Autophagy, One of the Main Steps in Periodontitis Pathogenesis and Evolution


 ,
,  ,
,
Abstract
1. Introduction
1.1. Periodontitis—Today’s Opinions and Tomorrow’s Perspectives
- –
- –
- Periodontitis is an inflammatory disease of the periodontal tissues with a more complex pathogenic mechanism [31,32]. Periodontium plays essential supporting roles and includes the mineralized bonelike cementum, the periodontal ligament, and the alveolar bone. The periodontal ligament is a connective tissue between the cementum and the alveolar bone [33,34]. The main symptoms of periodontitis include apical migration of gingival attachment, the formation of a periodontal pocket, and progressive periodontal bone loss. Periodontitis progression culminates with tooth loss [11,35].
1.2. Periodontitis Initiation and Progression Steps
1.2.1. Step 1: Bacterial Infection
1.2.2. Step 2: Inflammation
1.2.3. Step 3: Oxidative Stress
- ROS are able to activate NF-kB, initiating a signaling cascade involved in inflammatory and immune responses [62].
- ROS can activate JNK, initiating cell apoptosis [63].
- ROS are involved in inflammasome formation and activation, triggering cell death [64].
- ROS are the main contributors in the autophagy stage [65].
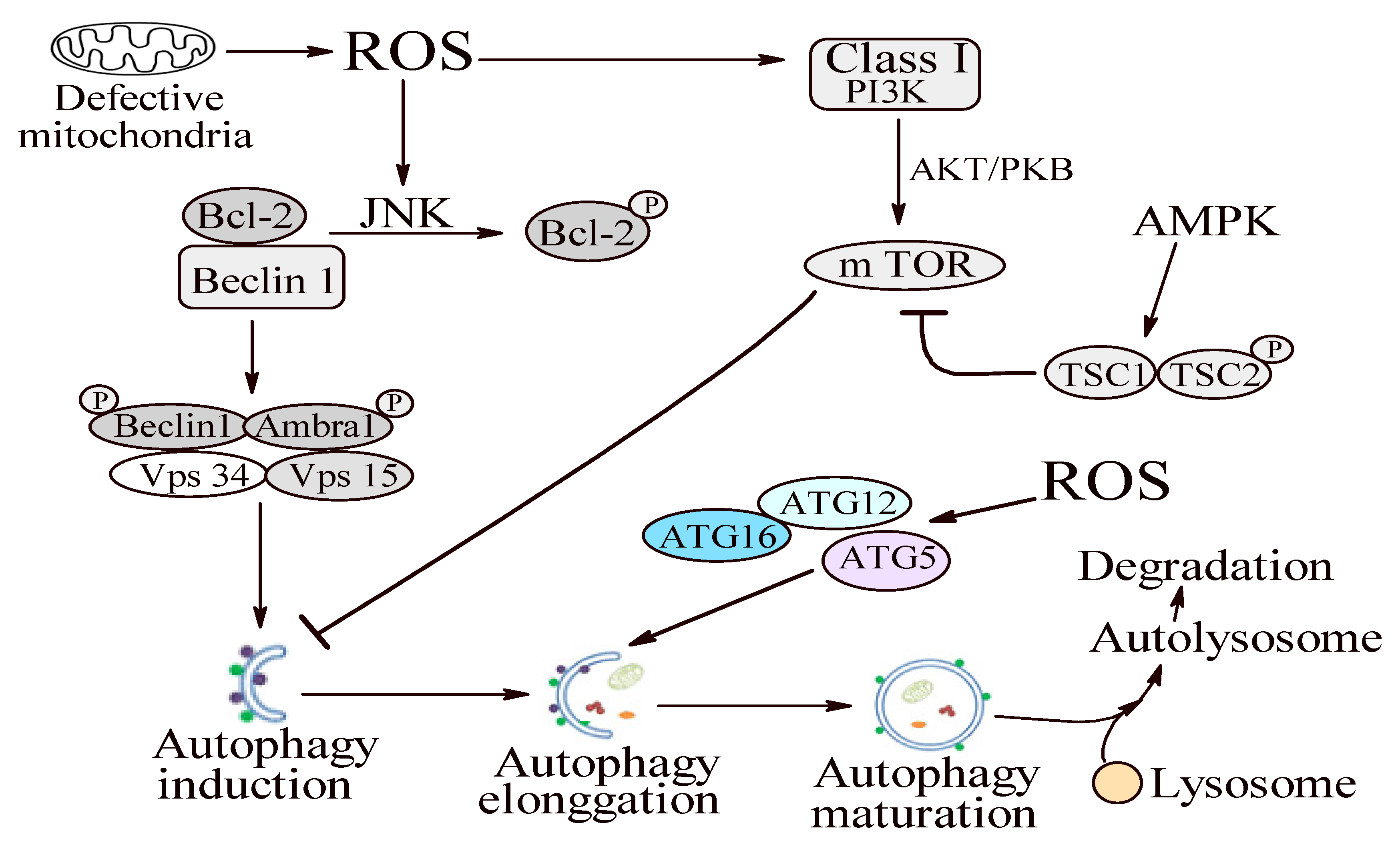
1.2.4. Step 4: Autophagy
1.3. Autophagy in Periodontitis Context
1.3.1. Oxidative Stress and Autophagy: A Double Sense Connection
1.3.2. Autophagy—A Possible Antioxidant Pathway?
1.3.3. Autophagy—A Janus God in Periodontitis
Periodontal Pathogen Invasion Control by Autophagy
Autophagy and Innate Immune Signaling Pathway Regulation
- a.
- The Inflammasome-Dependent Manner
- b.
- The Inflammasome-Independent Manner
Periodontal Cells Protection against Apoptosis
2. Conclusions
Funding
Conflicts of Interest
References
- Bluteau, G.; Luder, H.U.; De Bari, C.; Mitsia, T.A. Stem Cells for Tooth Engineerin. Eur. Cell. Mater. 2008, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Botelho, J.; Cavacas, M.A.; Machado, V.; Mendes, J.J. Dental stem cells: Recent progresses in tissue engineering and regenerative medicine. Ann. Med. 2017, 49, 644–651. [Google Scholar] [CrossRef]
- Eke, P.I.; Wei, L.; Borgnakke, W.S.; Thornton-Evans, G.; Zhang, X.; Lu, H.; McGuire, L.C.; Genco, R.J. Periodontitis prevalence in adults ≥ 65 years of age, in the USA. Periodontol. 2000 2016, 72, 76–95. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Ali, K.; Ardu, S.; Tredwin, C.; Hu, B. Autophagy in dental tissues: A double-edged sword. Cell Death Dis. 2016, 7, e2192. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q. Oxidative Stress and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 179–198. [Google Scholar] [CrossRef]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Rotilio, G.; Ciriolo, M.R. Under the ROS. thiol network is the principal suspect for autophagy commitment. Autophagy 2010, 6, 999–1005. [Google Scholar] [CrossRef]
- Romandini, M.; Laforí, A.; Romandini, P.; Baima, G.; Cordaro, M.J. Periodontitis and platelet count: A new potential link with cardiovascular and other systemic inflammatory diseases. Clin. Periodontol. 2018, 45, 1299–1310. [Google Scholar] [CrossRef]
