
Five Novel Non-Sialic Acid-Like Scaffolds Inhibit In Vitro H1N1 and H5N2 Neuraminidase Activity of Influenza a Virus

 and
and
Abstract
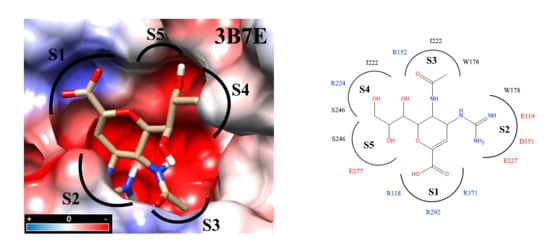
1. Introduction
2. Results
2.1. Analysis of the Drug-Like Structures
2.2. Analysis of the Amino Acid Sequences of Influenza a Viruses
2.3. Molecular Docking
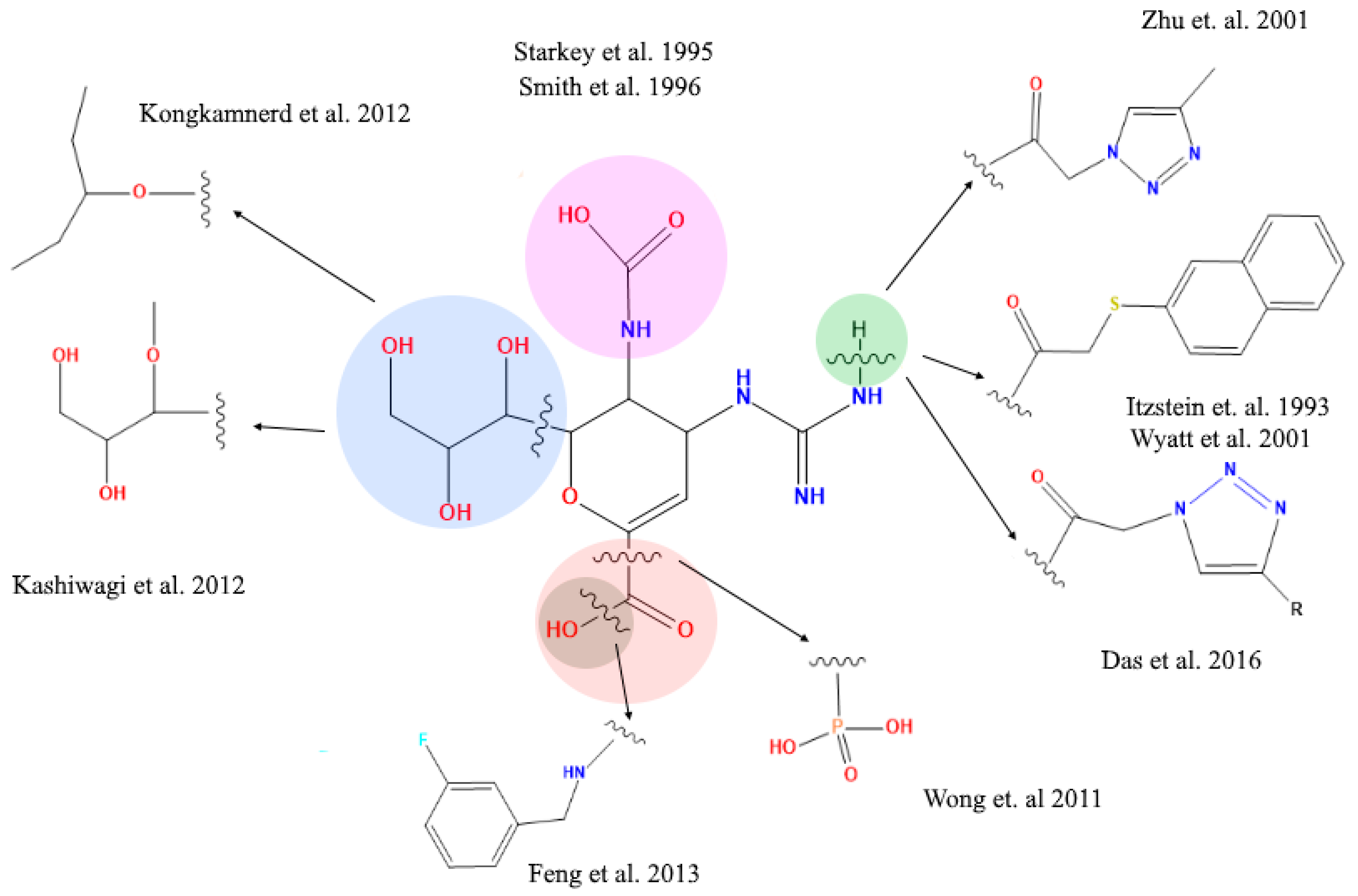
2.4. Analysis of Functional Substituent Groups on Zanamivir
2.5. Determination of Neuraminidase Activity
2.6. Determination of Kinetic Parameters
2.7. Determination of Half Maximal Inhibitory Concentration (IC50)
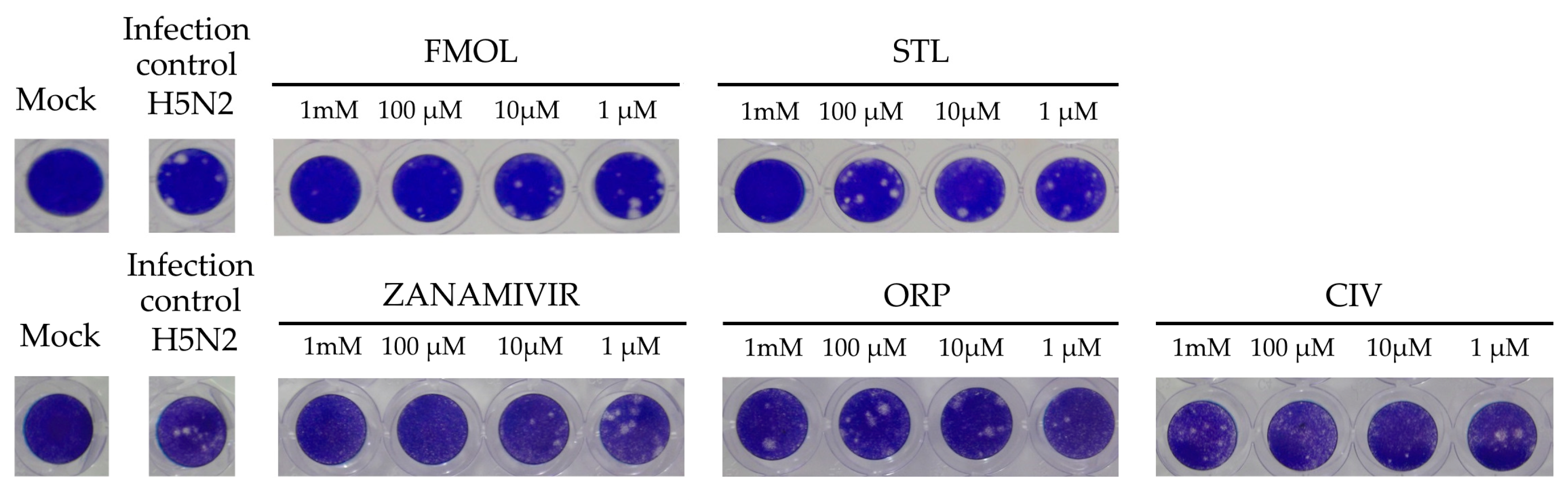
2.8. Inhibition of Viral Replication
3. Discussion
4. Materials and Methods
4.1. Molecular Docking
4.2. Cells and Viral Strains
4.3. Small Organic Molecules
4.4. Measurement of NA Activities
4.5. NA Enzyme Kinetics Assay
4.6. Toxicity Assays
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dawood, F.S.; Iuliano, A.D.; Reed, C.; Meltzer, M.I.; Shay, D.K.; Cheng, P.Y.; Bandaranayake, D.; Breiman, R.F.; Brooks, W.A.; Buchy, P.; et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: A modelling study. Lancet Infect. Dis. 2012, 12, 687–695. [Google Scholar] [CrossRef]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26 (Suppl. S4), D49–D53. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.J.; Gregory, V.; Douglas, A.R.; Lin, Y.P. The evolution of human influenza viruses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.B.; Saunders, V.A. Virology: Principles and Applications; John Wiley & Sons: Chichester, UK; Hoboken, NJ, USA, 2007. [Google Scholar]
- Palese P, S.M. Orthomyxoviridae: The viruses and their replication. Fields Virol. 2007, 1647–1690. [Google Scholar]
- von Itzstein, M.; Wu, W.Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Van Phan, T.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef]
- Li, W.; Escarpe, P.A.; Eisenberg, E.J.; Cundy, K.C.; Sweet, C.; Jakeman, K.J.; Merson, J.; Lew, W.; Williams, M.; Zhang, L.; et al. Identification of GS 4104 as an orally bioavailable prodrug of the influenza virus neuraminidase inhibitor GS 4071. Antimicrob. Agents Chemother. 1998, 42, 647–653. [Google Scholar] [CrossRef]
- Colman, P.M. Influenza virus neuraminidase: Structure, antibodies, and inhibitors. Protein Sci. 1994, 3, 1687–1696. [Google Scholar] [CrossRef]
- Taylor, N.R.; von Itzstein, M. Molecular modeling studies on ligand binding to sialidase from influenza virus and the mechanism of catalysis. J. Med. Chem. 1994, 37, 616–624. [Google Scholar] [CrossRef]
