
Probing the Highly Disparate Dual Inhibitory Mechanisms of Novel Quinazoline Derivatives against Mycobacterium tuberculosis Protein Kinases A and B


Abstract
1. Introduction
2. Results and Discussion
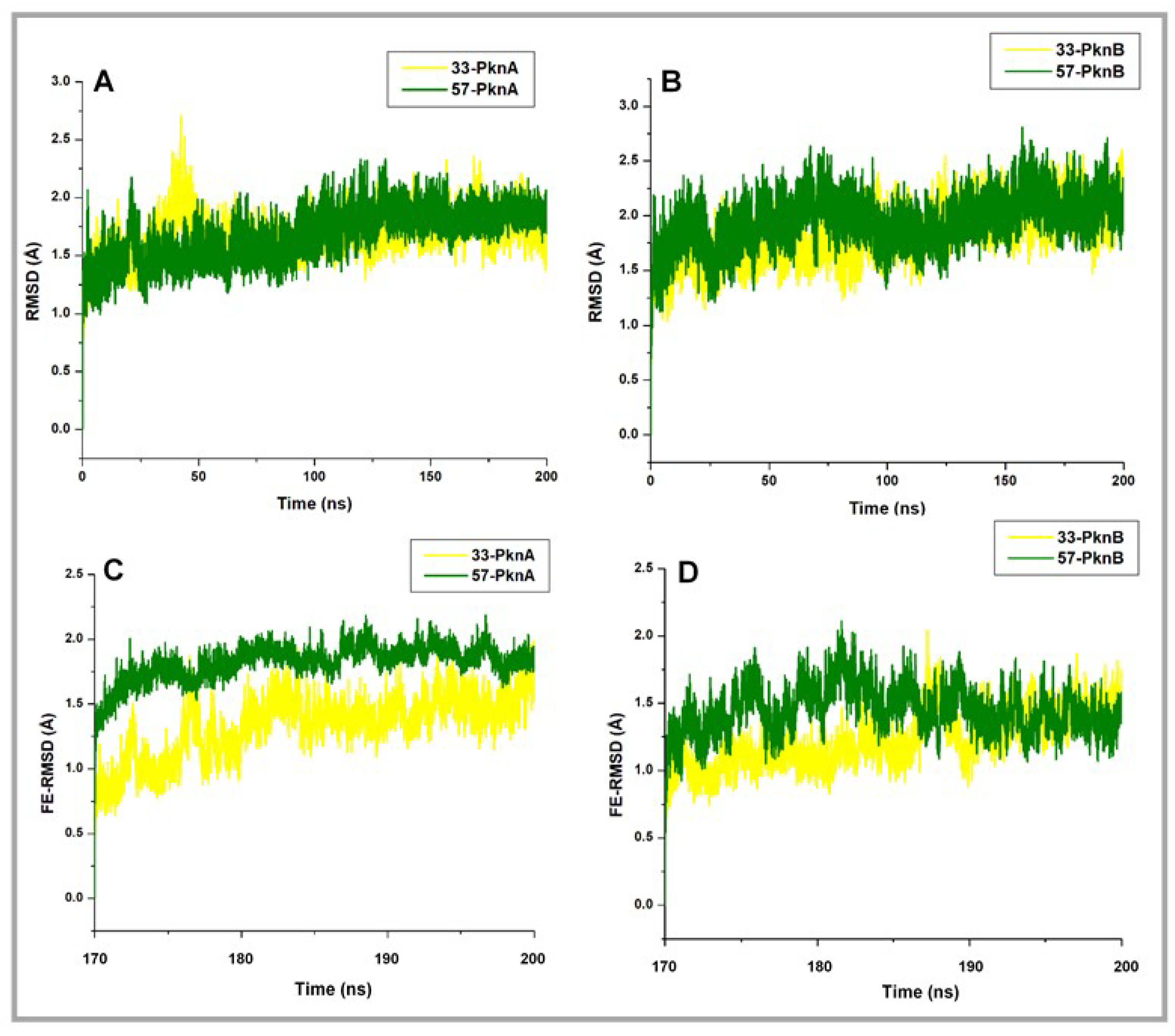
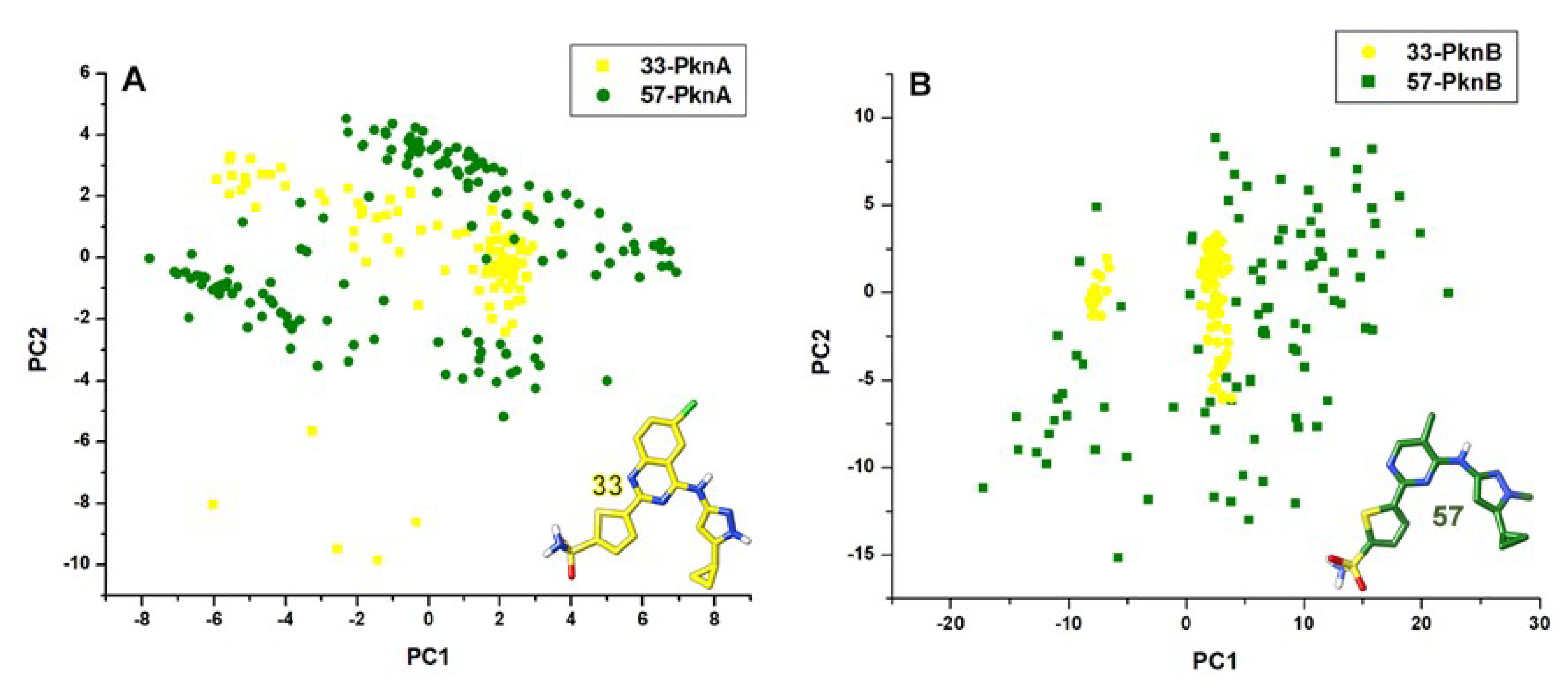
2.1. Conformational Stability and Ligand-Induced Variations
2.2. Dynamic Cross Correlation Matrix (DCCM)
2.3. Systematic Analyses of Binding Dynamics to Understand Differential Inhibitory Mechanisms
2.4. Estimation of Binding Affinities and Per-Residue Energy Decomposition
3. Computational Methods
3.1. Preparation of Ligand and Protein Starting Structures
3.2. Docking and MD Simulations
3.3. Binding Energy Calculations and Decomposition of Per-Residue Energy
3.4. Analyses of Dynamic Cross-Correlation Matrix (DCCM)
3.5. Principal Component Analyses (PCA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Khan, M.Z.; Kaur, P.; Nandicoori, V.K. Targeting the messengers: Serine/threonine protein kinases as potential targets for antimycobacterial drug development. IUBMB Life 2018. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2018; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Pal, S.K.; Pandey, D.; Fakir, N.A.; Rathod, S.; Sinha, D.; SivaKumar, S.; Sinha, P.; Periera, M.; Balgam, S.; et al. PknB remains an essential and a conserved target for drug development in susceptible and MDR strains of M. Tuberculosis. Ann. Clin. Microbiol. Antimicrob. 2017. [Google Scholar] [CrossRef] [PubMed]
- Av-Gay, Y.; Everett, M. The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 2000, 5, 238–244. [Google Scholar] [CrossRef]
- Hanks, S.K.; Hunter, T. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E.; et al. Deciphering the biology of mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Thangamani, L.; Manivel, G.; Kumar, P.; Piramanayagam, S. Molecular docking, molecular dynamics and MM/PBSA studies of FDA approved drugs for protein kinase a of Mycobacterium tuberculosis; application insights of drug repurposing. Inform. Med. Unlocked 2019. [Google Scholar] [CrossRef]
- Carette, X.; Platig, J.; Young, D.C.; Helmel, M.; Young, A.T.; Wang, Z.; Potluri, L.P.; Moody, C.S.; Zeng, J.; Prisic, S.; et al. Multisystem analysis of Mycobacterium tuberculosis reveals kinase-dependent remodeling of the pathogen-environment interface. MBio 2018. [Google Scholar] [CrossRef]
