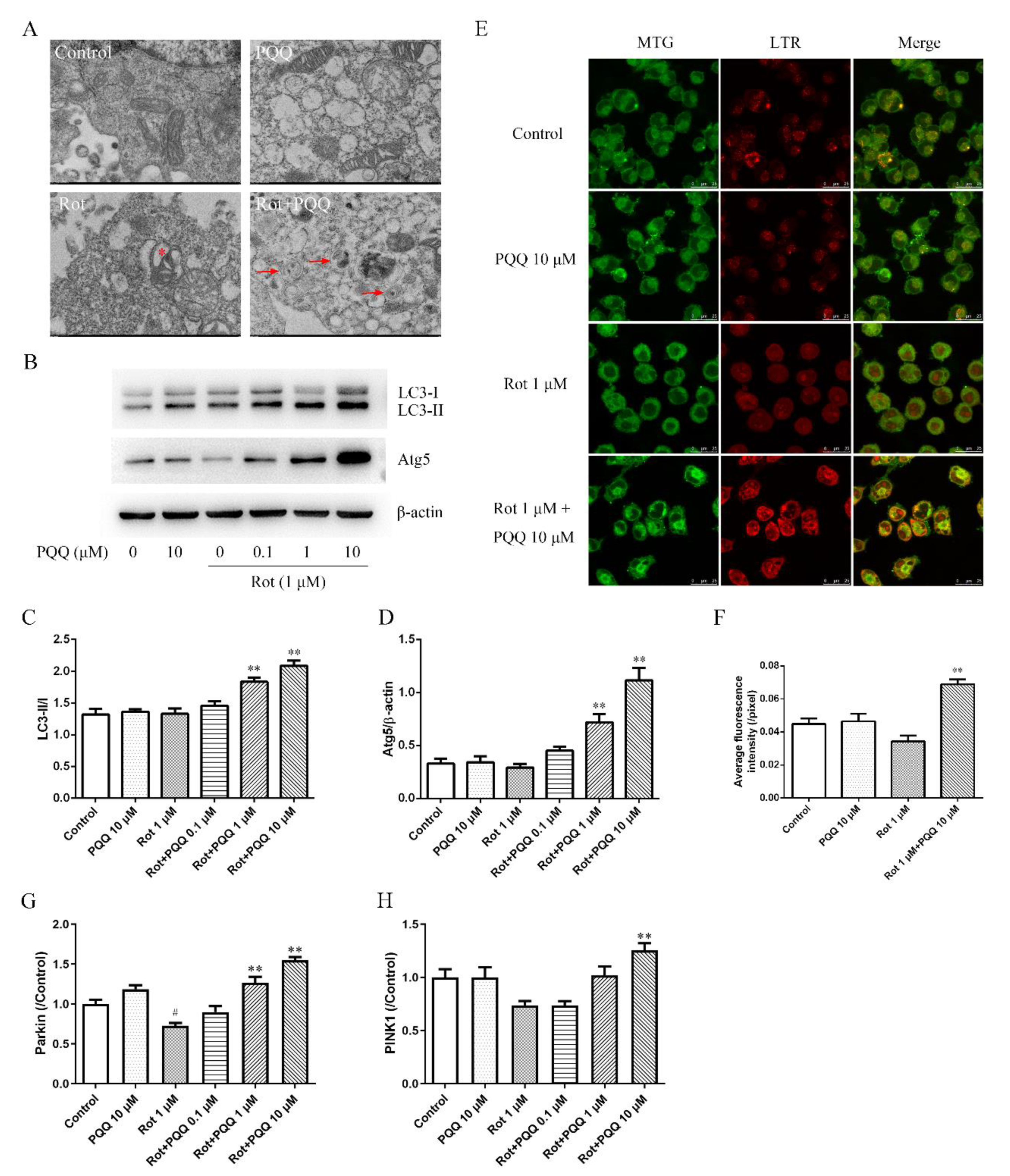
2.3. PQQ Enhanced Autophagy in Rotenone-Injured BV2 Microglia
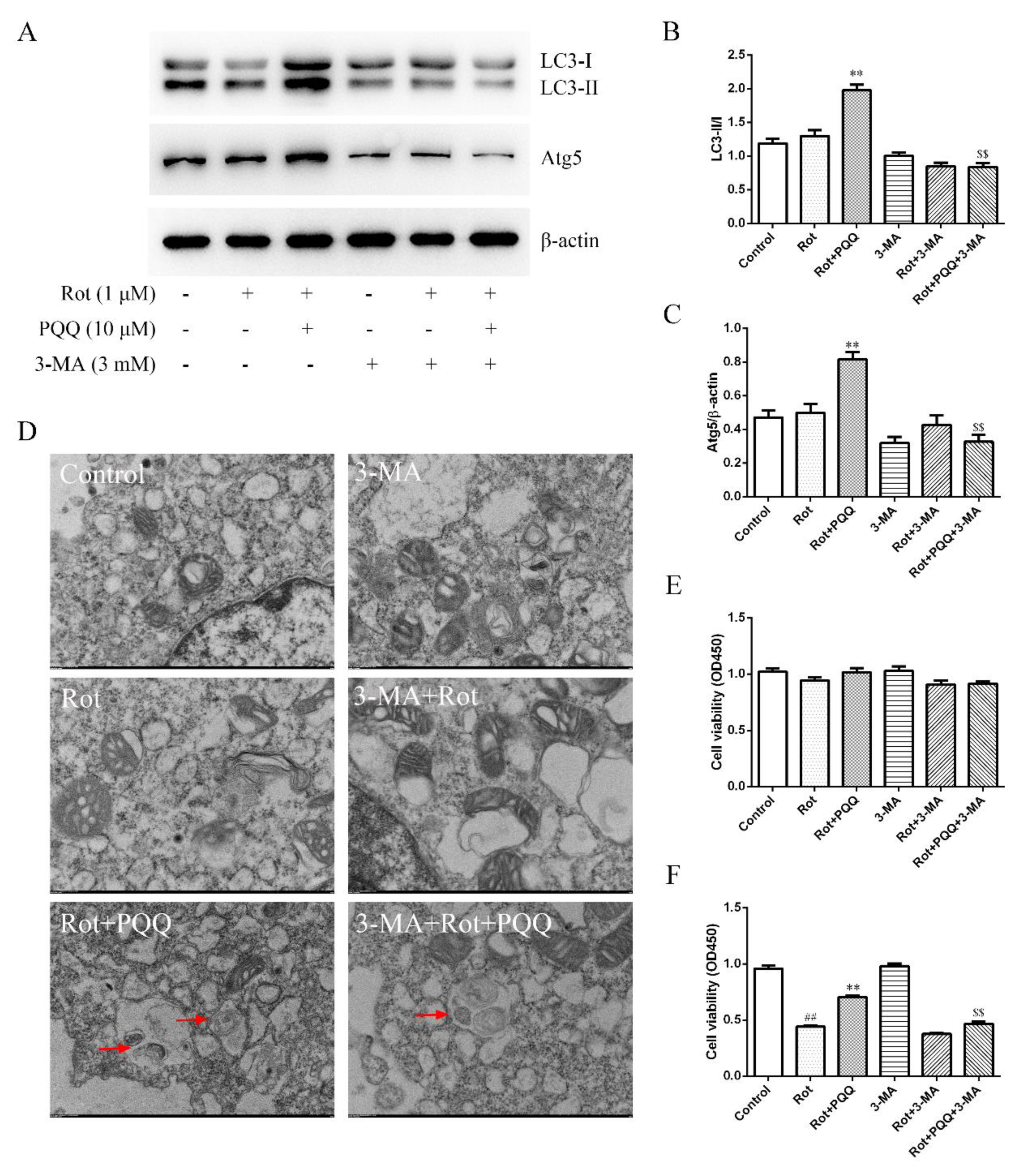
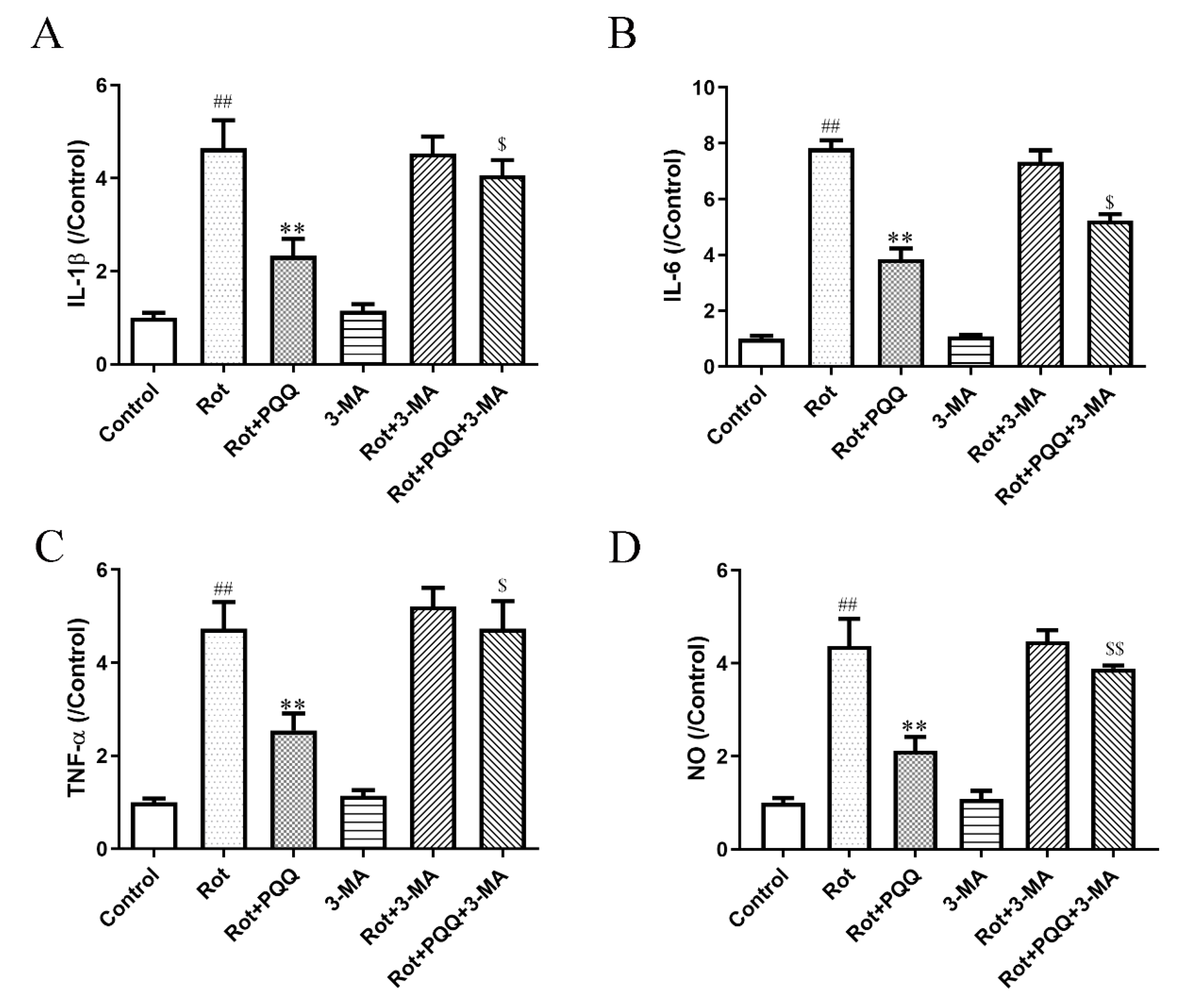
2.4. Autophagy Inhibitor Attenuated the Effect of PQQ on Rotenone-Induced Inflammation
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Culture and Treatment
4.3. Cell Viability Measurement
4.4. Cell Toxicity Assay
4.5. Cell Proliferation Assay
4.6. ELISA for Pro-Inflammatory Factors
4.7. NO Release Measurement
4.8. Detection of the Autophagic Degradation of Mitochondria (Mitophagy) by Immunofluorescence
4.9. Western Blotting Analysis
4.10. Transmission Electron Microscopy (TEM)
4.11. qRT-PCR
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 38, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells as Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yu, L.; Kong, L.; Ma, R.; Zhang, J.; Zhu, Q.; Zhu, J.; Hao, D. Pyrroloquinoline quinone (PQQ) inhibits lipopolysaccharide induced inflammation in part via downregulated NF-kappaB and p38/JNK activation in microglial and attenuates microglia activation in lipopolysaccharide treatment mice. PLoS ONE 2014, 9, e109502. [Google Scholar]
- Zhang, Q.; Chen, S.; Yu, S.; Qin, J.; Zhang, J.; Cheng, Q.; Ke, K.; Ding, F. Neuroprotective effects of pyrroloquinoline quinone against rotenone injury in primary cultured midbrain neurons and in a rat model of Parkinson’s disease. Neuropharmacology 2016, 108, 238–251. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, J.; Jiang, C.; Qin, J.; Ke, K.; Ding, F. Involvement of ERK1/2 pathway in neuroprotective effects of pyrroloquinoline quinine against rotenone-induced SH-SY5Y cell injury. Neuroscience 2014, 270, 183–191. [Google Scholar] [CrossRef]
- Qin, J.; Wu, M.; Yu, S.; Gao, X.; Zhang, J.; Dong, X.; Ji, J.; Zhang, Y.; Zhou, L.; Zhang, Q.; et al. Pyrroloquinoline quinone-conferred neuroprotection in rotenone models of Parkinson’s disease. Toxicol. Lett. 2015, 238, 70–82. [Google Scholar] [CrossRef]