- El-Shinnawi, U.; Soory, M. Actions of Adjunctive Nutritional Antioxidants in Periodontitis and Prevalent Systemic Inflammatory Diseases. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 261–276. [Google Scholar] [CrossRef]
- Kinane, D.F.; Galicia, J.C.; Gorr, S.U.; Stathopoulou, P.G.; Benakanakere, M. P. gingivalis interactions with epithelial cells. Front. Biosci. 2008, 13, 966–984. [Google Scholar] [CrossRef]
- Varela-López, A.; Navarro-Hortal, M.D.; Giampieri, F.; Bullón, P.; Battino, M.; Quiles, J.L. Nutraceuticals in periodontal health: A systematic review on the role of vitamins in periodontal health maintenance. Molecules 2018, 23, 1226. [Google Scholar] [CrossRef]
- Armitage, G.C. Periodontal diagnoses and classification of periodontal diseases. Periodontol. 2000 2004, 34, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Takane, M.; Sugano, N.; Iwasaki, H.; Iwano, Y.; Shimizu, N.; Ito, K. New biomarker evidence of oxidative DNA damage in whole saliva from clinically healthy and periodontally diseased individuals. J. Periodontol. 2002, 73, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, Y.; Sugano, N.; Tanaka, H.; Ito, K. Detection of periodontopathic bacteria and an oxidative stress marker in saliva from periodontitis patients. Oral Microbiol. Immunol. 2005, 20, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Canakci, C.F.; Canakci, V.; Tatar, A.; Eltas, A.; Sezer, U.; Cicek, Y.; Oztas, S. Increased salivary level of 8-hydroxydeoxyguanosine is a marker of premature oxidative mitochondrial DNA damage in gingival tissue of patients with periodontitis. Arch. Immunol. Ther. Exp. 2009, 57, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Gornitsky, M.; Velly, A.M.; Yu, H.; Benarroch, M.; Schipper, H.M. Salivary DNA, lipid, and protein oxidation in nonsmokers with periodontal disease. Free Radic. Biol. Med. 2009, 46, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Sezer, U.; Cicek, Y.; Canakci, C.F. Increased salivary levels of 8-hydroxydeoxyguanosine may be a marker for disease activity for periodontitis. Dis. Markers 2012, 32, 165–172. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Calenic, B.; Mocanu, B.; Didilescu, A.; Mohora, M.; Spinu, T.; Greabu, M. Salivary biomarkers: Relationship between oxidative stress and alveolar bone loss in chronic periodontitis. Acta Odontol. Scand. 2014, 72, 42–47. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ngo, L.Q.; Promsudthi, A.; Surarit, R. Salivary oxidative stress biomarkers in chronic periodontitis and acute coronary syndrome. Clin. Oral Investig. 2017, 21, 2345–2353. [Google Scholar] [CrossRef]
- Dede, F.O.; Ozden, F.O.; Avci, B. 8-hydroxy-deoxyguanosine levels in gingival crevicular fluid and saliva in patients with chronic periodontitis after initial periodontal treatment. J. Periodontol. 2013, 84, 821–828. [Google Scholar] [CrossRef]
- Akalin, F.A.; Baltacioglu, E.; Alver, A.; Karabulut, E. Lipid peroxidation levels and total oxidant status in serum, saliva and gingival crevicular fluid in patients with chronic periodontitis. J. Clin. Periodontol. 2007, 34, 558–565. [Google Scholar] [CrossRef]