- Gamblin, S.J.; Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 2010, 285, 28403–28409. [Google Scholar] [CrossRef]
- Russell, R.J.; Haire, L.F.; Stevens, D.J.; Collins, P.J.; Lin, Y.P.; Blackburn, G.M.; Hay, A.J.; Gamblin, S.J.; Skehel, J.J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 2006, 443, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, J.Z. Selective binding of antiinfluenza drugs and their analogues to ‘open’ and ‘closed’ conformations of H5N1 neuraminidase. J. Phys. Chem. B 2010, 114, 12958–12964. [Google Scholar] [CrossRef] [PubMed]
- Lackenby, A.; Besselaar, T.G.; Daniels, R.S.; Fry, A.; Gregory, V.; Gubareva, L.V.; Huang, W.; Hurt, A.C.; Leang, S.K.; Lee, R.T.C.; et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors and status of novel antivirals, 2016–2017. Antivir. Res. 2018, 157, 38–46. [Google Scholar] [CrossRef]
- Toledo-Rueda, W.; Rosas-Murrieta, N.H.; Munoz-Medina, J.E.; Gonzalez-Bonilla, C.R.; Reyes-Leyva, J.; Santos-Lopez, G. Antiviral resistance markers in influenza virus sequences in Mexico, 2000–2017. Infect. Drug Resist. 2018, 11, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martinez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Colman, P.M.; Hoyne, P.A.; Lawrence, M.C. Sequence and structure alignment of paramyxovirus hemagglutinin-neuraminidase with influenza virus neuraminidase. J. Virol. 1993, 67, 2972–2980. [Google Scholar] [CrossRef]
- Wang, M.; Qi, J.; Liu, Y.; Vavricka, C.J.; Wu, Y.; Li, Q.; Gao, G.F. Influenza A virus N5 neuraminidase has an extended 150-cavity. J. Virol. 2011, 85, 8431–8435. [Google Scholar] [CrossRef][Green Version]
- Gubareva, L.V.; Robinson, M.J.; Bethell, R.C.; Webster, R.G. Catalytic and framework mutations in the neuraminidase active site of influenza viruses that are resistant to 4-guanidino-Neu5Ac2en. J. Virol. 1997, 71, 3385–3390. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Xu, X.; Zhu, X.; Dwek, R.A.; Stevens, J.; Wilson, I.A. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J. Virol. 2008, 82, 10493–10501. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; McBride, R.; Nycholat, C.M.; Yu, W.; Paulson, J.C.; Wilson, I.A. Influenza virus neuraminidases with reduced enzymatic activity that avidly bind sialic Acid receptors. J. Virol. 2012, 86, 13371–13383. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Malbari, K.D.; Chintakrindi, A.S.; Ganji, L.R.; Gohil, D.J.; Kothari, S.T.; Joshi, M.V.; Kanyalkar, M.A. Structure-aided drug development of potential neuraminidase inhibitors against pandemic H1N1 exploring alternate binding mechanism. Mol. Divers. 2019, 23, 927–951. [Google Scholar] [CrossRef] [PubMed]