- Johnson, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology 2015, 46, 101–116. [Google Scholar] [CrossRef]
- Tabata, Y.; Imaizumi, Y.; Sugawara, M.; Andoh-Noda, T.; Banno, S.; Chai, M.; Sone, T.; Yamazaki, K.; Ito, M.; Tsukahara, K.; et al. T-type Calcium Channels Determine the Vulnerability of Dopaminergic Neurons to Mitochondrial Stress in Familial Parkinson Disease. Stem Cell Rep. 2018, 11, 1171–1184. [Google Scholar] [CrossRef]
- Lu, J.; Chen, S.; Shen, M.; He, Q.; Zhang, Y.; Shi, Y.; Ding, F.; Zhang, Q. Mitochondrial regulation by pyrroloquinoline quinone prevents rotenone-induced neurotoxicity in Parkinson’s disease models. Neurosci. Lett. 2018, 687, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid Type 2 (CB2) Receptors Activation Protects against Oxidative Stress and Neuroinflammation Associated Dopaminergic Neurodegeneration in Rotenone Model of Parkinson’s Disease. Front. Neurosci. 2016, 10, 321. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhou, T.; Chen, Y.; Lin, D.; Jing, X.; Peng, S.; Zheng, D.; Zeng, Z.; Lei, M.; Wu, X.; et al. Rifampicin inhibits rotenone-induced microglial inflammation via enhancement of autophagy. Neurotoxicology 2017, 63, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Maturana, M.G.; Pinheiro, A.S.; de Souza, T.L.; Follmer, C. Unveiling the role of the pesticides paraquat and rotenone on alpha-synuclein fibrillation in vitro. Neurotoxicology 2015, 46, 35–43. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Keller, C.W.; Lunemann, J.D. Autophagy and Autophagy-Related Proteins in CNS Autoimmunity. Front. Immunol. 2017, 8, 165. [Google Scholar] [CrossRef]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598. [Google Scholar] [CrossRef]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Carreira, R.S. Autophagy in health and disease. 5. Mitophagy as a way of life. Am. J. Physiol. Cell Physiol. 2010, 299, C203–C210. [Google Scholar] [CrossRef]