- Khalili, J.; Biloklytska, H.F. Salivary malondialdehyde levels in clinically healthy and periodontal diseased individuals. Oral Dis. 2008, 14, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Baltacıoğlu, E.; Kehribar, M.A.; Yuva, P.; Alver, A.; Atagün, O.S.; Karabulut, E.; Akalın, F.A. Total oxidant status and bone resorption biomarkers in serum and gingival crevicular fluid of patients with periodontitis. J. Periodontol. 2014, 85, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, S.; Lal, N.; Mahdi, A.A.; Mittal, M.; Singh, B.; Pandey, S. Evaluation of antioxidant enzymes activity and malondialdehyde levels in patients with chronic periodontitis and diabetes mellitus. J. Periodontol. 2014, 85, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Chen, H.S.; Chen, S.L.; Ho, Y.P.; Ho, K.Y.; Wu, Y.M.; Hung, C.C. Lipid peroxidation: A possible role in the induction and progression of chronic periodontitis. J. Periodontal. Res. 2005, 40, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.S.; Patil, V.P.; Gokhale, N.; Acharya, A.; Kangokar, P. Chronic periodontitis in type 2 diabetes mellitus: Oxidative stress as a common factor in periodontal tissue injury. J. Clin. Diagn. Res. 2016, 10, BC12–BC16. [Google Scholar] [CrossRef]
- Wei, D.; Zhang, X.L.; Wang, Y.Z.; Yang, C.X.; Chen, G. Lipid peroxidation levels, total oxidant status and superoxide dismutase in serum, saliva and gingival crevicular fluid in chronic periodontitis patients before and after periodontal therapy. Aust. Dent. J. 2010, 55, 70–78. [Google Scholar] [CrossRef]
- Gürsoy, U.K.; Könönen, E.; Tervahartiala, T.; Gürsoy, M.; Pitkänen, J.; Torvi, P.; Suominen, A.L.; Pussinen, P.; Sorsa, T. Molecular forms and fragments of salivary MMP-8 in relation to periodontitis. J. Clin. Periodontol. 2018, 45, 1421–1428. [Google Scholar] [CrossRef]
- Stadler, A.F.; Angst, P.; Arce, R.M.; Gomes, S.C.; Oppermann, R.V.; Susin, C. Gingival Crevicular Fluid Levels of Cytokines/Chemokines in Chronic Periodontitis: A Meta-Analysis. J. Clin. Periodontol. 2016, 43, 727–745. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, Q.; Xu, C.; Loo, W.T.Y.; Wang, M.; Wen, G.; Cheung, M.N.B.; Bai, L.; Dou, Y.; Chow, W.C. Comparative evaluation of cytokines in gingival crevicular fluid and saliva of patients with aggressive periodontitis. Int. J. Biol. Markers 2013, 28, 108–112. [Google Scholar] [CrossRef]
- Hendek, M.K.; Erdemir, E.O.; Kisa, U.; Ozcan, G. Effect of initial periodontal therapy on oxidative stress markers in gingival crevicular fluid, saliva, and serum in smokers and non-smokers with chronic periodontitis. J. Periodontol. 2015, 86, 273–282. [Google Scholar] [CrossRef]
- Chapple, I.L.; Brock, G.; Eftimiadi, C.; Matthews, J.B. Glutathione in gingival crevicular fluid and its relation to local antioxidant capacity in periodontal health and disease. Mol. Pathol. 2002, 55, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed]
- El-Awady, A.R.; Messer, R.L.; Gamal, A.Y.; Sharawy, M.M.; Wenger, K.H.; Lapp, C.A. Periodontal ligament fibroblasts sustain destructive immune modulators of chronic periodontitis. J. Periodontol. 2010, 81, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Morsczeck, C.; Gotz, W.; Schierholz, J.; Zeilhofer, F.; Kuhn, U.; Mohl, C.; Hoffmann, K.H. Isolation of precursor cells (PCs) from human dental follicle of wisdom teeth. Matrix Biol. 2005, 24, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Tonetti, M.S.; Van Dyke, T.E. Periodontitis and atherosclerotic cardiovascular disease: Consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J. Periodontol. 2013, 84 (Suppl. 4), S24–S29. [Google Scholar] [CrossRef]