- Kang, C.M.; Abbott, D.W.; Sang, T.P.; Dascher, C.C.; Cantley, L.C.; Husson, R.N. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: Substrate identification and regulation of cell shape. Genes Dev. 2005. [Google Scholar] [CrossRef]
- Shah, I.M.; Laaberki, M.H.; Popham, D.L.; Dworkin, J. A Eukaryotic-like Ser/Thr Kinase Signals Bacteria to Exit Dormancy in Response to Peptidoglycan Fragments. Cell 2008. [Google Scholar] [CrossRef]
- Chawla, Y.; Upadhyay, S.; Khan, S.; Nagarajan, S.N.; Forti, F.; Nandicoori, V.K. Protein kinase B (PknB) of Mycobacterium tuberculosis is essential for growth of the pathogen in vitro as well as for survival within the host. J. Biol. Chem. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ortega, C.; Liao, R.; Anderson, L.N.; Rustad, T.; Ollodart, A.R.; Wright, A.T.; Sherman, D.R.; Grundner, C. Mycobacterium tuberculosis Ser/Thr Protein Kinase B Mediates an Oxygen-Dependent Replication Switch. PLoS Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.; Saint-Joanis, B.; Barilone, N.; Jackson, M.; Gicquel, B.; Cole, S.T.; Alzari, P.M. The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J. Bacteriol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Molle, V.; Kremer, L. Division and cell envelope regulation by Ser/Thr phosphorylation: Mycobacterium shows the way. Mol. Microbiol. 2010, 75, 1064–1077. [Google Scholar] [CrossRef] [PubMed]
- Barthe, P.; Mukamolova, G.V.; Roumestand, C.; Cohen-Gonsaud, M. The structure of PknB extracellular PASTA domain from mycobacterium tuberculosis suggests a ligand-dependent kinase activation. Structure 2010. [Google Scholar] [CrossRef]
- Gee, C.L.; Papavinasasundaram, K.G.; Blair, S.R.; Baer, C.E.; Falick, A.M.; King, D.S.; Griffin, J.E.; Venghatakrishnan, H.; Zukauskas, A.; Wei, J.R.; et al. A phosphorylated pseudokinase complex controls cell wall synthesis in mycobacteria. Sci. Signal. 2012. [Google Scholar] [CrossRef]
- Pereira, S.F.F.; Goss, L.; Dworkin, J. Eukaryote-Like Serine/Threonine Kinases and Phosphatases in Bacteria. Microbiol. Mol. Biol. Rev. 2011. [Google Scholar] [CrossRef]
- Bellinzoni, M.; Wehenkel, A.M.; Durán, R.; Alzari, P.M. Novel mechanistic insights into physiological signaling pathways mediated by mycobacterial Ser/Thr protein kinases. Genes Immun. 2019, 20, 383–393. [Google Scholar] [CrossRef]
- Prisic, S.; Dankwa, S.; Schwartz, D.; Chou, M.F.; Locasale, J.W.; Kang, C.M.; Bemis, G.; Church, G.M.; Steene, H.; Husson, R.N. Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. USA 2010. [Google Scholar] [CrossRef]