- Trempe, J.F.; Sauve, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Menade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 2013, 340, 1451–1455. [Google Scholar] [CrossRef]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, H.; Guo, X.; Ge, D.; Shi, Y.; Lu, X.; Lu, J.; Chen, J.; Ding, F.; Zhang, Q. Involvement of Akt/mTOR in the Neurotoxicity of Rotenone-Induced Parkinson’s Disease Models. Int. J. Environ. Res. Public Health 2019, 16, 3811. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, D.; Hu, Q.; Wang, G. Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS ONE 2013, 8, e72046. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Y.; Zhang, S.P.; Cao, C.; Loh, Y.P.; Cheng, Y. Aberrations in Peripheral Inflammatory Cytokine Levels in Parkinson Disease: A Systematic Review and Meta-analysis. JAMA Neurol. 2016, 73, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef]
- Bassani, T.B.; Vital, M.A.; Rauh, L.K. Neuroinflammation in the pathophysiology of Parkinson’s disease and therapeutic evidence of anti-inflammatory drugs. Arq. Neuropsiquiatr. 2015, 73, 616–623. [Google Scholar] [CrossRef]
- Lin, T.K.; Cheng, C.H.; Chen, S.D.; Liou, C.W.; Huang, C.R.; Chuang, Y.C. Mitochondrial dysfunction and oxidative stress promote apoptotic cell death in the striatum via cytochrome c/caspase-3 signaling cascade following chronic rotenone intoxication in rats. Int. J. Mol. Sci. 2012, 13, 8722–8739. [Google Scholar] [CrossRef]
- Emmrich, J.V.; Hornik, T.C.; Neher, J.J.; Brown, G.C. Rotenone induces neuronal death by microglial phagocytosis of neurons. FEBS J. 2013, 280, 5030–5038. [Google Scholar] [CrossRef]