- Román-Malo, L.; Bullon, P. Influence of the Periodontal Disease, the Most Prevalent Inflammatory Event, in Peroxisome Proliferator-Activated Receptors Linking Nutrition and Energy Metabolism. Int. J. Mol. Sci. 2017, 18, 1438. [Google Scholar] [CrossRef]
- Mendes, L.; Azevedo, N.F.; Felino, A.; Pinto, M.G. Relationship between invasion of the periodontium by periodontal pathogens and periodontal disease: A systematic review. Virulence 2015, 6, 208–215. [Google Scholar] [CrossRef]
- Bai, Y.; Wei, Y.; Wu, L.; Wei, J.; Wang, X. C/EBP β mediates endoplasmic reticulum stress regulated inflammatory response and extracellular matrix degradation in LPS-stimulated human periodontal ligament cells. Int. J. Mol. Sci. 2016, 17, 385. [Google Scholar] [CrossRef]
- Isacco, C.G.; Ballini, A.; de Vito, D.; Nguyen, K.C.D.; Cantore, S.; Bottalico, L.; Quagliuolo, L.; Boccellino, M.; Di Domenico, M.; Santacroce, L. Rebalance the oral microbiota as efficacy tool in endocrine, metabolic, and immune disorders. Endocr. Metab. Immune Disord. Drug Targets 2020. [Google Scholar] [CrossRef]
- Inchingolo, F.; Martelli, F.S.; Isacco, C.G.; Borsani, E.; Cantore, S.; Corcioli, F.; Boddi, A.; Nguyễn, K.C.D.; De Vito, D.; Aityan, S.K. Chronic Periodontitis and Immunity, Towards the Implementation of a Personalized Medicine: A Translational Research on Gene Single Nucleotide Polymorphisms (SNPs) Linked to Chronic Oral Dysbiosis in 96 Caucasian Patients. Biomedicines 2020, 8, 115. [Google Scholar] [CrossRef]
- Inchingolo, F.; Dipalma, G.; Cirulli, N.; Cantore, S.; Saini, R.S.; Altini, V.; Santacroce, L.; Ballini, A.; Saini, R. Microbiological results of improvement in periodontal condition by administration of oral probiotics. J. Biol. Regul. Homeost. Agents 2018, 32, 1323–1328. [Google Scholar] [PubMed]
- Ballini, A.; Cantore, S.; Farronato, D.; Cirulli, N.; Inchingolo, F.; Papa, F.; Malcangi, G.; Inchingolo, A.D.; Dipalma, G.; Sardaro, N. Periodontal disease and bone pathogenesis: The crosstalk between cytokines and porphyromonas gingivalis. J. Biol. Regul. Homeost. Agents 2015, 29, 273–281. [Google Scholar] [PubMed]
- Ballini, A.; Dipalma, G.; Isacco, C.G.; Boccellino, M.; Di Domenico, M.; Santacroce, L.; Nguyễn, K.C.D.; Salvatore Scacco, S.; Calvani, M.; Anna Boddi, A. Oral Microbiota and Immune System Crosstalk: A Translational Research. Biology 2020, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, F. Unhealthy Behaviours: An International Comparison. PLoS ONE 2015, 10, e0141834. [Google Scholar] [CrossRef] [PubMed]
- Lamont, R.J.; Hajishengallis, G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol. Med. 2015, 21, 172–183. [Google Scholar] [CrossRef]
- Zhu, X.Q.; Lu, W.; Chen, Y.; Cheng, X.F.; Qiu, J.Y.; Xu, Y.; Sun, Y. Effects of Porphyromonas gingivalis lipopolysaccharidetolerized monocytes on inflammatory responses in neutrophils. PLoS ONE 2016, 11, e0161482. [Google Scholar] [CrossRef]