- Finley, J.B.; Atigadda, V.R.; Duarte, F.; Zhao, J.J.; Brouillette, W.J.; Air, G.M.; Luo, M. Novel aromatic inhibitors of influenza virus neuraminidase make selective interactions with conserved residues and water molecules in the active site. J. Mol. Biol. 1999, 293, 1107–1119. [Google Scholar] [CrossRef]
- Hsu, K.C.; Hung, H.C.; Horng, J.T.; Fang, M.Y.; Chang, C.Y.; Li, L.T.; Chen, I.J.; Chen, Y.C.; Chou, D.L.; Chang, C.W.; et al. Parallel screening of wild-type and drug-resistant targets for anti-resistance neuraminidase inhibitors. PLoS ONE 2013, 8, e56704. [Google Scholar] [CrossRef] [PubMed]
- Anuwongcharoen, N.; Shoombuatong, W.; Tantimongcolwat, T.; Prachayasittikul, V.; Nantasenamat, C. Exploring the chemical space of influenza neuraminidase inhibitors. PeerJ 2016, 4, e1958. [Google Scholar] [CrossRef]
- Collins, P.J.; Haire, L.F.; Lin, Y.P.; Liu, J.; Russell, R.J.; Walker, P.A.; Skehel, J.J.; Martin, S.R.; Hay, A.J.; Gamblin, S.J. Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature 2008, 453, 1258–1261. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Maring, C.J.; Stoll, V.S.; Zhao, C.; Sun, M.; Krueger, A.C.; Stewart, K.D.; Madigan, D.L.; Kati, W.M.; Xu, Y.; Carrick, R.J.; et al. Structure-based characterization and optimization of novel hydrophobic binding interactions in a series of pyrrolidine influenza neuraminidase inhibitors. J. Med. Chem. 2005, 48, 3980–3990. [Google Scholar] [CrossRef] [PubMed]
- Stoll, V.; Stewart, K.D.; Maring, C.J.; Muchmore, S.; Giranda, V.; Gu, Y.G.; Wang, G.; Chen, Y.; Sun, M.; Zhao, C.; et al. Influenza neuraminidase inhibitors: Structure-based design of a novel inhibitor series. Biochemistry 2003, 42, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Luo, M. Structural biology: Antiviral drugs fit for a purpose. Nature 2006, 443, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Varghese, J.N.; Colman, P.M.; van Donkelaar, A.; Blick, T.J.; Sahasrabudhe, A.; McKimm-Breschkin, J.L. Structural evidence for a second sialic acid binding site in avian influenza virus neuraminidases. Proc. Natl. Acad. Sci. USA 1997, 94, 11808–11812. [Google Scholar] [CrossRef]