- Meeske, A.J.; Riley, E.P.; Robins, W.P.; Uehara, T.; Mekalanos, J.J.; Kahne, D.; Walker, S.; Kruse, A.C.; Bernhardt, T.G.; Rudner, D.Z. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 2016. [Google Scholar] [CrossRef]
- Narayan, A.; Sachdeva, P.; Sharma, K.; Saini, A.K.; Tyagi, A.K.; Singh, Y. Serine threonine protein kinases of mycobacterial genus: Phylogeny to function. Physiol. Genom. 2007. [Google Scholar] [CrossRef] [PubMed]
- Young, T.A.; Delagoutte, B.; Endrizzi, J.A.; Falick, A.M.; Alber, T. Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases. Nat. Struct. Biol. 2003. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Datta, P.; Kundu, M.; Basu, J. The serine/threonine kinase PknB of Mycobacterium tuberculosis phosphorylates PBPA, a penicillin-binding protein required for cell division. Microbiology 2006. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.M.; Ng, H.L.; Alber, T. A Conserved Dimer and Global Conformational Changes in the Structure of apo-PknE Ser/Thr Protein Kinase from Mycobacterium tuberculosis. J. Mol. Biol. 2006. [Google Scholar] [CrossRef]
- Wang, T.; Bemis, G.; Hanzelka, B.; Zuccola, H.; Wynn, M.; Moody, C.S.; Green, J.; Locher, C.; Liu, A.; Gao, H.; et al. Mtb PKNA/PKNB Dual Inhibition Provides Selectivity Advantages for Inhibitor Design to Minimize Host Kinase Interactions. ACS Med. Chem. Lett. 2017. [Google Scholar] [CrossRef]
- Villarino, A.; Duran, R.; Wehenkel, A.; Fernandez, P.; England, P.; Brodin, P.; Cole, S.T.; Zimny-Arndt, U.; Jungblut, P.R.; Cerveñansky, C.; et al. Proteomic identification of M. tuberculosis protein kinase substrates: PknB recruits GarA, a FHA domain-containing protein, through activation loop-mediated interactions. J. Mol. Biol. 2005. [Google Scholar] [CrossRef]
- Wehenkel, A.; Bellinzoni, M.; Graña, M.; Duran, R.; Villarino, A.; Fernandez, P.; Andre-Leroux, G.; England, P.; Takiff, H.; Cerveñansky, C.; et al. Mycobacterial Ser/Thr protein kinases and phosphatases: Physiological roles and therapeutic potential. Biochimica et Biophysica Acta Proteins Proteomics 2008, 1784, 193–202. [Google Scholar] [CrossRef]
- Lombana, T.N.; Echols, N.; Good, M.C.; Thomsen, N.D.; Ng, H.L.; Greenstein, A.E.; Falick, A.M.; King, D.S.; Alber, T. Allosteric activation mechanism of the Mycobacterium tuberculosis receptor Ser/Thr protein Kinase, PknB. Structure 2010. [Google Scholar] [CrossRef]