- Cheng, Q.; Chen, J.; Guo, H.; Lu, J.L.; Zhou, J.; Guo, X.Y.; Shi, Y.; Zhang, Y.; Yu, S.; Zhang, Q.; et al. Pyrroloquinoline quinone promotes mitochondrial biogenesis in rotenone-induced Parkinson’s disease model via AMPK activation. Acta Pharmacol. Sin. 2020. [Google Scholar] [CrossRef]
- Su, P.; Zhang, J.; Wang, D.; Zhao, F.; Cao, Z.; Aschner, M.; Luo, W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience 2016, 319, 155–167. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Bussi, C.; Peralta Ramos, J.M.; Arroyo, D.S.; Gaviglio, E.A.; Gallea, J.I.; Wang, J.M.; Celej, M.S.; Iribarren, P. Autophagy down regulates pro-inflammatory mediators in BV2 microglial cells and rescues both LPS and alpha-synuclein induced neuronal cell death. Sci. Rep. 2017, 7, 43153. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Dodson, M.; Ravi, S.; Redmann, M.; Ouyang, X.; Darley Usmar, V.M.; Zhang, J. Bioenergetic adaptation in response to autophagy regulators during rotenone exposure. J. Neurochem. 2014, 131, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Huang, M.; Yao, Y.M. Autophagy and proinflammatory cytokines: Interactions and clinical implications. Cytokine Growth Factor Rev. 2018, 43, 38–46. [Google Scholar] [CrossRef]
- Harris, J. Autophagy and IL-1 Family Cytokines. Front. Immunol. 2013, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Wang, Z.; Han, N.; Zhao, K.; Li, Y.; Chi, Y.; Wang, B. Protective effects of pyrroloquinoline quinine against oxidative stress-induced cellular senescence and inflammation in human renal tubular epithelial cells via Keap1/Nrf2 signaling pathway. Int. Immunopharmacol. 2019, 72, 445–453. [Google Scholar] [CrossRef]
- Lin, X.; Yang, F.; Huang, J.; Jiang, S.; Tang, Y.; Li, J. Ameliorate effect of pyrroloquinoline quinone against cyclophosphamide-induced nephrotoxicity by activating the Nrf2 pathway and inhibiting the NLRP3 pathway. Life Sci. 2020, 256, 117901. [Google Scholar] [CrossRef]