- Prachanronarong, K.L.; Özen, A.; Thayer, K.M.; Yilmaz, L.S.; Zeldovich, K.B.; Bolon, D.N.; Kowalik, T.F.; Jensen, J.D.; Finberg, R.W.; Wang, J.P.; et al. Molecular basis for differential patterns of drug resistance in influenza N1 and N2 neuraminidase. J. Chem. Theory Comput. 2016, 12, 6098–6108. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, J.; Li, W.; Wang, X.; Xu, J.; Xie, J.; Tao, K.; Shen, L.; Zhang, R. Molecular docking of potential inhibitors for influenza H7N9. Comput. Math. Methods Med. 2015, 2015, 480764. [Google Scholar] [CrossRef]
- Starkey, I.D.; Mahmoudian, M.; Noble, D.; Smith, P.W.; Cherry, P.C.; Howes, P.D.; Sollis, S.L. Synthesis and influenza virus sialidase inhibitory activity of the 5-desacetamido analogue of 2,3-didehydro-2,4-dideoxy-4-guanidinyl-N-acetylneuraminic acid (GG167). Tetrahedron Lett. 1995, 36, 299–302. [Google Scholar] [CrossRef]
- Smith, P.W.; Starkey, I.D.; Howes, P.D.; Sollis, S.L.; Keeling, S.P.; Cherry, P.C.; von Itzstein, M.; Wu, W.Y.; Jin, B. Synthesis and influenza virus sialidase inhibitory activity of analogues of 4-guanidino-Neu5Ac2en (GG167) with modified 5-substituents. Eur. J. Med. Chem. 1996, 31, 143–150. [Google Scholar] [CrossRef]
- Shie, J.J.; Fang, J.M.; Lai, P.T.; Wen, W.H.; Wang, S.Y.; Cheng, Y.S.; Tsai, K.C.; Yang, A.S.; Wong, C.H. A practical synthesis of zanamivir phosphonate congeners with potent anti-influenza activity. J. Am. Chem. Soc. 2011, 133, 17959–17965. [Google Scholar] [CrossRef]
- Feng, E.; Shin, W.-J.; Zhu, X.; Li, J.; Ye, D.; Wang, J.; Zheng, M.; Zuo, J.-P.; No, K.T.; Liu, X.; et al. Structure-based design and synthesis of C-1- and C-4-modified analogs of zanamivir as neuraminidase inhibitors. J. Med. Chem. 2013, 56, 671–684. [Google Scholar] [CrossRef]
- Kongkamnerd, J.; Milani, A.; Cattoli, G.; Terregino, C.; Capua, I.; Beneduce, L.; Gallotta, A.; Pengo, P.; Fassina, G.; Miertus, S.; et al. A screening assay for neuraminidase inhibitors using neuraminidases N1 and N3 from a baculovirus expression system. J. Enzym. Inhib. Med. Chem. 2012, 27, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.W.; Sollis, S.L.; Howes, P.D.; Cherry, P.C.; Starkey, I.D.; Cobley, K.N.; Weston, H.; Scicinski, J.; Merritt, A.; Whittington, A.; et al. Dihydropyrancarboxamides related to zanamivir: A new series of inhibitors of influenza virus sialidases. 1. Discovery, synthesis, biological activity, and structure-activity relationships of 4-guanidino- and 4-amino-4H-pyran-6-carboxamides. J. Med. Chem. 1998, 41, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Adak, A.K.; Ponnapalli, K.; Lin, C.H.; Hsu, K.C.; Yang, J.M.; Hsu, T.A.; Lin, C.C. Design and synthesis of 1,2,3-triazole-containing N-acyl zanamivir analogs as potent neuraminidase inhibitors. Eur. J. Med. Chem. 2016, 123, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, P.G.; Coomber, B.A.; Evans, D.N.; Jack, T.I.; Fulton, H.E.; Wonacott, A.J.; Colman, P.; Varghese, J. Sialidase inhibitors related to zanamivir. Further SAR studies of 4-amino-4H-pyran-2-carboxylic acid-6-propylamides. Bioorg. Med. Chem. Lett. 2001, 11, 669–673. [Google Scholar] [CrossRef]