- Xu, M.; Yu, L.; Wan, B.; Yu, L.; Huang, Q. Predicting inactive conformations of protein kinases using active structures: Conformational selection of Type-II inhibitors. PLoS ONE 2011. [Google Scholar] [CrossRef]
- Baer, C.E.; Iavarone, A.T.; Alber, T.; Sassetti, C.M. Biochemical and spatial coincidence in the provisional Ser/Thr protein kinase interaction network of Mycobacterium tuberculosis. J. Biol. Chem. 2014. [Google Scholar] [CrossRef]
- Nagarajan, S.N.; Upadhyay, S.; Chawla, Y.; Khan, S.; Naz, S.; Subramanian, J.; Gandotra, S.; Nandicoori, V.K. Protein Kinase a (PknA) of Mycobacterium tuberculosis is independently activated and is critical for growth in vitro and survival of the pathogen in the host. J. Biol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Wong, D.; Zheng, X.; Poirier, V.; Bach, H.; Hmama, Z.; Av-Gay, Y. Protein kinase and phosphatase signaling in Mycobacterium tuberculosis physiology and pathogenesis. Biochimica et Biophysica Acta Proteins Proteomics 2010, 1804, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.M.; Bouloc, N.; Buxton, R.S.; Chugh, J.; Lougheed, K.E.A.; Osborne, S.A.; Saxty, B.; Smerdon, S.J.; Taylor, D.L.; Whalley, D. Substituted aminopyrimidine protein kinase B (PknB) inhibitors show activity against Mycobacterium tuberculosis. Bioorganic Med. Chem. Lett. 2012. [Google Scholar] [CrossRef] [PubMed]
- Nott, T.J.; Kelly, G.; Stach, L.; Li, J.; Westcott, S.; Patel, D.; Hunt, D.M.; Howell, S.; Buxton, R.S.; O’Hare, H.M.; et al. An intramolecular switch regulates phosphoindependent FHA domain interactions in Mycobacterium tuberculosis. Sci. Signal. 2009. [Google Scholar] [CrossRef]
- Ventura, M.; Rieck, B.; Boldrin, F.; Degiacomi, G.; Bellinzoni, M.; Barilone, N.; Alzaidi, F.; Alzari, P.M.; Manganelli, R.; O’Hare, H.M. GarA is an essential regulator of metabolism in Mycobacterium tuberculosis. Mol. Microbiol. 2013. [Google Scholar] [CrossRef]
- Veyron-Churlet, R.; Zanella-Cléon, I.; Cohen-Gonsaud, M.; Molle, V.; Kremer, L. Phosphorylation of the Mycobacterium tuberculosis β-ketoacyl-acyl carrier protein reductase MabA regulates mycolic acid biosynthesis. J. Biol. Chem. 2010. [Google Scholar] [CrossRef]
- Vilchèze, C.; Molle, V.; Carrère-Kremer, S.; Leiba, J.; Mourey, L.; Shenai, S.; Baronian, G.; Tufariello, J.; Hartman, T.; Veyron-Churlet, R.; et al. Phosphorylation of KasB Regulates Virulence and Acid-Fastness in Mycobacterium tuberculosis. PLoS Pathog. 2014. [Google Scholar] [CrossRef]
