
A New Method for the Synthesis of 3-Thiocyanatopyrazolo[1,5-a]pyrimidines


Abstract
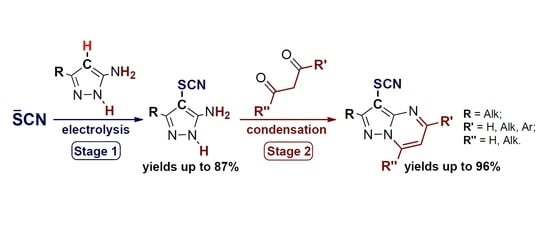
1. Introduction
2. Results and Discussion
2.1. Anodic C–H Thiocyanation of 5-Aminopyrazoles (Scheme 3, Stage 1)
2.1.1. CV Data
2.1.2. Effect of Electrolysis Conditions on the Yield of the Target Product
2.1.3. Synthesis of Target Products
2.2. Condensation of 4-Thiocyanato-5-Aminopyrazoles with 1,3-Dicarbonyl Compounds (or their Derivatives) (Scheme 3, Stage 2)
2.2.1. Effect of Conditions on the Target Product Yield
2.2.2. Synthesis of Target Products
3. Materials and Methods
3.1. General Information
3.2. Anodic C–H Thiocyanation of 5-Aminopyrazoles (Scheme 3, Stage 1)
3.2.1. Effect of Electrolysis Conditions on the Yield of the Target Product
3.2.2. Anodic Thiocyanation of Azoles 1a,b
3.2.3. Anodic Thiocyanation of Azoles 1a,b on a Larger Scale
3.3. Condensation of 4-Thiocyanato-5-Aminopyrazoles with 1,3-Dicarbonyl Compounds (or Their Derivatives) (Scheme 3, stage 2)
3.3.1. Effect of Condensation Conditions on the Yield of the Target Product
3.3.2. Synthesis of Target Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davies, H.M.L.; Morton, D. C–H Functionalization: Collaborative Methods to Redefine Chemical Logic. Angew. Chem. Int. Ed. 2014, 53, 10256–10258. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Morton, D. Recent Advances in C–H Functionalization. J. Org. Chem. 2016, 81, 343–350. [Google Scholar] [CrossRef]
- Charushin, V.N.; Chupakhin, O.N. Nucleophilic C–H functionalization of arenes: A contribution to green chemistry. Russ. Chem. Bull. 2019, 68, 453–471. [Google Scholar] [CrossRef]
- Petrosyan, V.A. Reactions of anodic and chemical aromatic substitution. Mendeleev Commun. 2011, 21, 115–121. [Google Scholar] [CrossRef]
- Schepochkin, A.V.; Chupakhin, O.N.; Charushin, V.N.; Petrosyan, V.A. Direct nucleophilic functionalization of C(sp2)–H-bonds in arenes and hetarenes by electrochemical methods. Russ. Chem. Rev. 2013, 82, 747–771. [Google Scholar] [CrossRef]
- Kärkäs, M.D. Electrochemical strategies for C–H functionalization and C–N bond formation. Chem. Soc. Rev. 2018, 47, 5786–5865. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.-L.; Fang, P.; Mei, T.-S. Recent Advances in Organic Electrochemical C–H Functionalization. Chin. J. Chem. 2018, 36, 338–352. [Google Scholar] [CrossRef]
- Nikoofar, K. A Brief on Thiocyanation of N-Activated Arenes and N-Bearing Heteroaromatic Compounds. Chem. Sci. Trans. 2013, 3, 691–700. [Google Scholar] [CrossRef][Green Version]
- Rezayati, S.; Ramazani, A. A review on electrophilic thiocyanation of aromatic and heteroaromatic compounds. Tetrahedron 2020, 131382. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Terent’Ev, A.O.; Ramenskaya, G.V.; Grammatikova, N.E.; Rodionova, G.M.; Ilovaiskii, A.I. Synthesis and Antifungal Activity of Arylthiocyanates. Pharm. Chem. J. 2013, 47, 422–425. [Google Scholar] [CrossRef]