- Lin, C.-H.; Chang, T.-C.; Das, A.; Fang, M.-Y.; Hung, H.-C.; Hsu, K.-C.; Yang, J.-M.; von Itzstein, M.; Mong, K.K.T.; Hsu, T.-A.; et al. Synthesis of acylguanidine zanamivir derivatives as neuraminidase inhibitors and the evaluation of their bio-activities. Org. Biomol. Chem. 2013, 11, 3943–3948. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.M.; Cadman, A.; Gor, D.; Dempsey, M.; Walters, M.; Candlin, A.; Tisdale, M.; Morley, P.J.; Owens, I.J.; Fenton, R.J.; et al. Zanamivir susceptibility monitoring and characterization of influenza virus clinical isolates obtained during phase II clinical efficacy studies. Antimicrob. Agents Chemother. 2000, 44, 78–87. [Google Scholar] [CrossRef][Green Version]
- Wetherall, N.T.; Trivedi, T.; Zeller, J.; Hodges-Savola, C.; McKimm-Breschkin, J.L.; Zambon, M.; Hayden, F.G. Evaluation of neuraminidase enzyme assays using different substrates to measure susceptibility of influenza virus clinical isolates to neuraminidase inhibitors: Report of the neuraminidase inhibitor susceptibility network. J. Clin. Microbiol. 2003, 41, 742–750. [Google Scholar] [CrossRef]
- Marathe, B.M.; Leveque, V.; Klumpp, K.; Webster, R.G.; Govorkova, E.A. Determination of neuraminidase kinetic constants using whole influenza virus preparations and correction for spectroscopic interference by a fluorogenic substrate. PLoS ONE 2013, 8, e71401. [Google Scholar] [CrossRef]
- Govorkova, E.A.; Leneva, I.A.; Goloubeva, O.G.; Bush, K.; Webster, R.G. Comparison of efficacies of RWJ-270201, zanamivir, and oseltamivir against H5N1, H9N2, and other avian influenza viruses. Antimicrob. Agents Chemother. 2001, 45, 2723–2732. [Google Scholar] [CrossRef]
- Weight, A.K.; Haldar, J.; Alvarez de Cienfuegos, L.; Gubareva, L.V.; Tumpey, T.M.; Chen, J.; Klibanov, A.M. Attaching zanamivir to a polymer markedly enhances its activity against drug-resistant strains of influenza a virus. J. Pharm. Sci. 2011, 100, 831–835. [Google Scholar] [CrossRef]