- Tamaki, N.; Hayashida, H.; Fukui, M.; Kitamura, M.; Kawasaki, K.; Nakazato, M.; Maeda, T.; Satio, T.; Ito, H.O. Oxidative stress and antibody levels to periodontal bacteria in adults: The Nagasaki Islands study. Oral Dis. 2014, 20, e49–e56. [Google Scholar] [CrossRef]
- McClean, C.; Harris, R.A.; Brown, M.; Brown, J.C.; Davison, G.W. Effects of exercise intensity on postexercise endothelial function and oxidative stress. Oxid. Med. Cell. Longev. 2015, 2015, 723679. [Google Scholar] [CrossRef]
- DiMeo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Choe, Y.; Yu, J.Y.; Son, Y.O.; Park, S.M.; Kim, J.G.; Shi, X.; Lee, J.C. Continuously generated H2O2 stimulates the proliferation and osteoblastic differentiation of human periodontal ligament fibroblasts. J. Cell. Biochem. 2012, 113, 1426–1436. [Google Scholar] [CrossRef]
- Liu, C.; Mo, L.; Niu, Y.; Li, X.; Zhou, X.; Xu, X. The Role of Reactive Oxygen Species and Autophagy in Periodontitis and Their Potential Linkage. Front. Physiol. 2017, 8, 439. [Google Scholar] [CrossRef] [PubMed]
- Cavalla, F.; Osorio, C.; Paredes, R.; Valenzuela, M.A.; Garcia-Sesnich, J.; Sorsa, T.; Tervahartiala, T.; Marcela Hernández, M. Matrix metalloproteinases regulate extracellular levels of SDF-1/CXCL12, IL-6, and VEGF in hydrogen peroxide-stimulated human periodontal ligament fibroblasts. Cytokine 2015, 73, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Burdon, R.H.; Gill, V.; Alliangana, D. Hydrogen peroxide in relation to proliferation and apoptosis in BHK-21 hamster fibroblasts. Free Radic. Res. 1996, 24, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; di Stefano, L.; di Meo, S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion 2013, 13, 71–82. [Google Scholar] [CrossRef]
- Netto, L.E.; Antunes, F. The roles of peroxiredoxin and thioredoxin in hydrogen peroxide sensing and in signal transduction. Mol. Cells 2016, 39, 65–71. [Google Scholar] [CrossRef]
- Waddington, R.J.; Moseley, R.; Embery, G. Reactive oxygen species: A potential role in the pathogenesis of periodontal diseases. Oral Dis. 2000, 6, 138–151. [Google Scholar] [CrossRef]
- Baltacioglu, E.; Yuva, P.; Aydin, G.; Alver, A.; Kahraman, C.; Karabulut, E.; Akalın, F.A. Lipid peroxidation levels and total oxidant/antioxidant status in serum and saliva from patients with chronic and aggressive periodontitis. Oxidative stress index: A new biomarker for periodontal disease? J. Periodontol. 2014, 85, 1432–1441. [Google Scholar] [CrossRef]
- Battino, M.; Bullon, P.; Wilson, M.; Newman, H. Oxidative injury and inflammatory periodontal diseases: The challenge of anti-oxidants to free radicals and reactive oxygen species. Crit. Rev. Oral Biol. Med. 1999, 10, 458–476. [Google Scholar] [CrossRef]
- Díaz, C.M.; Bullon, B.; Ruiz-Salmerón, R.J.; Fernández-Riejos, P.; Fernández-Palacín, A.; Battino, M.; Cordero, M.D.; Quiles, J.L.; Varela-López, A.; Bullón, P. Molecular inflammation and oxidative stress are shared mechanisms involved in both myocardial infarction and periodontitis. J. Periodontal. Res. 2020, 55, 519–528. [Google Scholar] [CrossRef]
- Pradeep, A.R.; Rao, N.S.; Bajaj, P.; Agarwal, E. 8-Isoprostane: A lipid peroxidation product in gingival crevicular fluid in healthy, gingivitis and chronic periodontitis subjects. Arch. Oral Biol. 2013, 58, 500–504. [Google Scholar] [CrossRef]