- Fedij, V.; Gajda-Christopher, A.; Huckabee-Brian, K.; Moon-Brian, S.; Porter-Kenneth, T.; Sobieray-Denis, M.; Stuk-Timothy, L.E.E.; Tait-Bradley, D.; Wemple-James, N. Methods of Making Dihydropyrone HIV Protease Inhibitors. U.S. Patent 6380400 B1, 30 April 2002. [Google Scholar]
- Chao, M.N.; Matiuzzi, C.E.; Storey, M.; Li, C.; Szajnman, S.H.; Docampo, R.; Moreno, S.N.J.; Rodriguez, J.B. Aryloxyethyl Thiocyanates Are Potent Growth Inhibitors of Trypanosoma cruzi and Toxoplasma gondii. ChemMedChem 2015, 10, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Fortes, M.P.; da Silva, P.B.; da Silva, T.G.; Kaufman, T.S.; Militão, G.C.; Silveira, C.D.C. Synthesis and preliminary evaluation of 3-thiocyanato-1H-indoles as potential anticancer agents. Eur. J. Med. Chem. 2016, 118, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Kokorekin, V.A.; Sigacheva, V.L.; Petrosyan, V.A. New data on heteroarene thiocyanation by anodic oxidation of NH4SCN. The processes of electroinduced nucleophilic aromatic substitution of hydrogen. Tetrahedron Lett. 2014, 55, 4306–4309. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Yaubasarova, R.R.; Neverov, S.V.; Petrosyan, V.A. Reactivity of electrogenerated thiocyanogen in the thiocyanation of pyrazolo[1,5-a]pyrimidines. Mendeleev Commun. 2016, 26, 413–414. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Yaubasarova, R.R.; Neverov, S.V.; Petrosyan, V.A.; Neverov, S.V. Electrooxidative C-H Functionalization of Heteroarenes. Thiocyanation of Pyrazolo[1,5-a]pyrimidines. Eur. J. Org. Chem. 2019, 2019, 4233–4238. [Google Scholar] [CrossRef]
- Yaubasarova, R.R.; Kokorekin, V.A.; Ramenskaya, G.V.; Petrosyan, V.A. Double electrooxidative C–H functionalization of (het)arenes with thiocyanate and 4-nitropyrazolate ions. Mendeleev Commun. 2019, 29, 334–336. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Mel’Nikova, E.I.; Yaubasarova, R.R.; Gorpinchenko, N.V.; Petrosyan, V.A. “Metal-free” electrooxidative C–H thiocyanation of arenes. Russ. Chem. Bull. 2019, 68, 2140–2141. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.-G.; Jiang, H.; Sun, L. A low-cost electrochemical thio- and selenocyanation strategy for electron-rich arenes under catalyst- and oxidant-free conditions. RSC Adv. 2018, 8, 22042–22045. [Google Scholar] [CrossRef]
- Fotouhi, L.; Nikoofar, K. Electrochemical thiocyanation of nitrogen-containing aromatic and heteroaromatic compounds. Tetrahedron Lett. 2013, 54, 2903–2905. [Google Scholar] [CrossRef]
- Gitkis, A.; Becker, J.Y. Anodic thiocyanation of mono- and disubstituted aromatic compounds. Electrochimica Acta 2010, 55, 5854–5859. [Google Scholar] [CrossRef]
- Cataldo, F. New Developments in the Study of the Structure of Parathiocyanogen: (SCN)x, An Inorganic Polymer. J. Inorg. Organomet. Polym. 1997, 7, 35–50. [Google Scholar] [CrossRef]