- Baer, A.; Kehn-Hall, K. Viral concentration determination through plaque assays: Using traditional and novel overlay systems. J. Vis. Exp. 2014, 93, e52065. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S.; Sakai-Tagawa, Y.; Kiso, M.; Goto, H.; Kawakami, C.; Mitamura, K.; Sugaya, N.; Suzuki, Y.; Kawaoka, Y. Enhanced expression of an alpha2,6-linked sialic acid on MDCK cells improves isolation of human influenza viruses and evaluation of their sensitivity to a neuraminidase inhibitor. J. Clin. Microbiol. 2005, 43, 4139–4146. [Google Scholar] [CrossRef] [PubMed]
- Matrosovich, M.; Matrosovich, T.; Garten, W.; Klenk, H.D. New low-viscosity overlay medium for viral plaque assays. Virol. J. 2006, 3, 63. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Pizzorno, A.; Abed, Y.; Boivin, G. Influenza virus resistance to neuraminidase inhibitors. Antivir. Res. 2013, 98, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Kode, S.S.; Pawar, S.D.; Tare, D.S.; Keng, S.S.; Hurt, A.C.; Mullick, J. A novel I117T substitution in neuraminidase of highly pathogenic avian influenza H5N1 virus conferring reduced susceptibility to oseltamivir and zanamivir. Vet. Microbiol. 2019, 235, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.S.; Jeong, J.H.; Kwon, J.J.; Ahn, S.J.; Lloren, K.K.S.; Kwon, H.I.; Chae, H.B.; Hwang, J.; Kim, M.H.; Kim, C.J.; et al. Screening for neuraminidase inhibitor resistance markers among avian influenza viruses of the N4, N5, N6, and N8 neuraminidase subtypes. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Little, K.; Leang, S.K.; Butler, J.; Baas, C.; Harrower, B.; Mosse, J.; Barr, I.G.; Hurt, A.C. Zanamivir-resistant influenza viruses with Q136K or Q136R neuraminidase residue mutations can arise during MDCK cell culture creating challenges for antiviral susceptibility monitoring. Eurosurveillance 2015, 20. [Google Scholar] [CrossRef]
- Roth, H.J.; Eger, K.; Troschütz, R. Pharmazeutische Chemie II: Arzneistoffanalyse-Reaktivität, Stabilität, Analytik, 3rd ed.; Georg Thieme Verlag, Stuttgart: New York, NY, USA, 1981; pp. 273–284. [Google Scholar]
- Brinkevich, S.D.; Boreko, E.I.; Savinova, O.V.; Pavlova, N.I.; Shadyro, O.I. Radical-regulating and antiviral properties of ascorbic acid and its derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 2424–2427. [Google Scholar] [CrossRef]