- Nibali, L.; Donos, N. Periodontitis and redox status: A review. Curr. Pharm. Des. 2013, 19, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Nakajima, A.; Sakon-Komazawa, S.; Piao, J.H.; Xue, X.; Okumura, K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 2006, 13, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxininteracting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Kabat, A.M.; Pott, J.; Maloy, K.J. The Mucosal Immune System and Its Regulation by Autophagy. Front. Immunol. 2016, 7, 240. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and Autophagy-Related Proteins in the Immune System. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef]
- Desideri, E.; Filomeni, G.; Ciriolo, M.R. Glutathione participates in the modulation of starvationinduced autophagy in carcinoma cells. Autophagy 2012, 8, 1769–1781. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef]
- Cardaci, S.; Filomeni, G.; Ciriolo, M.R. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J. Cell Sci. 2012, 125, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- He, Z.J.; Zhu, F.Y.; Li, S.S.; Zhong, L.; Tan, H.Y.; Wang, K. Inhibiting ROS-NF-kappaB-dependent autophagy enhanced brazilin-induced apoptosis in head and neck squamous cell carcinoma. Food Chem. Toxicol. 2017, 101, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, F.; Afrin, S.; Forbes-Hernandez, T.Y.; Gasparrini, M.; Cianciosi, D.; Reboredo-Rodriguez, P.; Varela-Lopez, A.; Quiles, J.L.; Battino, M. Autophagy in Human Health and Disease: Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 577–634. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Wang, B.; Dai, X.; Ding, W.; Yang, T.; Li, X.; Lewin, S.; Xu, L.; Lian, J.; He, F. HCC cells with high levels of Bcl-2 are resistant to ABT-737 via activation of the ROS-JNK-autophagy pathway. Free Radic. Biol. Med. 2014, 70, 194–203. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Liu, W.; Xue, P.; Zhang, Y.; Wang, Q.; Jin, Y. Increased autophagy is required to protect periodontal ligament stem cells from apoptosis in inflammatory microenvironment. J. Clin. Periodontol. 2016, 43, 618–625. [Google Scholar] [CrossRef]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human ATG12_ATG5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59–66. [Google Scholar] [CrossRef]
- Mai, S.; Muster, B.; Bereiter-Hahn, J.; Jendrach, M. Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence lifespan. Autophagy 2012, 8, 47–62. [Google Scholar] [CrossRef]
- Bullon, P.; Cordero, M.D.; Quiles, J.L.; Ramirez-Tortosa Mdel, C.; Gonzalez-Alonso, A.; Alfonsi, S.; García-Marín, R.; de Miguel, M.; Battino, M. Autophagy in periodontitis patients and gingival fibroblasts: Unraveling the link between chronic diseases and inflammation. BMC Med. 2012, 10, 122. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Zhang, J.; Kim, J.; Alexander, A.; Cai, S.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; Di Nardo, A.; Han, J.M. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Ji-Hoon Lee, J.H.; Paull, T.T. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Dermit, M.; Casado, P.; Rajeeve, V.; Wilkes, E.H.; Foxler, D.E.; Campbell, H.; Critchlow, S.; Sharp, T.V.; J G Gribben, J.G.; Unwin, R. Oxidative stress downstream of mTORC1 but not AKT causes a proliferative defect in cancer cells resistant to PI3K inhibition. Oncogene 2016, 36, 2762–2774. [Google Scholar] [CrossRef]