- Cauquis, G.; Pierre, G. Les proprietes electrochimiques de l’ion thiocyanate et du thiocyanogene au sein de l’acetonitrile et la thiocyanation par voie electrochimique. CR Acad. Sci. Paris 1968, 266, 883–886. [Google Scholar]
- Cherukupalli, S.; Karpoormath, R.; Chandrasekaran, B.; Hampannavar, G.A.; Thapliyal, N.; Palakollu, V.N. An insight on synthetic and medicinal aspects of pyrazolo[1,5-a]pyrimidine scaffold. Eur. J. Med. Chem. 2017, 126, 298–352. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, J.; Portilla, J.; Abonía, R.; Insuasty, B.; Nogueras, M.; Cobo, J. Synthesis of novel 5-amino-1-aroylpyrazoles. Tetrahedron Lett. 2008, 49, 5943–5945. [Google Scholar] [CrossRef]
- Frizzo, C.P.; Martins, M.A.P.; Marzari, M.R.B.; Campos, P.T.; Claramunt, R.M.; Garcia, M.A.; Sanz, D.; Alkorta, I.; Elguero, J. Structural studies of 2-methyl-7-substituted pyrazolo[1,5-a]pyrimidines. J. Heterocycl. Chem. 2010, 47, 1259–1268. [Google Scholar] [CrossRef]
- Martins, M.A.P.; Scapin, E.; Frizzo, C.P.; Rosa, F.A.; Bonacorso, H.G.; Zanatta, N. 2-methyl-7-substituted pyrazolo[1,5-a]pyrimidines: Highly regioselective synthesis and bromination. J. Braz. Chem. Soc. 2009, 20, 205–213. [Google Scholar] [CrossRef]
- Shipps, G.W.; Rosner, K.E.; Popovici-Muller, J.; Deng, Y.; Wang, T.; Curran, P.J. Pyrazolo[1,5-a]pyrimidine Compounds as Antiviral Agents. US Patent 7196111B2, 27 March 2007. [Google Scholar]
- Nenajdenko, V.G.; Krasovsky, A.L.; Hartulyari, A.S.; Balenkova, E.S. Efficient Syntheses of New CF3-containing Diazolopyrimidines. Synthesis 2004, 2002, 0133–0137. [Google Scholar] [CrossRef]
- Terranova, E.; Fadli, A.; Lagrange, A. Compositions for Dyeing Keratin Fibers Containing Pyrazolo (1,5-a) Pyrimidine Derivatives and Dyeing Processes. U.S. Patent 6099593, 8 August 2000. [Google Scholar]
- Maquestiau, A.; Taghret, H.; Eynde, J.-J.V. Preparation and Characterization of Pyrazolo[1,5-a]Pyrimidines. Bulletin des Sociétés Chimiques Belges 2010, 101, 131–136. [Google Scholar] [CrossRef]
- Ning, X. Substituted heteroaryl compounds as well as composition and application. CN Patent 104650092, 10 November 2017. [Google Scholar]
- Terranova, E.; Fadli, A.; Lagrange, A. Compositions for dyeing keratinous fibres, containing 3-aminopyrazolo [1,5-a]pyrimidines, method of dyeing and novel 3-aminopyrazolo [1,5-a]pyrimidines. U.S. Patent 6248137, 19 June 2001. [Google Scholar]
- Kokorekin, V.A.; Melnikova, E.I.; Yaubasarova, R.R.; Petrosyan, V.A. Electrooxidative C–H thiocyanation of hetarenes: Voltammetric assessment of thiocyanogen reactivity. Mendeleev Commun. 2020, 30, 70–72. [Google Scholar] [CrossRef]
- Ali, I.; Park, S.; Jung, M.E.; Lee, N.; Bibi, M.; Chae, C.H.; Yang, K.-M.; Kim, S.-J.; Choi, G.; Lee, K. Identification of TRD-35 as Potent and Selective DRAK2 Inhibitor. Bull. Korean Chem. Soc. 2020, 41, 567–569. [Google Scholar] [CrossRef]