- Wang, H.; Xu, R.; Shi, Y.; Si, L.; Jiao, P.; Fan, Z.; Han, X.; Wu, X.; Zhou, X.; Yu, F.; et al. Design, synthesis and biological evaluation of novel L-ascorbic acid-conjugated pentacyclic triterpene derivatives as potential influenza virus entry inhibitors. Eur. J. Med. Chem. 2016, 110, 376–388. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI viral genomes resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. Bioedit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Batool, S.; Mushtaq, G.; Kamal, W.; Kamal, M.A. Pharmacophore-based virtual screening for identification of novel neuraminidase inhibitors and verification of inhibitory activity by molecular docking. Med. Chem. 2016, 12, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Neri-Bazan, R.M.; Garcia-Machorro, J.; Mendez-Luna, D.; Tolentino-Lopez, L.E.; Martinez-Ramos, F.; Padilla, M., II; Aguilar-Faisal, L.; Soriano-Ursua, M.A.; Trujillo-Ferrara, J.G.; Fragoso-Vazquez, M.J.; et al. Design, in silico studies, synthesis and in vitro evaluation of oseltamivir derivatives as inhibitors of neuraminidase from influenza A virus H1N1. Eur. J. Med. Chem. 2017, 128, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Racaniello, V.R.; Palese, P. Isolation of influenza C virus recombinants. J. Virol. 1979, 32, 1006–1014. [Google Scholar] [CrossRef]
- Potier, M.; Mameli, L.; Belisle, M.; Dallaire, L.; Melancon, S.B. Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-alpha-D-N-acetylneuraminate) substrate. Anal. Biochem. 1979, 94, 287–296. [Google Scholar] [CrossRef]
- Leang, S.K.; Hurt, A.C. Fluorescence-based Neuraminidase Inhibition Assay to Assess the Susceptibility of Influenza Viruses to The Neuraminidase Inhibitor Class of Antivirals. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Mungall, B.A.; Xu, X.; Klimov, A. Assaying susceptibility of avian and other influenza A viruses to zanamivir: Comparison of fluorescent and chemiluminescent neuraminidase assays. Avian Dis. 2003, 47, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Tu, V.; Abed, Y.; Barbeau, X.; Carbonneau, J.; Fage, C.; Lague, P.; Boivin, G. The I427T neuraminidase (NA) substitution, located outside the NA active site of an influenza A(H1N1)pdm09 variant with reduced susceptibility to NA inhibitors, alters NA properties and impairs viral fitness. Antivir. Res. 2017, 137, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Govorkova, E.A.; Ilyushina, N.A.; McClaren, J.L.; Naipospos, T.S.; Douangngeun, B.; Webster, R.G. Susceptibility of highly pathogenic H5N1 influenza viruses to the neuraminidase inhibitor oseltamivir differs in vitro and in a mouse model. Antimicrob. Agents Chemother. 2009, 53, 3088–3096. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.L.; Ilyushina, N.A.; Salomon, R.; Hoffmann, E.; Webster, R.G.; Govorkova, E.A. Neuraminidase inhibitor-resistant recombinant A/Vietnam/1203/04 (H5N1) influenza viruses retain their replication efficiency and pathogenicity in vitro and in vivo. J. Virol. 2007, 81, 12418–12426. [Google Scholar] [CrossRef]