- Su, R.; Jin, X.; Zhang, W.; Li, Z.; Liu, X.; Ren, J. Particulate matter exposure induces the autophagy of macrophages via oxidative stress-mediated PI3K/AKT/mTOR pathway. Chemosphere 2017, 167, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Stafford, P.; Higham, J.; Pinnock, A.; Murdoch, C.; Douglas, C.W.; Stafford, G.P.; Lambert, D.V. Gingipain-dependent degradation of mammalian target of rapamycin pathway proteins by the periodontal pathogen Porphyromonas gingivalis during invasion. Mol. Oral Microbiol. 2013, 28, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Ekuni, D.; Tomofuji, T.; Irie, K.; Kunitomo, M.; Uchida, Y.; Fukuhara, D.; Manabu Morita, M. Visualization of oxidative stress induced by experimental periodontitis in keap1-dependent oxidative stress detector-luciferase mice. Int. J. Mol. Sci. 2016, 17, 1907. [Google Scholar] [CrossRef]
- Sima, C.; Aboodi, G.M.; Lakschevitz, F.S.; Sun, C.; Goldberg, M.B.; Glogauer, M. Nuclear factor erythroid 2-related factor 2 down-regulation in oral neutrophils is associated with periodontal oxidative damage and severe chronic periodontitis. Am. J. Pathol. 2016, 186, 1417–1426. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, H.M.; Shin, D.M.; Kim, W.; Yuk, J.M.; Jin, H.S.; Lee, S.H.; Cha, G.H.; Kim, J.M.; Zee-Won Lee, Z.W. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 2012, 11, 457–468. [Google Scholar] [CrossRef]
- Jiang, M.; Li, Z.; Zhu, G. The role of autophagy in the pathogenesis of periodontal disease. Oral Dis. 2020, 26, 259–269. [Google Scholar] [CrossRef]
- Belanger, M.; Rodrigues, P.H.; Dunn, W.A., Jr.; Progulske-Fox, A. Autophagy: A highway for Porphyromonas gingivalis. Autophagy 2006, 2, 165–170. [Google Scholar] [CrossRef]
- Park, M.H.; Jeong, S.Y.; Na, H.S.; Chung, J. Porphyromonas gingivalis induces autophagy in THP-1-derived macrophages. Mol. Oral Microbiol. 2017, 32, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Blasi, I.; Korostoff, J.; Dhingra, A.; Reyes-Reveles, J.; Shenker, B.J.; Shahabuddin, N.; Alexander, D.; Lally, E.T.; Bragin, A.; Boesze-Battaglia, K. Variants of Porphyromonas gingivalis lipopolysaccharide alter lipidation of autophagic protein, microtubule-associated protein 1 light chain 3, LC3. Mol. Oral Microbiol. 2016, 31, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Lee, H.K. Pattern recognition receptors and autophagy. Front. Immunol. 2014, 5, 300. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef] [PubMed]
- Benetti, E.; Chiazza, F.; Patel, N.S.; Collino, M. The NLRP3 Inflammasome as a Novel Player of the Intercellular Crosstalk in Metabolic Disorders. Mediat. Inflamm. 2013, 678627. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef]