- Effenberger, F.; Maier, R.; Schönwälder, K.-H.; Ziegler, T. Enolether, XIII. Die Acylierung von Enolethern mit reaktiven Carbonsäure-chloriden. Eur. J. Inorg. Chem. 1982, 115, 2766–2782. [Google Scholar] [CrossRef]
- Buback, M.; Tost, W.; Hübsch, T.; Voß, E.; Tietze, L.F. Inter- and Intramolecular Hetero Diels-Alder Reactions, Part XXVI. Diastereoselectivity and Kinetics of Intermolecular Hetero Diels-Alder Reactions under High Pressure. A Significant Pressure-Induced Increase in Stereoselectivity. Eur. J. Inorg. Chem. 1989, 122, 1179–1186. [Google Scholar] [CrossRef]
- Volgraf, M.; Sellers, B.D.; Jiang, Y.; Wu, G.; Ly, C.Q.; Villemure, E.; Pastor, R.M.; Yuen, P.-W.; Lu, A.; Luo, X.; et al. Discovery of GluN2A-Selective NMDA Receptor Positive Allosteric Modulators (PAMs): Tuning Deactivation Kinetics via Structure-Based Design. J. Med. Chem. 2016, 59, 2760–2779. [Google Scholar] [CrossRef] [PubMed]
Sample Availability





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
| Entry | Conditions | Yield of 3a, % |
| 1 | Optimal 1 | 83 |
| 2 | Pt electrodes instead of GC | 72 |
| 3 | Undivided cell | 42 |
| 4 | KSCN or NaSCN instead of NH4SCN | 65 |
| 5 | MeCN instead of MeCN-H2O | 61 |
| 6 | ЕAn = 1.10 V instead of 0.90 V | 63 |
| 7 | ЕAn = 0.70 V instead of 0.90 V 2 | 80 |
 | |||
| Entry | Substrate 1 | Product 3 | Yield, % |
| 1 1 |  |  | 83 |
| 2 2 | 74 | ||
| 3 3 | 69 | ||
| 4 1 |  |  | 87 |
| 5 2 | 78 | ||
| 6 3 | 71 | ||
 | ||
| Entry | Conditions | Yield of 5aa, % |
| 1 | Optimal 1 | 77 |
| 2 | Without HCl | traces |
| 3 | AcOH instead of HCl | 36 |
| 4 | H2SO4 instead of HCl | 39 |
| 5 | 5 mL HCl instead of 2.5 mL | 75 |
| 6 | H2O-EtOH (1:4) instead of H2O | 67 |
| 7 | EtOH instead of H2O | 65 |
 | ||||
| Entry | Substrate 3 | Substrate 4 | Product 5 | Yield, % |
| 1 1 |  |  |  | 77 |
| 2 1 |  |  |  | 84 |
| 3 1 |  |  |  | 96 |
| 4 1 |  |  |  | 92 |
| 5 2 |  |  |  | 89 |
| 6 2 |  |  |  | 78 |
| 7 2 |  |  |  | 71 |
| 8 2 |  |  |  | 87 |
| 9 3 |  |  |  | 84 |
| 10 3 |  |  |  | 91 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kokorekin, V.A.; Neverov, S.V.; Kuzina, V.N.; Petrosyan, V.A. A New Method for the Synthesis of 3-Thiocyanatopyrazolo[1,5-a]pyrimidines. Molecules 2020, 25, 4169. https://doi.org/10.3390/molecules25184169
Kokorekin VA, Neverov SV, Kuzina VN, Petrosyan VA. A New Method for the Synthesis of 3-Thiocyanatopyrazolo[1,5-a]pyrimidines. Molecules. 2020; 25(18):4169. https://doi.org/10.3390/molecules25184169
Chicago/Turabian StyleKokorekin, Vladimir A., Sergey V. Neverov, Vera N. Kuzina, and Vladimir A. Petrosyan. 2020. "A New Method for the Synthesis of 3-Thiocyanatopyrazolo[1,5-a]pyrimidines" Molecules 25, no. 18: 4169. https://doi.org/10.3390/molecules25184169
APA StyleKokorekin, V. A., Neverov, S. V., Kuzina, V. N., & Petrosyan, V. A. (2020). A New Method for the Synthesis of 3-Thiocyanatopyrazolo[1,5-a]pyrimidines. Molecules, 25(18), 4169. https://doi.org/10.3390/molecules25184169