- Zhang, Q.; Ding, M.; Cao, Z.; Zhang, J.; Ding, F.; Ke, K. Pyrroloquinoline quinine protects rat brain cortex against acute glutamate-induced neurotoxicity. Neurochem. Res. 2013, 38, 1661–1671. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, C.; Tao, R.; Xu, X.; Xu, L.; Cheng, H.; Wang, Y.; Zhang, D. Pyrroloquinoline Quinone Decelerates Rheumatoid Arthritis Progression by Inhibiting Inflammatory Responses and Joint Destruction via Modulating NF-kappaB and MAPK Pathways. Inflammation 2016, 39, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Chowanadisai, W.; Bauerly, K.A.; Tchaparian, E.; Wong, A.; Cortopassi, G.A.; Rucker, R.B. Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGC-1alpha expression. J. Biol. Chem. 2010, 285, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wang, B.; Li, W.; Wang, L.; Song, X.; Guo, C.; Li, Y.; Liu, F.; Zhu, F.; Wang, Q.; et al. Systemic application of 3-methyladenine markedly inhibited atherosclerotic lesion in ApoE(-/-) mice by modulating autophagy, foam cell formation and immune-negative molecules. Cell Death Dis. 2016, 7, e2498. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Loeffler, D.A. Influence of Normal Aging on Brain Autophagy: A Complex Scenario. Front. Aging Neurosci. 2019, 11, 49. [Google Scholar] [CrossRef]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59–66. [Google Scholar] [CrossRef]
- Su, L.Y.; Luo, R.; Liu, Q.; Su, J.R.; Yang, L.X.; Ding, Y.Q.; Xu, L.; Yao, Y.G. Atg5- and Atg7-dependent autophagy in dopaminergic neurons regulates cellular and behavioral responses to morphine. Autophagy 2017, 13, 1496–1511. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Ivankovic, D.; Chau, K.Y.; Schapira, A.H.; Gegg, M.E. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J. Neurochem. 2016, 136, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Cherra, S.J., 3rd; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef]