- Székely, R.; Wáczek, F.; Szabadkai, I.; Németh, G.; Hegymegi-Barakonyi, B.; Eros, D.; Szokol, B.; Pató, J.; Hafenbradl, D.; Satchell, J.; et al. A novel drug discovery concept for tuberculosis: Inhibition of bacterial and host cell signalling. Immunol. Lett. 2008. [Google Scholar] [CrossRef]
- Ortiz-Lombardía, M.; Pompeo, F.; Boitel, B.; Alzari, P.M. Crystal structure of the catalytic domain of the PknB serine/threonine kinase from Mycobacterium tuberculosis. J. Biol. Chem. 2003. [Google Scholar] [CrossRef]
- Gupta, V.; Naqvi, A.; Tanwar, J.; Mishra, K. Molecular Modeling study of Protein Kinase PKnB from Mycobacterium tuberculosis with derivatives of 1, 3, 4- Thiadiazoles. Nat. Preced. 2011. [Google Scholar] [CrossRef]
- Xu, J.; Wang, J.; Zhou, J.; Xu, C.; Huang, B.; Xing, Y.; Wang, B.; Luo, R.; Wang, Y.; You, X.; et al. A novel protein kinase inhibitor IMB-YH-8 with anti-tuberculosis activity. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Ladbury, J.E. Hydrogen bonds in protein-ligand complexes. In Protein-Ligand Interactions: From Molecular Recognition to Drug Design; Wiley-VCH: Weinheim, Germany, 2005; ISBN 9783527601813. [Google Scholar]
- Majewski, M.; Ruiz-Carmona, S.; Barril, X. An investigation of structural stability in protein-ligand complexes reveals the balance between order and disorder. Commun. Chem. 2019. [Google Scholar] [CrossRef]
- Menéndez, C.A.; Accordino, S.R.; Gerbino, D.C.; Appignanesi, G.A. Hydrogen bond dynamic propensity studies for protein binding and drug design. PLoS ONE 2016. [Google Scholar] [CrossRef] [PubMed]
- Prisic, S.; Husson, R.N. Mycobacterium tuberculosis Serine/Threonine Protein Kinases. Microbiol. Spectr. 2014. [Google Scholar] [CrossRef]
- Alber, T. Signaling mechanisms of the Mycobacterium tuberculosis receptor Ser/Thr protein kinases. Curr. Opin. Struct. Biol. 2009, 19, 650–657. [Google Scholar] [CrossRef][Green Version]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galziskaya, O. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Olotu, F.A.; Munsamy, G.; Soliman, M.E.S. Does Size Really Matter? Probing the Efficacy of Structural Reduction in the Optimization of Bioderived Compounds—A Computational “Proof-of-Concept”. Comput. Struct. Biotechnol. J. 2018, 16, 573–586. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, L.; Zhang, H.; Feng, W.; Tan, T. Lid closure mechanism of Yarrowia lipolytica lipase in methanol investigated by molecular dynamics simulation. J. Chem. Inf. Model. 2014. [Google Scholar] [CrossRef]