- Anton, Z.; Landajuela, A.; Hervas, J.H.; Montes, L.R.; Hernandez-Tiedra, S.; Velasco, G.; Goñi, F.M.; Alonso, A. Human Atg8-cardiolipin interactions in mitophagy: Specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy 2016, 12, 2386–2403. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Bo-Gie, Y.; Takas, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J. Autophagy controls IL-1β secretion by targeting pro-IL-1β for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Castillo, E.F.; Dekonenko, A.; Arko-Mensah, J.; Mandell, M.A.; Dupont, N.; Jiang, S.; Delgado-Vargas, M.; Timmins, G.S.; Bhattacharya, D.; Yang, H. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, E3168–E3176. [Google Scholar] [CrossRef]


{kind=link}
| Marker | Type of Marker | Pattern |
|---|---|---|
| 8-hydroxy-2-deoxyguanosine (8-OHdG) | Oxidative stress DNA damage marker | Increased in saliva [12,13,14,15,16,17,18] No significant change in saliva [13] |
| Malondialdehyde (MDA) | Oxidative stress protein damage marker | Increased in saliva [17,18,19,20,21,22,23] |
| Protein carbonylation | Oxidative stress protein damage marker | Increased in saliva [15,18] |
| Salivary total antioxidant capacity | Antioxidant | Decreased in saliva [17] |
| Uric acid | Antioxidant | Decreased in saliva [17] |
| Reduced and oxidized glutathione (GSH and GSSG) | Antioxidant | Decreased in saliva [24] |
| Superoxide dismutase (SOD) | Enzymatic antioxidant | Decreased in saliva [23,25] Increased in saliva [26] |
| Glutathione peroxidase (GPX) | Enzymatic antioxidant | Decreased in saliva [17,23] No significant change in saliva [24] |
| matrix metalloproteinases-8 | Bone loss marker | Increased in saliva [17,27] |
| C-terminal telopeptide of type I collagen (CTX I) | Bone loss marker | Increased in saliva [17] |
| IL-1β | Cytokines | Increased [28,29] |
| IL-6 | Cytokines | Increased [28,29] |
| IFN-γ | Cytokines | Increased [28,29] |
| Marker | Type of Marker | Pattern |
|---|---|---|
| Malondialdehyde (MDA) | Oxidative stress protein damage marker | Increased [20,26] |
| 8-hydroxy-2-deoxyguanosine (8-OHdG) | Oxidative stress DNA damage marker | Increased [30] |
| Superoxide dismutase (SOD) | Enzymatic antioxidant | Increased [26] |
| Reduced and oxidized glutathione (GSH and GSSG) | Antioxidant | Decreased [31] |
| IL-1β | Cytokines | Increased [28,29] |
| IL-6 | Cytokines | Increased [28,29] |
| IFN-γ | Cytokines | Increased [28,29] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greabu, M.; Giampieri, F.; Imre, M.M.; Mohora, M.; Totan, A.; Pituru, S.M.; Ionescu, E. Autophagy, One of the Main Steps in Periodontitis Pathogenesis and Evolution. Molecules 2020, 25, 4338. https://doi.org/10.3390/molecules25184338
Greabu M, Giampieri F, Imre MM, Mohora M, Totan A, Pituru SM, Ionescu E. Autophagy, One of the Main Steps in Periodontitis Pathogenesis and Evolution. Molecules. 2020; 25(18):4338. https://doi.org/10.3390/molecules25184338
Chicago/Turabian StyleGreabu, Maria, Francesca Giampieri, Marina Melescanu Imre, Maria Mohora, Alexandra Totan, Silviu Mirel Pituru, and Ecaterina Ionescu. 2020. "Autophagy, One of the Main Steps in Periodontitis Pathogenesis and Evolution" Molecules 25, no. 18: 4338. https://doi.org/10.3390/molecules25184338
APA StyleGreabu, M., Giampieri, F., Imre, M. M., Mohora, M., Totan, A., Pituru, S. M., & Ionescu, E. (2020). Autophagy, One of the Main Steps in Periodontitis Pathogenesis and Evolution. Molecules, 25(18), 4338. https://doi.org/10.3390/molecules25184338