- Henn, I.H.; Bouman, L.; Schlehe, J.S.; Schlierf, A.; Schramm, J.E.; Wegener, E.; Nakaso, K.; Culmsee, C.; Berninger, B.; Krappmann, D.; et al. Parkin mediates neuroprotection through activation of IkappaB kinase/nuclear factor-kappaB signaling. J. Neurosci. 2007, 27, 1868–1878. [Google Scholar] [CrossRef]
- Tran, T.A.; Nguyen, A.D.; Chang, J.; Goldberg, M.S.; Lee, J.K.; Tansey, M.G. Lipopolysaccharide and tumor necrosis factor regulate Parkin expression via nuclear factor-kappa B. PLoS ONE 2011, 6, e23660. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5′–3′) |
|---|---|
| Parkin | Forward-aaggggattgcgactcact Reverse-cttttgtccaccctgtaggc |
| PINK1 | Forward-gcgaagccatcttaagcaaa Reverse-tgggaccatctctggatctt |
| 18S rRNA | Forward-tagagggacaagtggcgttc Reverse-cgctgagccagtcagtgt |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Zhou, J.; Shen, M.; Xu, H.; Yu, S.; Cheng, Q.; Ding, F. Pyrroloquinoline Quinone Inhibits Rotenone-Induced Microglia Inflammation by Enhancing Autophagy. Molecules 2020, 25, 4359. https://doi.org/10.3390/molecules25194359
Zhang Q, Zhou J, Shen M, Xu H, Yu S, Cheng Q, Ding F. Pyrroloquinoline Quinone Inhibits Rotenone-Induced Microglia Inflammation by Enhancing Autophagy. Molecules. 2020; 25(19):4359. https://doi.org/10.3390/molecules25194359
Chicago/Turabian StyleZhang, Qi, Jing Zhou, Mi Shen, Hui Xu, Shu Yu, Qiong Cheng, and Fei Ding. 2020. "Pyrroloquinoline Quinone Inhibits Rotenone-Induced Microglia Inflammation by Enhancing Autophagy" Molecules 25, no. 19: 4359. https://doi.org/10.3390/molecules25194359
APA StyleZhang, Q., Zhou, J., Shen, M., Xu, H., Yu, S., Cheng, Q., & Ding, F. (2020). Pyrroloquinoline Quinone Inhibits Rotenone-Induced Microglia Inflammation by Enhancing Autophagy. Molecules, 25(19), 4359. https://doi.org/10.3390/molecules25194359