- Singh, S.K.; Das, A. The n → π* interaction: A rapidly emerging non-covalent interaction. Phys. Chem. Chem. Phys. 2015. [Google Scholar] [CrossRef]
- Olotu, F.A.; Soliman, M.E.S. From mutational inactivation to aberrant gain-of-function: Unraveling the structural basis of mutant p53 oncogenic transition. J. Cell. Biochem. 2018, 119. [Google Scholar] [CrossRef]
- Akher, F.B.; Farrokhzadeh, A.; Olotu, F.A.; Agoni, C.; Soliman, M.E.S. The irony of chirality–unveiling the distinct mechanistic binding and activities of 1-(3-(4-amino-5-(7-methoxy-5-methylbenzo [b] thiophen-2-yl)-7 H-pyrrolo [2, 3-d] pyrimidin-7-yl) pyrrolidin-1-yl) prop-2-en-1-one enantiomers as irreversible covalent FGFR4 inhibitors. Org. Biomol. Chem. 2019, 17, 1176–1190. [Google Scholar] [CrossRef]
- Frisch, M.J.G.; Trucks, W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian, 16; Scientific Research Publisher: Wuhan, China, 2016. [Google Scholar]
- Case, D.A. Amber 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Seifert, E. OriginPro 9.1: Scientific Data Analysis and Graphing Software—Software Review. J. Chem. Inf. Model. 2014, 54, 1552. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Discovery Studio 2016 Client; San Diego Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Sittel, F.; Jain, A.; Stock, G. Principal component analysis of molecular dynamics: On the use of Cartesian vs. internal coordinates. J. Chem. Phys. 2014, 141. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Kirschner, K.N. Principal component and clustering analysis on molecular dynamics data of the ribosomal L11 23S subdomain. J. Mol. Model. 2013, 19, 539–549. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Post Equilibration | PknA | 33-PknA | 57-PknA | PknB | 33-PknB | 57-PknB |
|---|---|---|---|---|---|---|
| Whole Structure | ||||||
| Whole RMSD | 2.42 ± 0.16 | 1.40 ± 0.2 | 1.14 ± 0.11 | 2.01 ± 0.30 | 1.39 ± 0.25 | 1.44 ± 0.19 |
| FE-RMSD (Å) | 2.38 ± 0.35 | 1.33 ± 0.23 | 1.82 ± 0.13 | 1.95 ± 0.29 | 1.24 ± 0.20 | 1.45 ± 0.18 |
| FE-RMSF (Å) | 1.41 ± 0.69 | 0.89 ± 0.40 | 1.00 ± 0.42 | 1.11 ± 0.34 | 0.79 ± 0.46 | 1.05 ± 0.55 |
| Ki (nM) [26] | <8 | >4000 | <1 | >4000 | ||
| Nucleotide Binding Pockets | ||||||
| FE-RMSD (Å) | 2.31 ± 0.31 | 1.51 ± 0.17 | 2.00 ± 0.23 | 2.42 ± 0.11 | 1.59 ± 0.26 | 2.34 ± 0.43 |
| FE-RoG (Å) | 13.10 ± 0.28 | 9.27 ± 0.13 | 10.26 ± 0.11 | 12.34 ± 0.03 | 8.54 ± 0.09 | 11.93 ± 0.14 |