- Eisenbrand, G.; Pool-Zobel, B.; Baker, V.; Balls, M.; Blaauboer, B.J.; Boobis, A.; Carere, A.; Kevekordes, S.; Lhuguenot, J.C.; Pieters, R.; et al. Methods of in vitro toxicology. Food Chem. Toxicol 2002, 40, 193–236. [Google Scholar] [CrossRef]
Sample Availability: Samples of all compounds are commercially available from chemical substance supply companies or commercial fine chemical catalogues sine none was synthesized by the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Influenza A Virus Strain | |||
|---|---|---|---|---|
| A/PR/18/34 H1N1 | A/CHK/MX/1994 H5N2 | |||
| Ki (mM) | IC50 (mM) | Ki (mM) | IC50 (mM) | |
| AAMOL | 15.3 | 57 ± 1.1 * | 24.37 | 73 ± 1.18 * |
| CIV | 26.43 | 32 ± 1.5 * | 17.03 | 17.5 ± 1.2 * |
| ORP | 10.5 | 23 ± 1.2 * | 3.4 | 15.3 ± 1.13 * |
| FMOL | ND 1 | ND 1 | ND 1 | 6.4 ± 1.1 * |
| STL | ND 1 | ND 1 | ND 1 | 24.8 ± 1.3 * |
| Zanamivir | 2.58 × 10 −6 | 1.19 × 10−6 ± 0.043 | 1.59 × 10−5 | 2.84 × 10−6 ± 0.047 |
| Molecule | Influenza A Virus Strain A/CHK/MX/1994 H5N2 |
|---|---|
| IC50 (mM) | |
| AAMOL | ND |
| FMOL | 0.059 ± 1.28 * |
| STL | 0.243 ± 1.59 * |
| CIV | - |
| ORP | 0.157 ± 1.50 * |
| Zanamivir | 7.41 × 10−4 ± 1.24 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Márquez-Domínguez, L.; Reyes-Leyva, J.; Herrera-Camacho, I.; Santos-López, G.; Scior, T. Five Novel Non-Sialic Acid-Like Scaffolds Inhibit In Vitro H1N1 and H5N2 Neuraminidase Activity of Influenza a Virus. Molecules 2020, 25, 4248. https://doi.org/10.3390/molecules25184248
Márquez-Domínguez L, Reyes-Leyva J, Herrera-Camacho I, Santos-López G, Scior T. Five Novel Non-Sialic Acid-Like Scaffolds Inhibit In Vitro H1N1 and H5N2 Neuraminidase Activity of Influenza a Virus. Molecules. 2020; 25(18):4248. https://doi.org/10.3390/molecules25184248
Chicago/Turabian StyleMárquez-Domínguez, Luis, Julio Reyes-Leyva, Irma Herrera-Camacho, Gerardo Santos-López, and Thomas Scior. 2020. "Five Novel Non-Sialic Acid-Like Scaffolds Inhibit In Vitro H1N1 and H5N2 Neuraminidase Activity of Influenza a Virus" Molecules 25, no. 18: 4248. https://doi.org/10.3390/molecules25184248
APA StyleMárquez-Domínguez, L., Reyes-Leyva, J., Herrera-Camacho, I., Santos-López, G., & Scior, T. (2020). Five Novel Non-Sialic Acid-Like Scaffolds Inhibit In Vitro H1N1 and H5N2 Neuraminidase Activity of Influenza a Virus. Molecules, 25(18), 4248. https://doi.org/10.3390/molecules25184248