| Structural Fluctuation (Å) | ||||||
|---|---|---|---|---|---|---|
| Structural Components | PknA | 33-PknA | 57-PknA | PknB | 33-PknB | 57-PknB |
| P-loop | 2.36 ± 0.25 | 1.50 ± 0.36 | 1.46 ± 0.36 | 2.14 ± 0.14 | 0.92 ± 0.22 | 1.14 ± 0.37 |
| Helix C | 1.59 ± 0.51 | 1.11 ± 0.30 | 1.35 ± 0.50 | 1.52 ± 0.43 | 0.88 ± 0.32 | 1.48 ± 0.57 |
| Catalytic loop | 0.88 ± 0.10 | 0.70 ± 0.12 | 0.71 ± 0.13 | 0.69 ± 0.06 | 0.48 ± 0.04 | 0.66 ± 0.22 |
| DFG-motif | 1.41 ± 0.14 | 0.55 ± 0.02 | 0.72 ± 0.10 | 1.01 ± 0.04 | 0.48 ± 0.02 | 0.92 ± 0.05 |
| Binding Free Energy Analysis | ||||
|---|---|---|---|---|
| Energy Components (kcal mol−1) | 33-PknA | 57-PknA | 33-PknB | 57-PknB |
| ∆EvdW | −46.38 ± 0.18 | −28.25 ± 0.10 | −51.02 ± 0.12 | −29.79 ± 0.11 |
| ∆Eele | −24.64 ± 0.22 | −11.18 ± 0.08 | −29.15 ± 0.19 | −24.99 ± 0.42 |
| ∆Ggas | −71.02 ± 0.26 | −39.43 ± 0.10 | −80.17 ± 0.20 | −54.78 ± 0.83 |
| ∆Gele,sol(GB) | 28.36 ± 0.13 | 29.89 ± 0.05 | 31.70 ± 0.13 | 35.26 ± 0.45 |
| ∆Gnp,sol | −5.85 ± 0.01 | −3.0 ± 0.01 | −6.26 ± 0.01 | −5.57 ± 0.07 |
| ∆Gsol | 22.51 ± 0.13 | 26.89 ± 0.05 | 25.44 ± 0.13 | 29.99 ± 0.39 |
| ∆H | −48.51 ± 0.16 | −12.54 ± 0.06 | −54.73 ± 0.14 | −24.79 ± 0.52 |
| T∆S | −2.76 ± 0.08 | −7.46 ± 0.03 | −4.73 ± 0.06 | −3.89 ± 0.26 |
| ∆Gbind | −45.75 ± 0.24 | −20.03 ± 0.15 | −50.0 ± 0.14 | −20.9 ± 0.15 |
| Ki (nM) | <8 | >4000 | <1 | >4000 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olotu, F.A.; Soliman, M.E. Probing the Highly Disparate Dual Inhibitory Mechanisms of Novel Quinazoline Derivatives against Mycobacterium tuberculosis Protein Kinases A and B. Molecules 2020, 25, 4247. https://doi.org/10.3390/molecules25184247
Olotu FA, Soliman ME. Probing the Highly Disparate Dual Inhibitory Mechanisms of Novel Quinazoline Derivatives against Mycobacterium tuberculosis Protein Kinases A and B. Molecules. 2020; 25(18):4247. https://doi.org/10.3390/molecules25184247
Chicago/Turabian StyleOlotu, Fisayo A., and Mahmoud E. Soliman. 2020. "Probing the Highly Disparate Dual Inhibitory Mechanisms of Novel Quinazoline Derivatives against Mycobacterium tuberculosis Protein Kinases A and B" Molecules 25, no. 18: 4247. https://doi.org/10.3390/molecules25184247
APA StyleOlotu, F. A., & Soliman, M. E. (2020). Probing the Highly Disparate Dual Inhibitory Mechanisms of Novel Quinazoline Derivatives against Mycobacterium tuberculosis Protein Kinases A and B. Molecules, 25(18), 4247. https://doi.org/10.3390/molecules25184247